The field of genomic epidemiology focuses on using the genetic sequences of pathogens to understand patterns of transmission and spread. Viruses mutate very quickly and accumulate changes during the process of transmission from one infected individual to another. The novel coronavirus which is responsible for the emerging COVID-19 pandemic mutates at an average of about two mutations per month. After someone is exposed they will generally incubate the virus for ~5 days before symptoms develop and transmission occurs. Other research has shown that the “serial interval” of SARS-CoV-2 is ~7 days. You can think of a transmission chain as looking something like:
![]()
where, on average, we have 7 days from one infection to the next. As the virus transmits, it will mutate at this rate of two mutations per month. This means, that on average every other step in the transmission chain will have a mutation and so would look something like:
![]()
These mutations are generally really simple things. An ‘A’ might change to a ‘T’, or a ‘G’ to a ‘C’. This changes the genetic code of the virus, but it’s hard for a single letter change to do much to make the virus behave differently. However, with advances in technology, it’s become readily feasible to sequence the genome of the novel coronavirus. This works by taking a swab from someone’s nose and extracting the RNA in the sample and then determining the ‘letters’ of this RNA genome using chemistry and very powerful cameras. Each person’s coronavirus infection will yield a sequence of 30,000 ‘A’, ‘T’, ‘G’ or ‘C’ letters. We can use these sequences to reconstruct which infection is connected to which infection. As an example, if we sequenced three of these infections and found:
![]()
We could take the “genomes” ATTT, ATCT and GTCT and infer that the infection with sequence ATTT lead to the infection with sequence ATCT and this infection lead to the infection with sequence GTCT. This approach allows us learn about epidemiology and transmission in a completely novel way and can supplement more traditional contact tracing and case-based reporting.
For a few years now, we’ve been working on the Nextstrain software platform, which aims to make genomic epidemiology as rapid and as useful as possible. We had previously applied this to outbreaks like Ebola, Zika and seasonal flu. Owing to advances in technology and open data sharing, the genomes of 140 SARS-CoV-2 coronaviruses have been shared from all over the world via gisaid.org. As these genomes are shared, we download them from GISAID and incorporate them into a global map as quickly as possible and have an always up-to-date view of the genomic epidemiology of novel coronavirus at nextstrain.org/ncov.
The big picture looks like this at the moment:
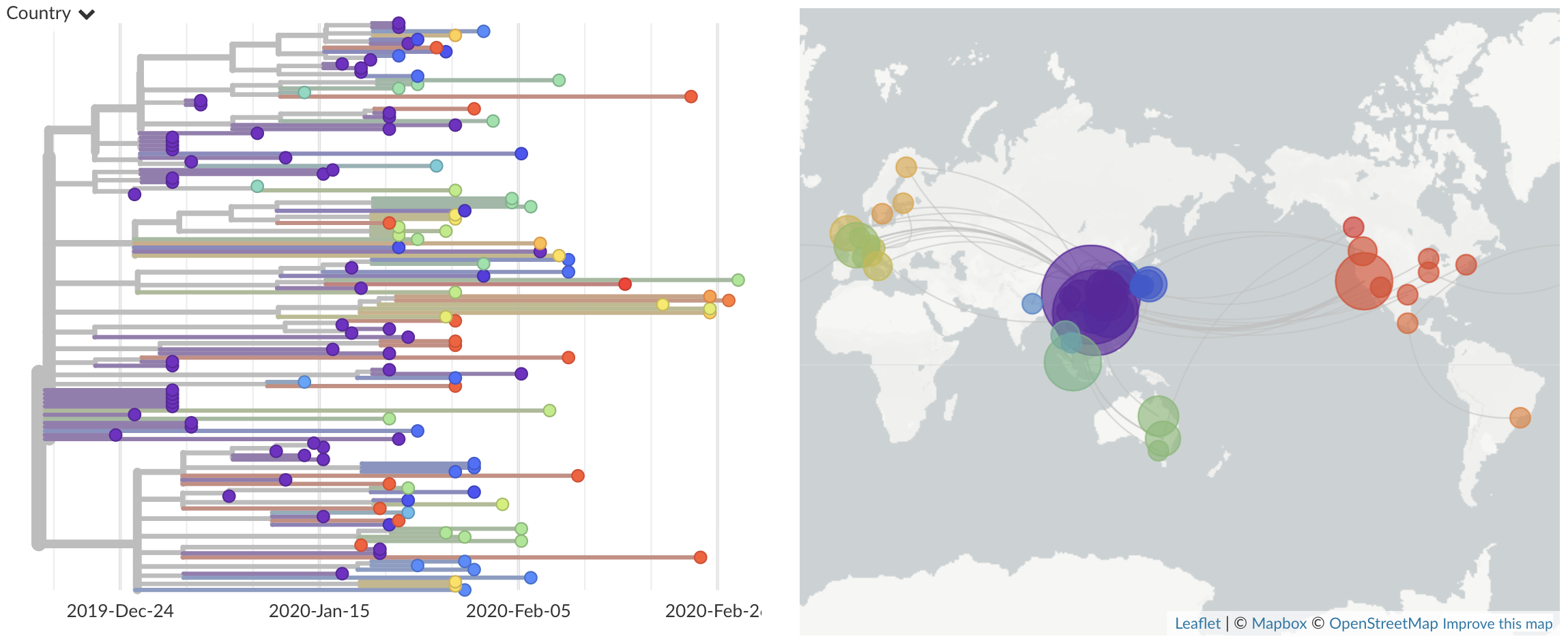
where we can see the earliest infections in Wuhan, China in purple on the left side of the tree. All these genomes from Wuhan have a common ancestor in late Nov or early Dec, suggesting that this virus has emerged recently in the human population.
