Background
Avian influenza A(H7N9) has resulted in annual epidemics over the past five years in China. H7N9 has a high mortality rate of around 40% and the currently ongoing fifth epidemic is the largest yet, with 460 infections reported by February 27th, 2017 (WHO report). Transmission is still predominantly from poultry, where H7N9 viruses continue to circulate, and human-to-human transmission is thought to be rare. The CDC considers H7N9 to have “the greatest potential to cause a pandemic” of all influenza A viruses. For more information, see this CDC information page.
Nextstrain
Nextstrain now has the ability to display phylogenies and geographical data for both the NA and HA genes, drawn from over 1200 samples covering all five human epidemics of H7N9. This analysis was possible thanks to the data sharing of the influenza research community through GISAID. It is our hope that making these analyses available to the community will aid understanding of this epidemic as it unfolds. Please note that the analysis currently presented in nextstrain is preliminary and further research is required.
Phylogeny & Geographic Distribution
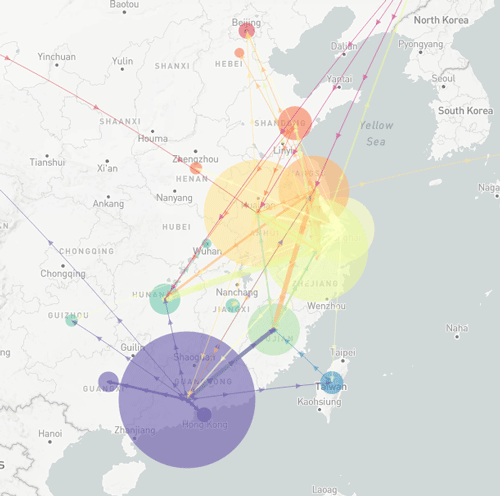
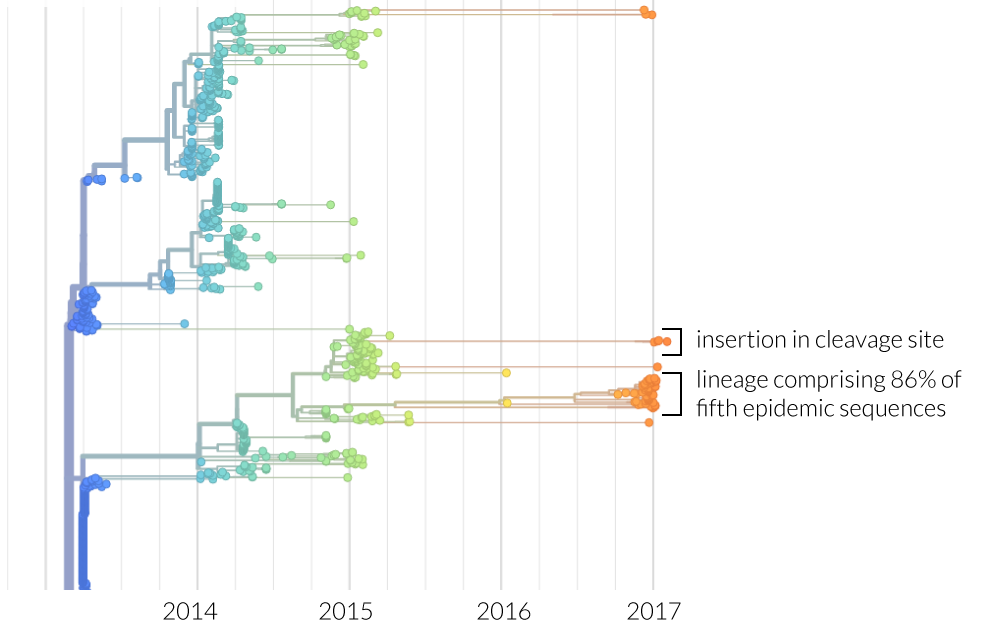 The HA phylogeny indicates that the expansion of a single lineage contributes 86% of sequences from the current epidemic. Temporal analysis indicates that this lineage originated during the 2015 (third) epidemic, however only one isolate from this lineage was sampled during the fourth epidemic. Inference of the geographical distribution of H7N9 indicate frequent jumps throughout the eastern coast of China, with limited dispersion elsewhere. Host jumps have also been inferred, however incomplete sampling restricts our ability to comment further.
The HA phylogeny indicates that the expansion of a single lineage contributes 86% of sequences from the current epidemic. Temporal analysis indicates that this lineage originated during the 2015 (third) epidemic, however only one isolate from this lineage was sampled during the fourth epidemic. Inference of the geographical distribution of H7N9 indicate frequent jumps throughout the eastern coast of China, with limited dispersion elsewhere. Host jumps have also been inferred, however incomplete sampling restricts our ability to comment further.
Insertion in the Protease Cleavage Site
Highly pathogenic avian influenza viruses are often characterized by insertions in the host protease cleavage site, which enhance the cleavage of HA protein to HA1 and HA2 - a process necessary for infection. We find a lineage consisting of four isolates with a four amino-acid insertion (KRTA) in this region, in agreement with Iuliano et al. This insertion is not present in any other isolates, and this lineage contributes less than 10% of the sequences from the current epidemic. Interestingly, this lineage appears to have diverged from the lineage causing most fifth epidemic infections in mid-2014. Despite the lack of expansion during this epidemic, the potential for a highly pathogenic H7N9 variant is worrying and warrants closer inspection.
