In 2016 and 2017, mumps outbreaks were reported in several countries, and the CDC reported 5,629 cases within the United States. Washington state has among the highest incidence rates in the country, reporting 891 confirmed cases between October 2016 and September 2017. We are collaborating with the Washington State Department of Health to sequence mumps virus samples collected from throughout the outbreak. We will use these data to determine the number and size of distinct transmission clusters, describe where distinct transmission clusters were likely introduced from, and describe how the virus spread within the state. This analysis is greatly aided by recent mumps virus sequencing efforts by the British Columbia CDC and the Broad Institute, as pooling data provides critical context.
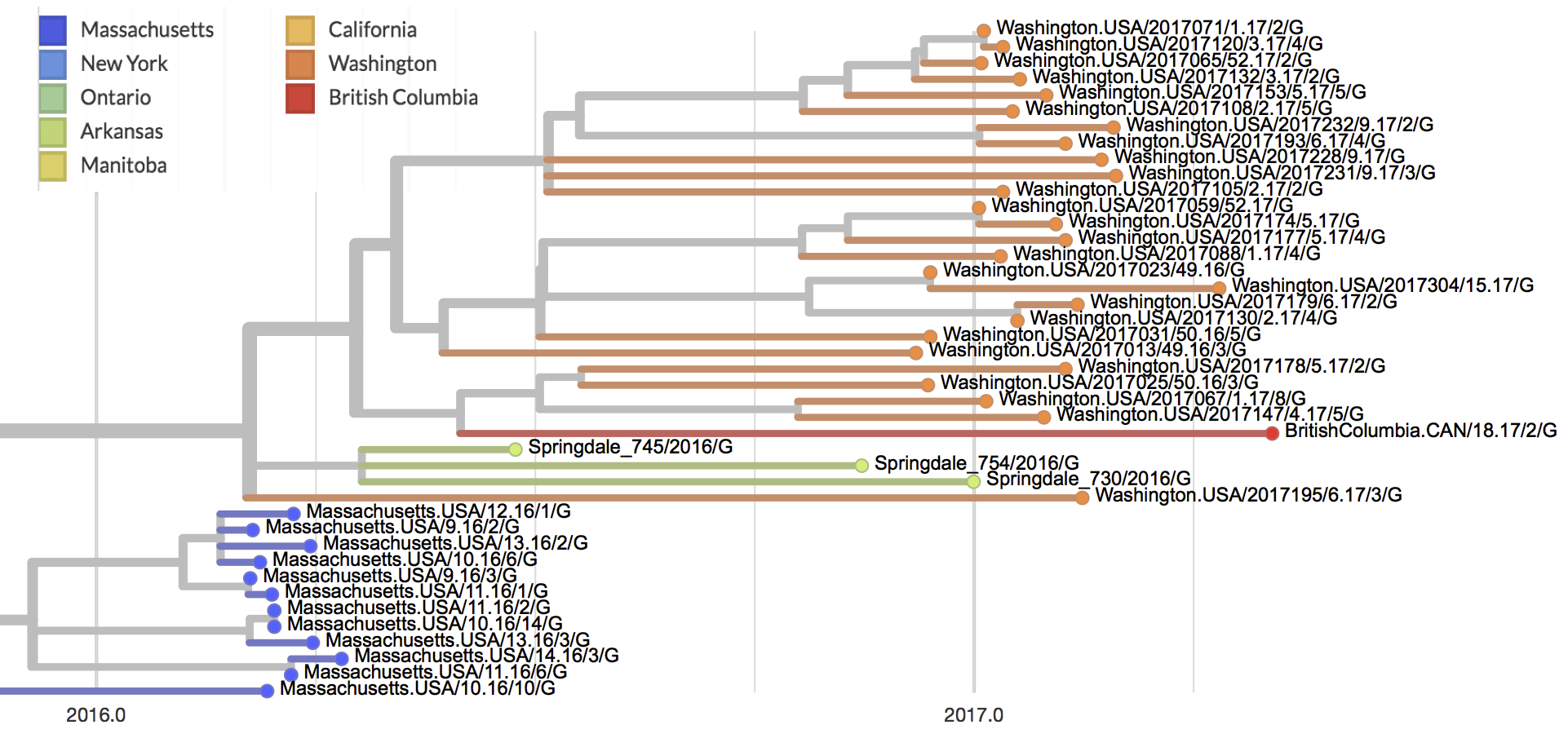
We recently completed sequencing the first batch of mumps virus genomes provided by the Washington State Department of Health and have released the first 27 draft genomes on nextstrain.org/mumps. Protocols for sample preparation and sequencing are available at github.com/blab/mumps-seq. The 27 sequences from Washington state were collected between December 2016 and April 2017. The vast majority of these sequences (25 out of 27) cluster together within a single large clade, which we will refer to as the primary outbreak clade. This finding indicates that the majority of transmission within Washington likely occurred due to a single introduction of mumps followed by sustained person-to-person transmission. This conclusion may change as we sequence further viruses, which may provide evidence for additional clades of circulating viruses. The primary outbreak clade is closely related to all sequenced viruses sampled from the Arkansas outbreak, and is nested within the diversity of viruses sampled during Massachusetts outbreaks in 2016. Thus, we hypothesize that mumps outbreaks within the US are likely related. We also find a single genome from Washington state that clusters outside of the primary outbreak clade, which could represent a separate introduction of Mumps virus to Washington which did not yield sustained person-to-person transmission. We note that further sequencing and more sophisticated genomic analysis is required to confidently determine the total number of introductions that occurred, and how each introduction contributed to observed transmission. Finally, the primary outbreak clade also includes a single sequence from British Columbia, providing evidence for some degree of transmission between the US and Canada.
We are aiming to sequencing ~100 clinical samples in total, and will continue sequencing and adding data to nextstrain.org/mumps in the coming months. Stay tuned!