We just had a paper published in MBE on comparing different phylogenetic models. Generally, its much easier to estimate the parameters of a particular model (including a level of uncertainty on these estimates) than it is to assess which model of several is the “best” fit. More complicated models will necessarily better fit the data at hand. However, overly complicated models will be brittle and not generalize well when new data is confronted. Thus, there is a trade-off between number of effective parameters and a model’s likelihood. Assessing this trade-off can be quite difficult computationally.
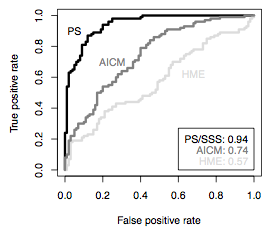
Here, Guy Baele put in a lot of effort to implement very general methods of “thermodynamic integration” (as it’s known in the phylogenetics literature) or “path sampling” (as it’s known in the statistics literature) into BEAST. The basic idea is very similar to what Ed and I did with the food web analysis paper, calculate a marginal likelihood by comparing the posterior likelihood across MCMC chains at different “temperatures”. The MBE paper shows that this estimate is much more accurate and repeatable than the very popular harmonic mean estimator of marginal likelihood.
For the paper, I did most of the work involving measuring Akaike information criteria (AIC) in a Bayesian Monte Carlo context, and thus called AICM. This measure has the computational advantage that, like the harmonic mean estimator, it can be computed directly from MCMC runs produced by BEAST or another piece of software, without resorting to a second, more-complicated, analysis. We find that AICM does not match path sampling for accuracy, but definitely beats the harmonic mean. It’s currently implemented in BEAST XML and will be built in to the next version of Tracer.