Reassortment
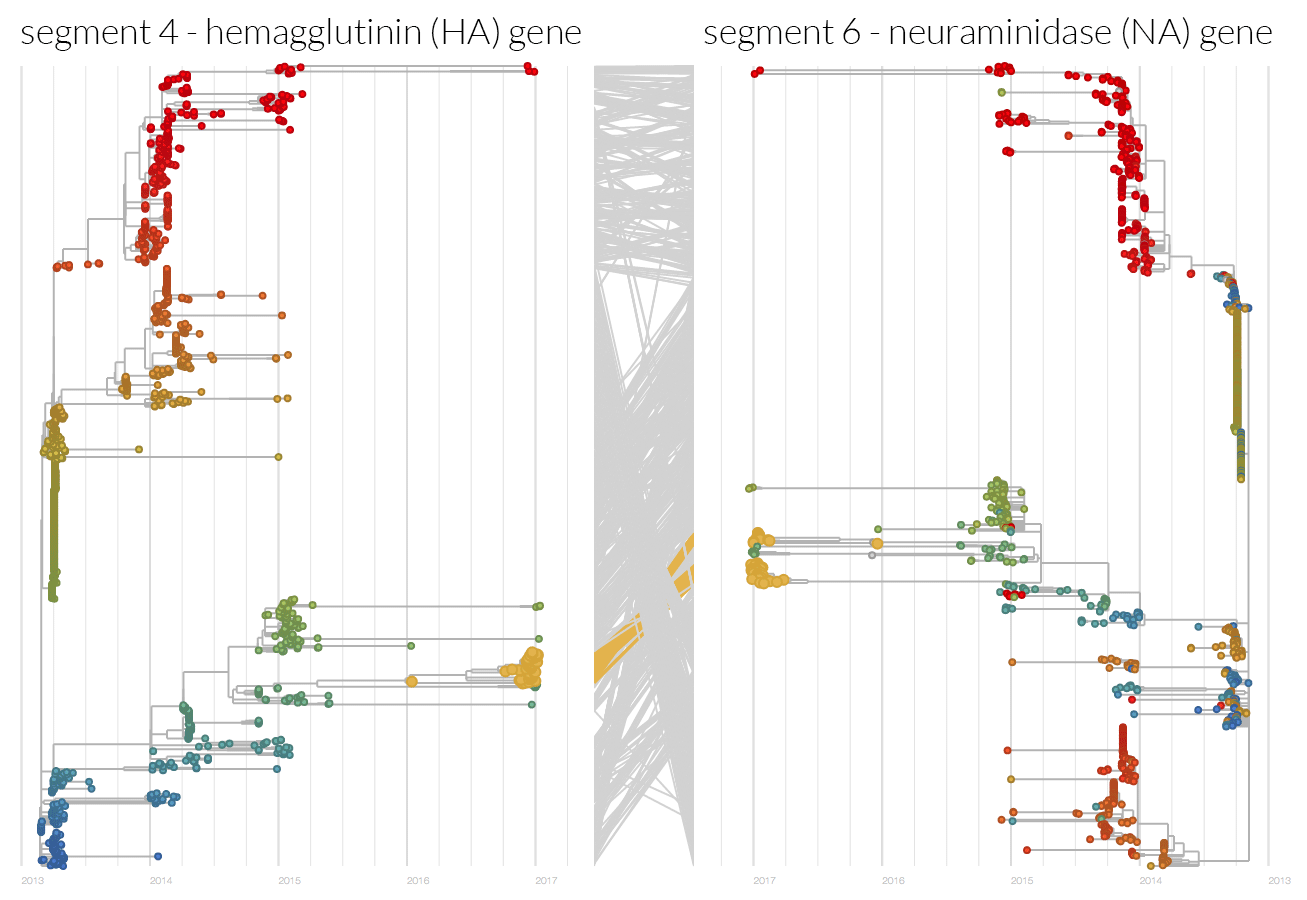 Reassortment led to the origin of human H7N9 and continues to play a role during the epidemics. The above figure shows that the current dominant clade in the HA phylogeny, which contributes the majority of fifth epidemic cases, is comprised of at least two NA clades due to reassortment. The isolates containing the protease cleavage site insertion (in HA) are monophyletic in both HA and NA segments.
Reassortment led to the origin of human H7N9 and continues to play a role during the epidemics. The above figure shows that the current dominant clade in the HA phylogeny, which contributes the majority of fifth epidemic cases, is comprised of at least two NA clades due to reassortment. The isolates containing the protease cleavage site insertion (in HA) are monophyletic in both HA and NA segments.
Acknowledgements
A full list of labs and authors who have made data available for analysis in these samples is available in this spreadsheet. All figures from nextstrain. Many thanks to Gytis Dudas, Richard Neher and Trevor Bedford for assistance.