The influenza virus shows a remarkable capacity to evolve to escape human immunity. Many other viruses, like measles, do not have this capacity. After infection with measles, a person gains life-long immunity to the virus, and hence measles has become constrained to be a childhood infection. Continual antigenic evolution in influenza necessitates frequent vaccine updates to provide sufficient protection to circulating strains.
Antigenic differences between strains are commonly quantified using the hemagglutination inhibition (HI) assay, which measures the ability of antibodies created against one strain to interfere with virus from another strain. The resulting HI data is represented as a sparse matrix of comparisons between viruses from strains A, B, C… and sera from strains X, Y, Z… Taken by itself, this matrix is difficult to work with. Experienced virologists can pick up the loss of reactivity between groups of viruses in the noisy HI data, but these patterns are not fully quantified.
In our new paper, available on the arXiv, we extend techniques of multidimensional scaling (MDS) pioneered by Derek Smith and colleagues for the analysis of influenza antigenic data. Here, we attempted to bring the MDS antigenic model into a fully Bayesian framework and refer to the revised technique as Bayesian MDS (BMDS). In this model, viruses and sera are represented as 2D coordinates on an antigenic map in which their pairwise distances yield expectations for the HI titers, with antigenically similar viruses lying close to one another and antigenically distant viruses lying far apart.
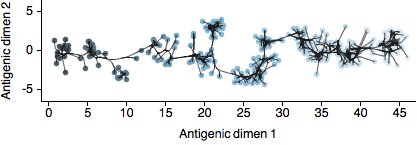
By placing antigenic cartography in a Bayesian context, we are able to integrate other data sources, most notably sequence data. In this case, genetic sequences provide an evolutionary tree relating virus strains and we assume that antigenic location evolves along this tree in a 2D diffusion process. This process imposes a prior on antigenic locations in which evolutionary similar viruses have a prior expectation of lying close to one another on the map. In the paper, we use this BMDS / diffusion model to investigate patterns of antigenic evolution in 4 circulating lineages of influenza and show that antigenic drift determines to a large degree incidence patterns across time and across lineages.
The paper is also up on GitHub, which I’ll keep updating as it goes through the review process. The BMDS model is implemented in the software package BEAST and is available in the latest source code. I hope to provide tutorials on running the BMDS model in the not-to-distant future. Mirrored as a guest post on Haldane’s Sieve.