Today, my paper on migration patterns in the flu virus was published in PLoS Pathogens. This was fun work to do, requiring approaches from multiple disciplines. While the basics of the migration model came from population genetics and coalescent theory, fitting this model to sequence data required a lot of heavy-lifting computation implemented by Peter Beerli in the program Migrate. I originally wrote my program PACT to deal with the enormous (2000+ tips) phylogenetic trees produced by this analysis. Additionally, a lot of epidemiology went in to making realistic simulations on which to hone the methods.
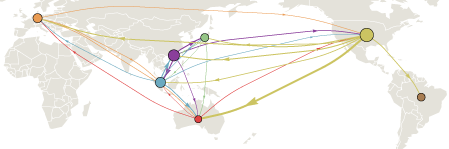
The common ancestor of all contemporaneous H3N2 flu can be traced back to a single infection occurring somewhere in the world approximately 2-5 years before hand. This infection, by luck and by virtue of its genotype, becomes the progenitor of the entire worldwide flu population. The main goal of this analysis was to trace this progenitor lineage through time. We found that this lineage existed primarily in China and Southeast Asia, but also, surprisingly, in the USA. The occasional presence of this progenitor lineage in USA has important public health implications.