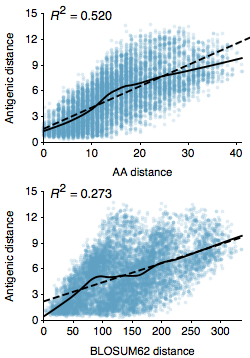
As part of the “Integrating influenza antigenic dynamics…” paper I calculated the relationship between amino acid sequence distance between influenza A/H3N2 viruses and their degree of antigenic distance from one another (Figure 1). As influenza strains accumulate genetic mutations, these mutations result in hemagglutinin (HA) proteins that are recognized to a lesser degree by previously acquired immunity. In a discussion with colleagues earlier today, we were trying to identify a sequence predictor for antigenic distance in influenza. A major question was whether to use raw amino acid distance between strains, or to modify distance based on biochemical properties via a BLOSUM matrix. The logic here is that we might expect mutations that have a larger biochemical effect to be more likely to result in antigenic change.
{kind=link}
However, here I’m showing the correlation for raw amino acid (Hamming) distance and also the correlation for BLOSUM62 distance between pairs of A/H3N2 strains separated by at most 10 years. These are random pairs of viruses from 1968 to 2011 and I’m only looking at the HA1 portion of the hemagglutinin protein. We see significantly improved correlations for amino acid distance over BLOSUM62 distance, with an R^2 of 0.52 vs 0.27. In addition, the average error of prediction for amino acid distance is 1.61 antigenic units, while the average error of prediction for BLOSUM62 distance is 2.01 antigenic units. Thus, it appears that the BLOSUM matrix just adds noise to the correlation, without imparting appreciable signal.