
We have a new preprint up on bioRxiv describing within-host evolution of H5N1 avian influenza viruses sampled from humans and domestic ducks in Cambodia!
Why should we care about avian flu in Cambodia?
We’ve been collaborating with the Institut Pasteur du Cambodge (IPC) to try to understand how H5N1 avian influenza viruses evolve during cross-species transmission. H5N1 viruses are highly pathogenic avian influenza viruses that naturally circulate in aquatic birds, but can cross species barrier and cause spillover infections in humans. Although H5N1 viruses aren’t currently capable of transmitting among humans efficiently, laboratory studies suggest that only a few mutations might be required to render them human-adapted. Influenza viruses generate lots of genetic diversity within a single infected host, leading to concern that continued spillover infection might one day facilitate human adaptation. Unfortunately, assessing cross-species transmission risk is really difficult, and the data we have currently comes from animal experiments and modelling studies. Because spillover infection is rare, it has been difficult to study how H5N1 viruses might evolve during natural infection, in either humans or birds.
H5 avian influenza viruses are endemic in Cambodia, and are frequently detected in domestic birds in live bird markets throughout the country. The Institut Pasteur du Cambodge conducts regular poultry market surveillance and outbreak investigation for avian influenza viruses, making it an incredible resource for studying avian influenza virus circulation and evolution. IPC and collaborators in China previously generated deep sequence data from a unique dataset of 8 humans and 5 domestic ducks infected with H5N1 and sampled in Cambodia between 2010 and 2014. This dataset provided a great opportunity to examine whether human adaptation occurs during natural spillover infection. Although a couple other studies have looked at within-host diversity in infected humans, data from infected poultry has been more difficult to come by. Because this dataset also included data from infected poultry collected in the same geographic location and time, we could compare the evolutionary patterns we observed in humans to those in birds.
What can within-host diversity tell us about the potential for H5N1 to adapt to humans?
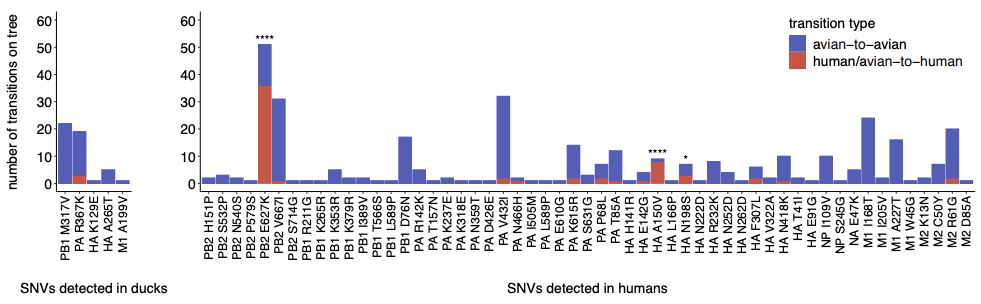
When we compared within-host evolution in these two hosts, we found that virus populations in both humans and ducks are mostly comprised of low-frequency variation (present in <10% of the population), that is shaped heavily by purifying selection, genetic drift, and demography. This is important because we didn’t see strong signatures of rampant positive selection in humans. However, we did detect a few putative human-adapting mutations in multiple, independent humans. Two human samples contained an E627K mutation in the polymerase subunit PB2, a well-known marker of mammalian adaptation that has been repeatedly shown to improve human replication in animal and cell culture models. We also found mutations in the receptor binding protein, HA, that have been phenotypically linked to improved human receptor binding. Two humans harbored an A150V mutation within-host, which contributes to receptor binding and was also identified in H5N1-infected humans in Vietnam, while 2 others harbored an HA Q238L, a mutation identified in ferret transmission studies as a determinant of human receptor binding and transmission. These results show that H5N1 viruses have the capacity to generate known makers of human adaptation during natural spillover infection. This is important because it suggests that molecular markers identified in laboratory studies also evolve in nature, at least in this genetic backbone, and may be useful for surveillance.