The first case in the USA was called “USA/WA1/2020”. This was from a traveller directly returning from Wuhan to Snohomish County on Jan 15, with a swab collected on Jan 19. This virus was rapidly sequenced by the US CDC Division of Viral Diseases and shared publicly on Jan 24 (huge props to the CDC for this). We can zoom into the tree to place WA1 among related viruses:
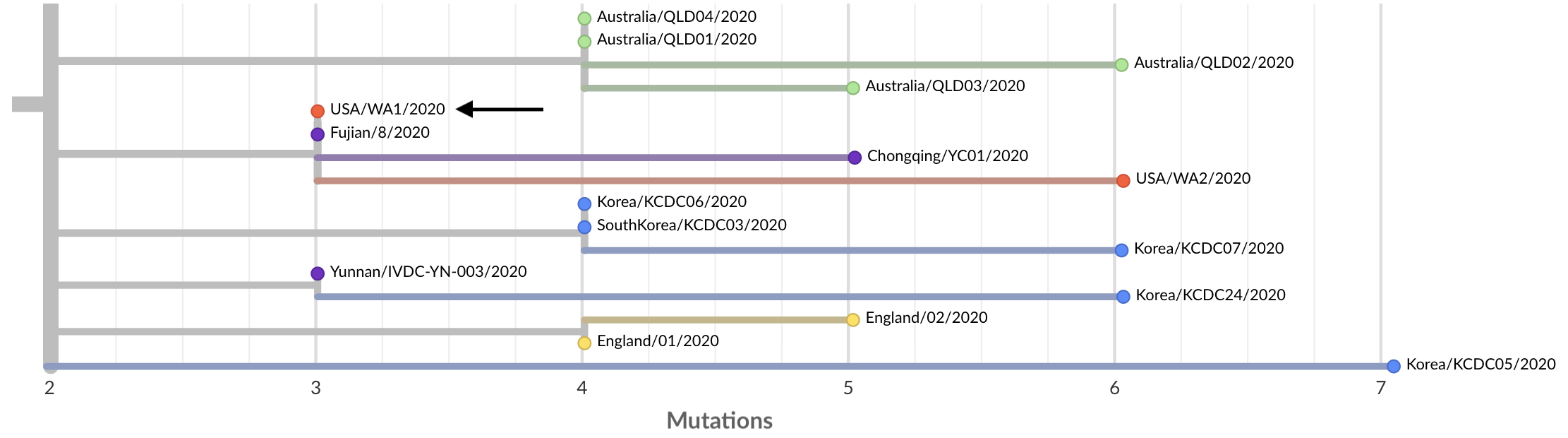
The virus has an identical genome to the virus Fujian/8/2020 sampled in Fujian on Jan 21, also labeled as a travel export from Wuhan, suggesting a close relationship between these two cases.
Last week the Seattle Flu Study started screening samples for COVID-19 as described here. Soon after starting screening we found a first positive in a sample from Snohomish County. The case was remarkable in that it was a “community case”, only the second recognized in the US, someone who had sought treatment for flu-like symptoms, been tested for flu and then sent home owing to mild disease. After this was diagnostically confirmed by Shoreline Public Health labs on Fri Feb 28 we were able to immediately get the sample USA/WA2/2020 on a sequencer and have a genome available on Sat Feb 29. The results were remarkable. The WA2 case was identical to WA1 except that it had three additional mutations.
