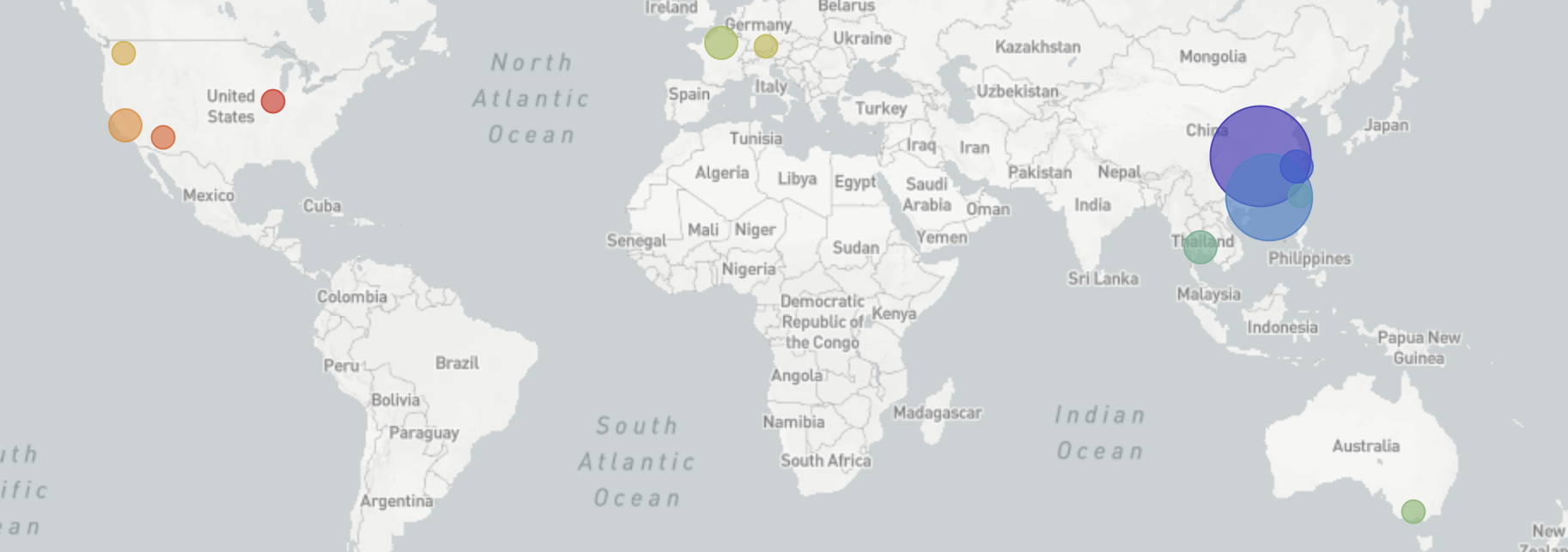
I started following what’s now referred to as “novel coronavirus (nCoV)” on Jan 6 when I started to notice reports of a cluster of viral pneumonia of unknown origin in Wuhan, China. Just 4 days later on Jan 10, a first genome was released on Virological.org only to be followed by five more the following day via GISAID.org. From very early on, it was clear that the nCoV genomes lacked the expected genetic diversity that would occur with repeated zoonotic events from a diverse animal reservoir. The simplest parsimonious explanation for this observation was that there was a single zoonotic spillover event into the human population in Wuhan between mid-Nov and mid-Dec and sustained human-to-human transmission from this point. However, at first I struggled to reconcile this lack of genetic diversity with WHO reports of “limited human-to-human” transmission. The conclusion of sustained human-to-human spread became difficult to ignore on Jan 17 when nCoV genomes from the two Thai travel cases that reported no market exposure showed the same limited genetic diversity. This genomic data represented one of the first and strongest indications of sustained epidemic spread. As this became clear to me, I spent the week of Jan 20 alerting every public health official I know.
At this moment there are 54 publicly shared viral genomes, with genomes being shared by public health and academic groups all over the world 3-6 days after sample collection. I can’t overstate how remarkable this is and what an inflection point it is for the field of genomic epidemiology. Seasonal influenza had been far ahead of the general curve, but there we were still generally seeing a ~1 month turnaround from sample collection to genome in the best of circumstances. Getting to a 3-6 day turnaround opens up huge new avenues in epidemiology.
Since the first nCoV genome was shared on Jan 10, we’ve been tracking viral transmission and evolution on nextstrain.org/ncov aiming to have ~1hr turnarounds from public deposition of genome data to inclusion in the live transmission tracking. We are also producing public situation reports describing what can be concluded from current genomic data. These reports have now been generously translated into 5 other languages by volunteers from Twitter. With groups all over the world working tirelessly to generate genomic data as rapidly as possible, I’m feeling a moral obligation to not hold up the analysis side. The entire Nextstrain team (shoutouts to Richard Neher, Emma Hodcroft, James Hadfield, Kairsten Fay, Thomas Sibley, Misja Ilcisin and Jover Lee 🙌) have come together to conduct analyses and tailor the platform for nCoV response. There’s also been a remarkable amount of sharing of pre-publication analyses on Virological.org and bioRxiv and scientific communication on Twitter. Although the situation is looking a bit dire at the moment, it’s been humbling to see scientists from all over the world break down traditional barriers to rapid scientific progress.