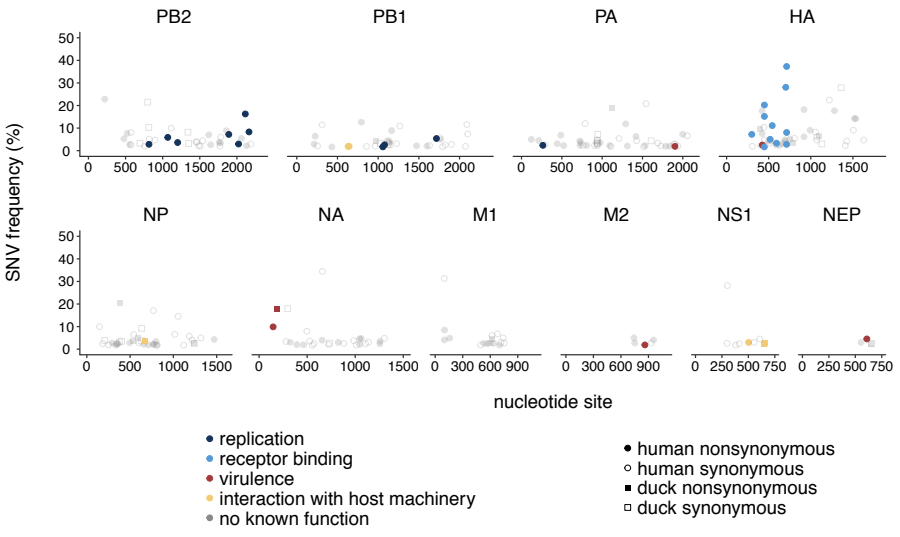
We next wanted to determine whether there were other mutations within-host that might be human-adaptive. To test this, we generated phylogenetic trees for all currently available H5N1 sequences and queried whether mutations we found in our dataset were enriched along branches leading to human infections. This analysis showed that both PB2 E627K and HA A150V were heavily enriched on phylogenetic branches leading to human infections, suggesting that they are likely human-adapting. However, we also found that about half of the mutations detected in our dataset are never detected on the H5N1 phylogeny. This suggests that fraction of variation generated within-host is likely deleterious, and purged from the H5N1 population over time.
What we learned and open questions
By studying within-host diversity, we were able to learn a few important things from this dataset. The first is that H5N1 viruses have very clear potential to generate human-adapting mutations within-host. The fact that we identify previously validated markers of mammalian adaptation and identify mutations that are enriched on spillover branches in nature support this. Importantly though, all of the putative human-adapting mutations we found remained at low frequencies in our samples, despite 5-14 days of infection. Our data therefore also underscore that even mutations that have been hypothesized to be strongly beneficial (PB2 E627K and HA Q238L) may remain at low frequencies in vivo. This suggests that factors like purifying selection, randomness, and short infection times counteract the adaptive potential of H5N1 viruses to evolve during any individual spillover infection. Although this result is somewhat nuanced, it makes sense given what we know about avian influenza. While animal experiments suggest that human transmissibility should be easy to evolve, H5N1 has never actually done so in nature. Although H5N1 has clear potential to evolve within-host, a combination purifying selection, randomness, and epistasis likely restrict its ability to evolve extensively during a single infection.
This study was small and only examined two H5N1 genetic backbones, so there are lots of open questions that remain. How would the patterns we observe in this data compare to spillover infections with other genetic backbones? Would our findings in poultry be the same if we had access to hundreds of samples, over many years of sample collection? Are there other mutations that elicit host-adapting phenotypes that are yet undiscovered? Are certain viral backbones more conducive to human adaptation than others? What environmental factors contribute to spillover? These are all challenging, open questions that I hope we can answer one day.
To look at the data and analyses…
If you’re interested in checking out how we did any of this or looking at the data yourself, all of the code for the figures and analysis of data described in the manuscript are freely available at github.com/blab/h5n1-cambodia. All of the raw sequence data is available from the SRA under accession number PRJNA547644, and the bioinformatic pipeline used to process the raw FASTQ files is available here. You can also find other useful data files, like the within-host variant calls and phylogenetic trees in the GitHub repo.
This has been an incredible opportunity to work with a large group of collaborators from all across the world to answer some interesting questions about avian influenza evolution. I’d like to give a special thank you to Paul Horwood, Philippe Dussart, Philippe Buchy, Erik Karlsson, Srey Viseth Horm and Sareth Rith from Institut Pasteur for getting this project off the ground, and for all of the amazing work they are doing for avian influenza surveillance in Cambodia. Thank you to Lifeng Li, Yongmei Liu, Huachen Zhu, and Yi Guan for generating the original sequence data and for sharing it with us. And of course, a huge thank you to Tom Friedrich and Trevor Bedford for working with me on this project during my transition between labs.