Marta Łuksza, Michael Lässig and I have done an analysis of Ebola virus evolution using the recently published Gire et al. dataset of 78 full genomes. We’ve put the manuscript up on the arXiv.
The Gire et al. paper examines Ebola sequences from Sierra Leone during May and June identifying 55 new mutations that have occurred during the West African outbreak. Most of these mutations are shared by just one or a small handful of cases, but there is a particular mutation at genomic site 10218 that first appears at the beginning of June that distinguishes two major clades of virus (shown in blue and red below).
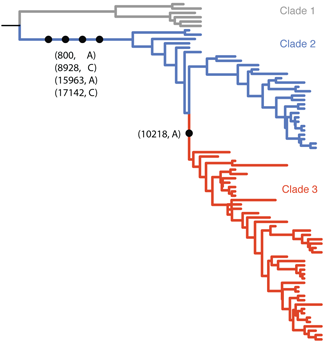
Previous phylodynamic studies have developed general methodologies to estimate epidemic growth rates from sequence data. Here, we developed a novel approach to look for differences in epidemic growth rate across genetic clades. We use this method to compare growth rates in clade 2 and clade 3 viruses, finding a significantly higher growth rate in clade 3 viruses. This suggests that clade 3 viruses spread more rapidly than clade 2 viruses during the May/June study period.
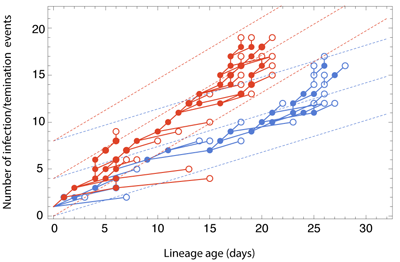
This result could be explained in two fashions, either the mutation at site 10218 has functional and evolutionary consequences that aid the spread of the virus or this mutation represents a neutral marker and the difference in growth rates is entirely due to epidemiological circumstance. In the former case, we expect natural selection to favor a virus that transmits more readily or a virus that has a longer duration of infection, perhaps due to attenuation (a common occurence in the process of host adaptation). In the latter case, we might expect that clade 3 viruses happened to arise in a more favorable local environment or underwent a superspreading event that rapidly boosted the number of infections. Either of these scenarios demands more detailed follow-up.
However, we would predict that if the mutation at site 10218 has adaptive significance then the observed difference in growth rate will be sustained, while if it is epidemiologically driven, then the observed difference will be transient. The mutation at site 10218 is now an event that occurred 5 months ago; either clade 3 has continued to take over or it has not. Sequence data from more recent infections would immediately tell us which scenario took place.
In general, I would hope that this methodology could be used to search for novel adaptive variants in near real-time as sequence data is released.