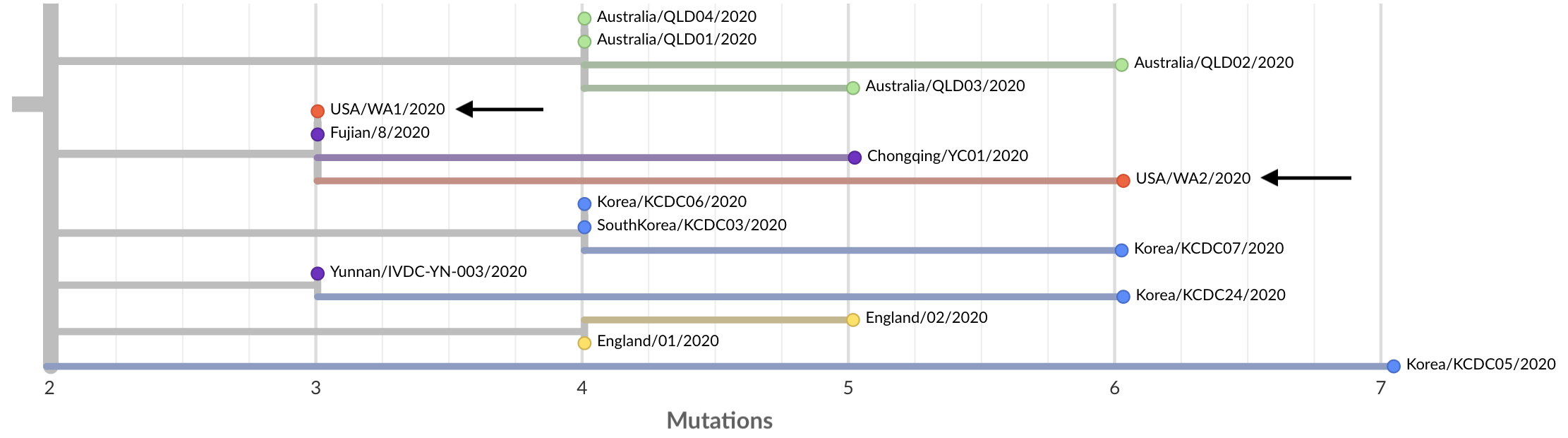
This tree structure is consistent with WA2 being a direct descendent of WA1. If this virus arrived in Snohomish County in mid-January with the WA1 traveler from Wuhan and circulated locally for 5 weeks, we’d expect exactly this pattern, where the WA2 genome is a copy of the WA1 genome except it has some mutations that have arisen over the 5 weeks that separate them.
Again, this tree structure is consistent with a transmission chain leading from WA1 to WA2, but we wanted to assess the probability of this pattern arising by chance instead of direct transmission. Scientists often try to approach this situation by thinking of a “null model”, ie if it was coincidence, how likely of a coincidence was it? Here, WA1 and WA2 share the same genetic variant at site 18060 in the virus genome, but only 2/59 sequenced viruses from China possess this variant. Given this low frequency, we’d expect probability of WA2 randomly having the same genetic variant at 2/59 = 3%. To me, this not quite conclusive evidence, but still strong evidence that WA2 is a direct descendent of WA1.
Additional evidence for the relationship between these cases comes from location. The Seattle Flu Study had screened viruses from all over the greater Seattle area, however, we got the positive hit in Snohomish County with cases less than 15 miles apart. This by itself would only be suggestive, but combined with the genetic data, is firmer evidence for continued transmission.
I’ve been referring to this scenario as “cryptic transmission”. This is a technical term meaning “undetected transmission”. Our best guess of a scenario looks something like:
![]()
We believe this may have occurred by the WA1 case having exposed someone else to the virus in the period between Jan 15 and Jan 19 before they were isolated. If this second case was mild or asymptomatic, contact tracing efforts by public health would have had difficulty detecting it. After this point, community spread occurred and was undetected due to the CDC narrow case definition that required direct travel to China or direct contact with a known case to even be considered for testing. This lack of testing was a critical error and allowed an outbreak in Snohomish County and surroundings to grow to a sizable problem before it was even detected.
Knowing that transmission was initiated on Jan 15 allows us to estimate the total number of infections that exist in this cluster today. Our preliminary analysis puts this at 570 with an 90% uncertainty interval of between 80 and 1500 infections.
