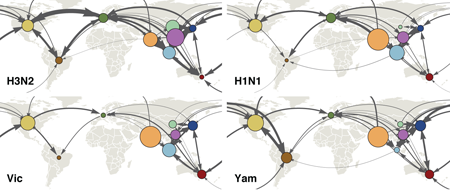
We’ve just had a paper published in Nature investigating the global circulation patterns of seasonal influenza viruses. This work represents a collaborative effort between myself, Colin Russell at Cambridge University, analysis partners Philipe Lemey and Steven Riley and data collaborators from all five WHO Influenza Collaborating Centers. Here, we analyze the most comprehensive dataset of seasonal influenza viruses compiled to date, representing ~9600 viruses from all four circulating influenza lineages sampled between 2000 and 2012. Circulation patterns of influenza A/H3N2 have been well characterized (see for example Russell et al. 2008, Bedford et al. 2010 and Lemey et al. 2014), showing little regional persistence and a powerful source-sink dynamic in which viruses emerge from the East and Southeast Asian tropics and spread to the rest of the world, rapidly replacing endemic lineages. In this new paper, we apply phylogeographic methods to show that the other three lineages of seasonal influenza, A/H1N1, B/Victoria and B/Yamagata, are traversing the world in quite a different fashion from H3N2.
We find that influenza B viruses in particular often remain endemic within a geographic region without spreading to other parts of the world. India and China still represent source populations from which influenza B viruses throughout the world descend, but this process occurs much more slowly in influenza B. Because geographic mixing is slower in influenza B than influenza H3N2, we observe distinct genetic lineages of influenza B associated with different geographic regions. We quantify this difference by measuring the regional persistence of different viruses, finding that H3N2 viruses persist within a region an average of 6 months, while B viruses persist an average of 12 or 13 months. Influenza H1N1 shows up in between H3N2 and B, with an average regional persistence of 9 months.
We review literature showing that influenza B viruses evolve antigenically more slowly than influenza A viruses, particularly H3N2, resulting in a greater proportion of childhood infections in B viruses. We find that ~60% of B infections are in children, while only ~30% of H3N2 infection are in children. We reason that much of the difference in rates of geographic spread between A and B viruses could be due to different travel patterns in children and adults. To test this hypothesis, we built a large-scale individual-based epidemiological model that includes age structure, antigenic evolution and geographic mixing. We show that in this model, including realistic differences in child vs adult travel patterns results in a very similar impact on geographic movement as observed in the empirical influenza A and B viral sequence data. Thus, we observe a quite interesting interaction between virus evolution, epidemiology and human behavior patterns that drives large-scale differences in virus geographic spread.
Working on this, it was great to be able to build so much on previous analytical code and results. Analysis code for geographically-annotated phylogenies was largely expanded on from the PACT tree traversal scripts originally developed for Bedford et al. 2010, while simulation code was expanded on from the individual-based Antigen model originally implemented for Bedford et al. 2012. Results on quantifying differences in antigenic drift rates between seasonal viruses from Bedford et al. 2014 were also incredibly helpful.
All analysis code and results (with over 50 figures!) are freely available in the GitHub repository associated with the paper. I’ve done my best to include Rake build files wherever possible to make this research as reproducible as possible. This repository contains code to subsample data, set up phylogenetic and phylogeographic BEAST XML files, conduct PACT analyses of BEAST output and set up Antigen simulations.