Back on Feb 8, I tweeted this thought experiment:
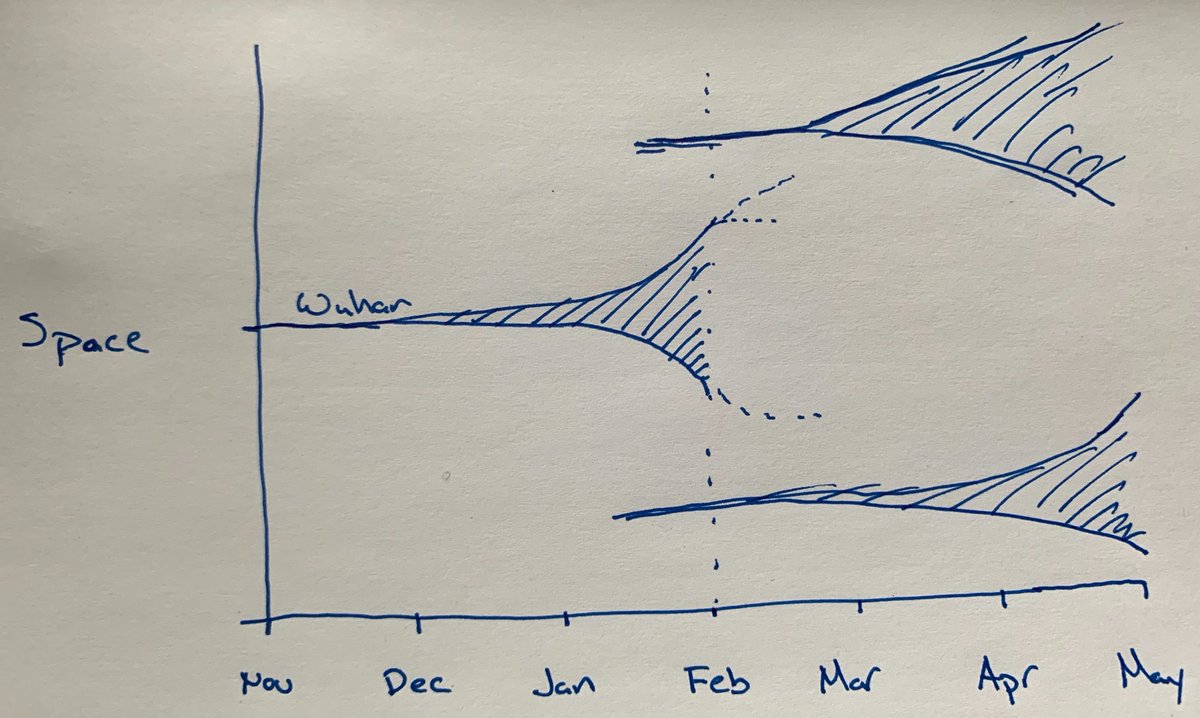
We know that Wuhan went from an index case in ~Nov-Dec 2019 to several thousand cases by mid-Jan 2020, thus going from initial seeding event to widespread local transmission in the span of ~9-10 weeks. We now believe that the Seattle area seeding event was ~Jan 15 and we’re now ~7 weeks later. I expect Seattle now to look like Wuhan around ~1 Jan, when they were reporting the first clusters of patients with unexplained viral pneumonia. We are currently estimating ~600 infections in Seattle, this matches my phylodynamic estimate of the number of infections in Wuhan on Jan 1. Three weeks later, Wuhan had thousands of infections and was put on large-scale lock-down. However, these large-scale non-pharmaceutical interventions to create social distancing had a huge impact on the resulting epidemic. China averted many millions of infections through these intervention measures and cases there have declined substantially.
This suggests that this is controllable. We’re at a critical junction right now, but we can still mitigate this substantially.
Some ways to implement non-pharmaceutical interventions include:
- Practicing social distancing, such as limiting attendance at events with large groups of people
- Working from home, if your job and employer allows it
- Staying home if you are feeling ill
- Take your temperature daily, if you develop a fever, self-isolate and call your doctor
- Implementing good hand washing practices - it is extremely important to wash hands regularly
- Covering coughs and sneezes in your elbow or tissue
- Avoiding touching your eyes, nose, and mouth with unwashed hands
- Disinfecting frequently touched surfaces, such as doorknobs
- Beginning some preparations in anticipation of social distancing or supply chain shortages, such as ensuring you have sufficient supplies of prescription medicines and ensuring you have about a 2 week supply of food and other necessary household goods.
- With these preparation in mind, it is important to not panic buy. Panic buying unnecessarily increases strain on supply chains and can make it difficult to ensure that everyone is able to get supplies that they need.
For more information please see: