Tracking and forecasting epidemic spread through viral genome sequencing
Trevor Bedford (@trvrb)
10 Oct 2019
BioHub Seminar Series
BioHub
Slides at: bedford.io/talks
We work at the interface of virology, evolution and epidemiology
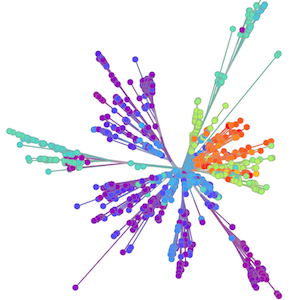
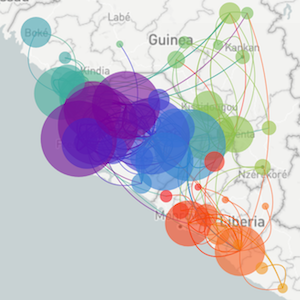

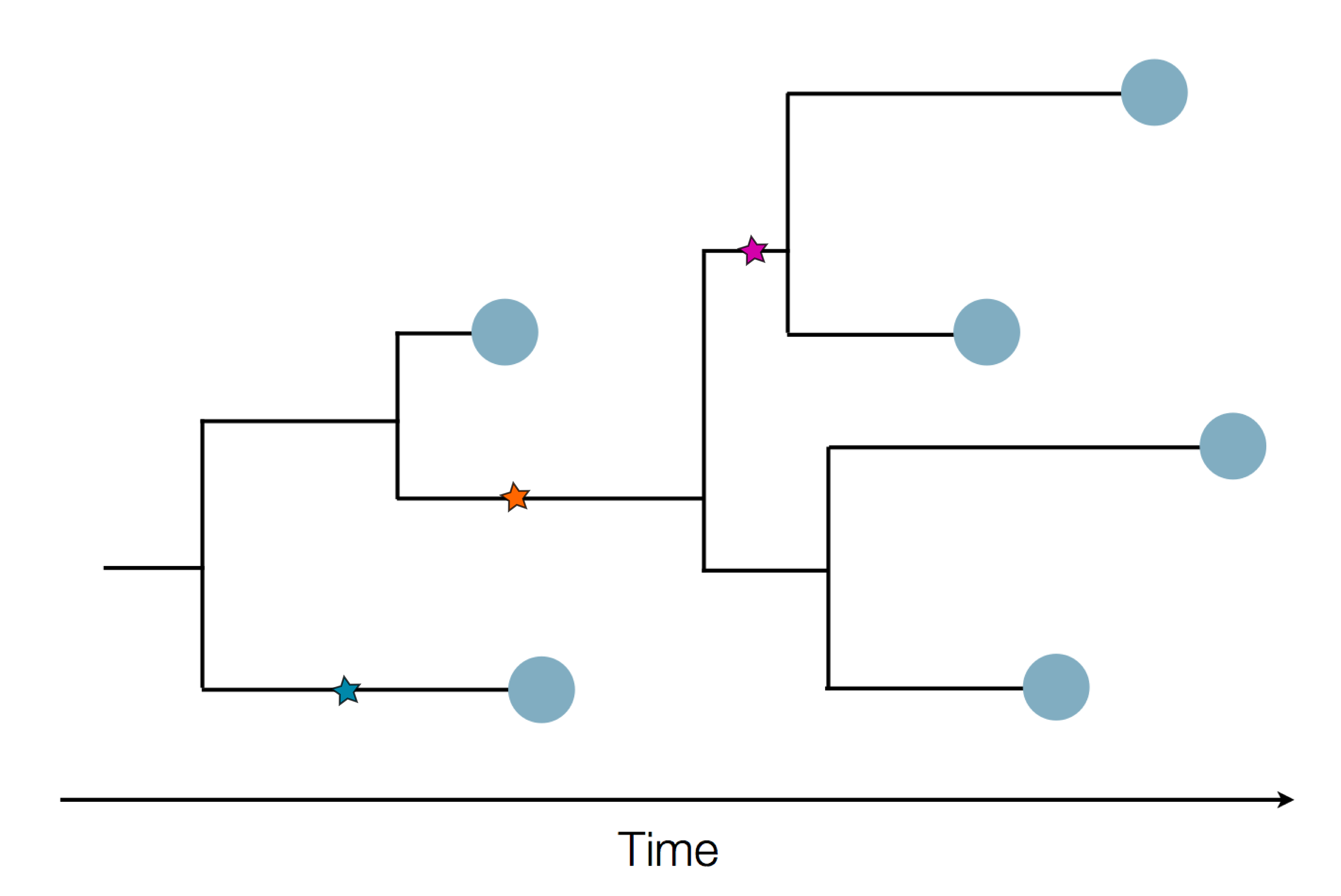
Sequencing to reconstruct pathogen spread
Epidemic process
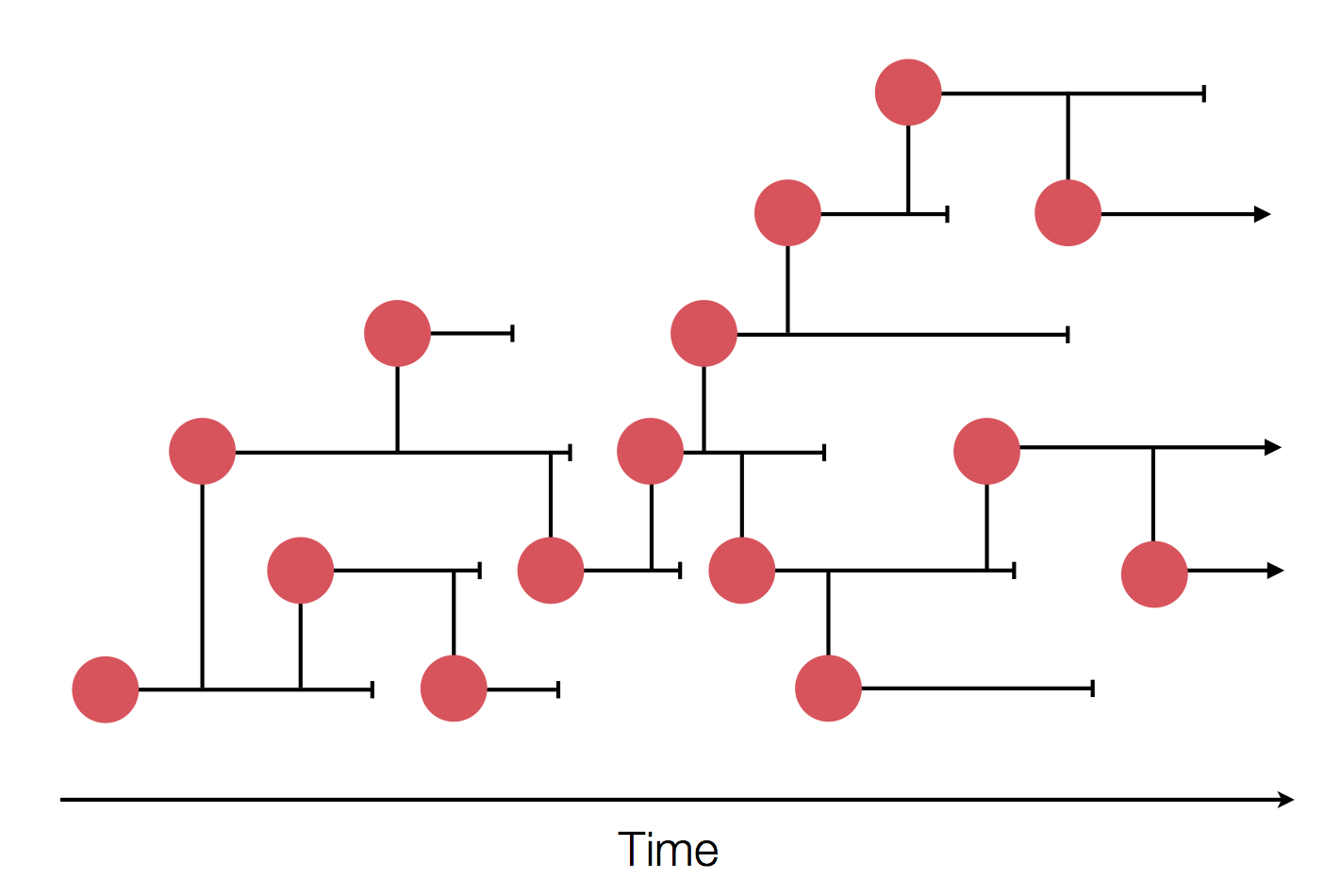
Sample some individuals
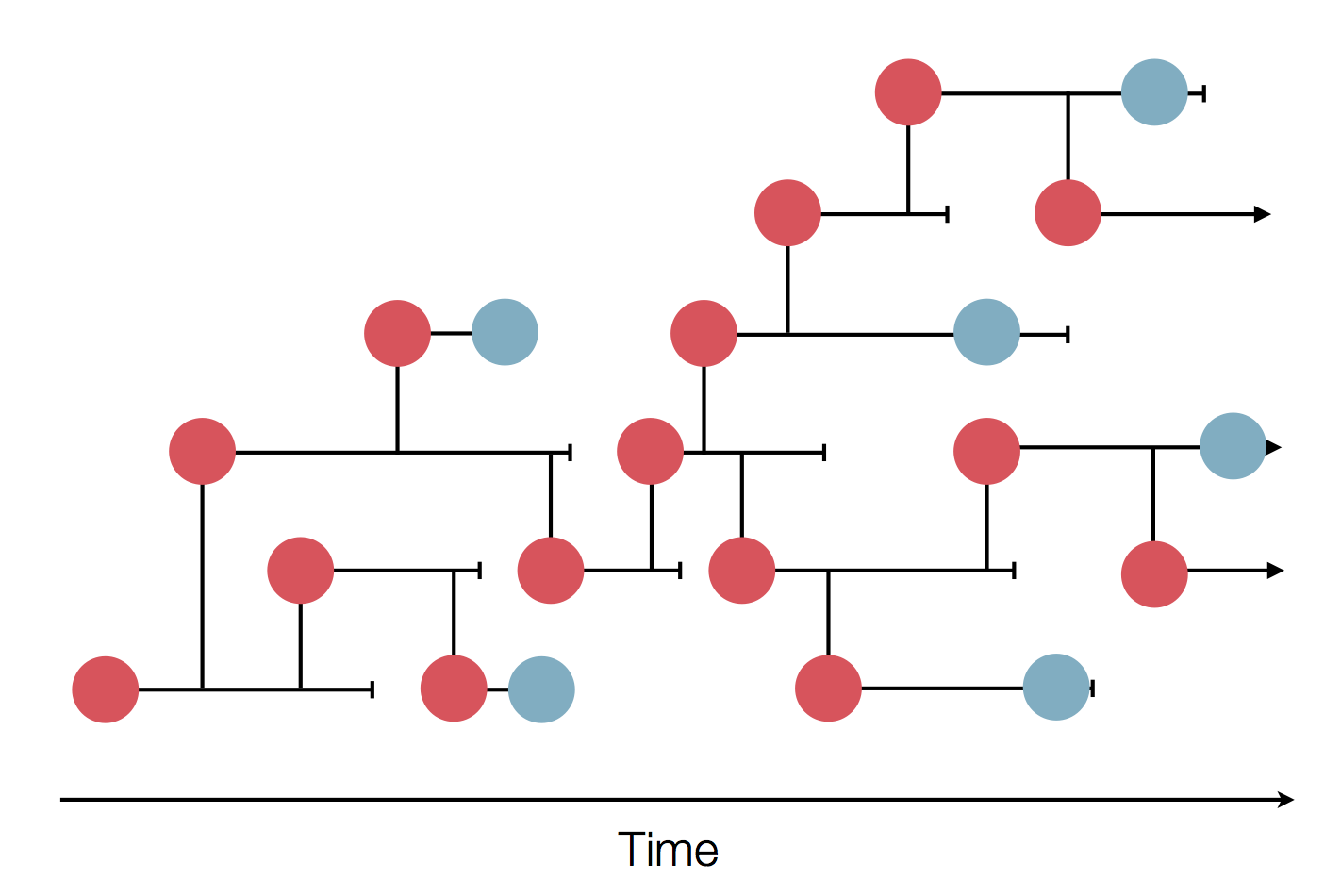
Sequence and determine phylogeny
Sequence and determine phylogeny
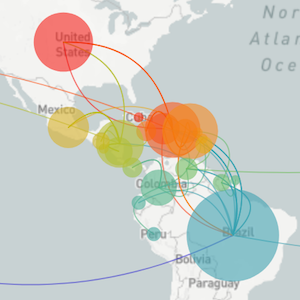
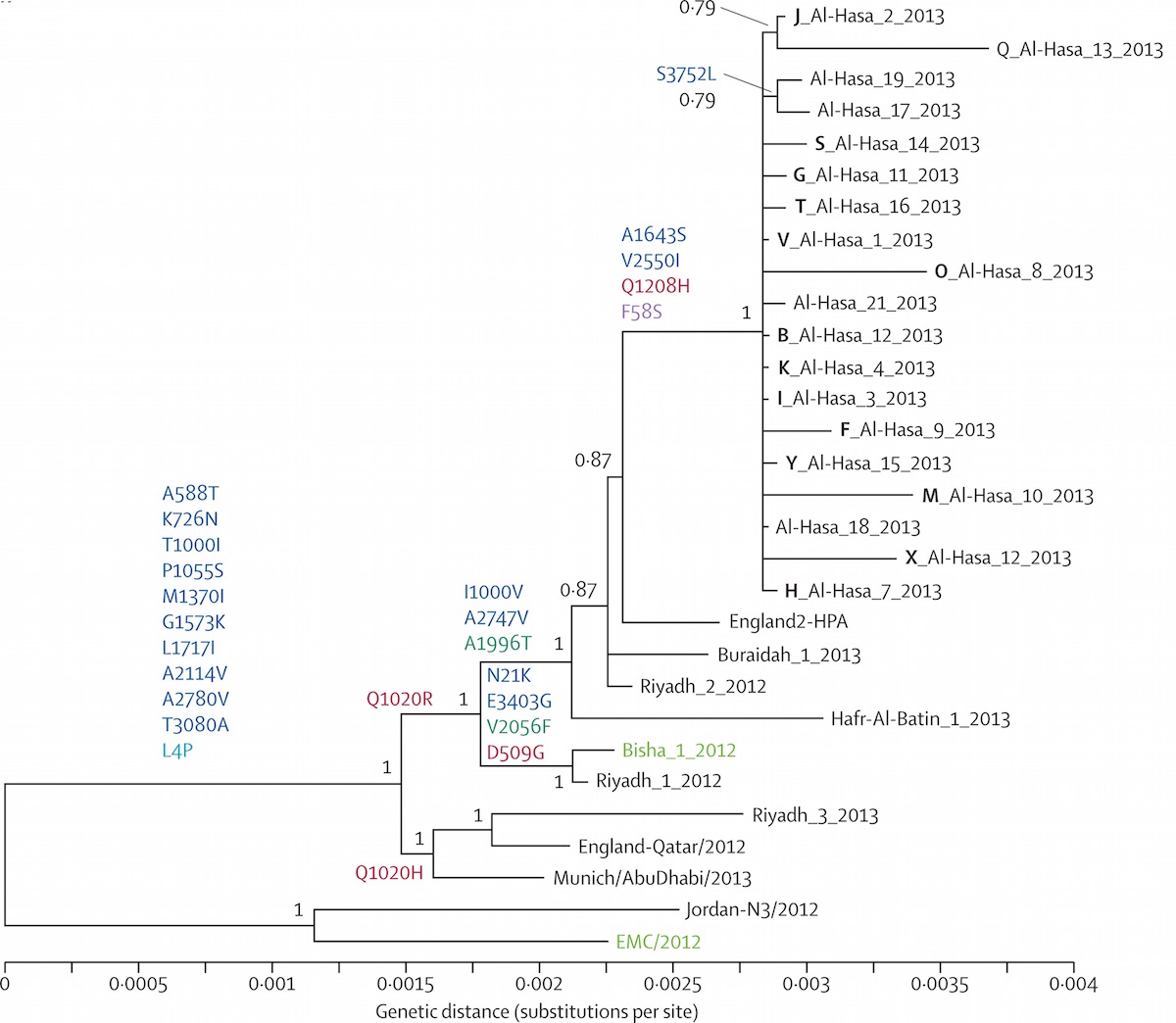
Localized Middle Eastern MERS-CoV phylogeny
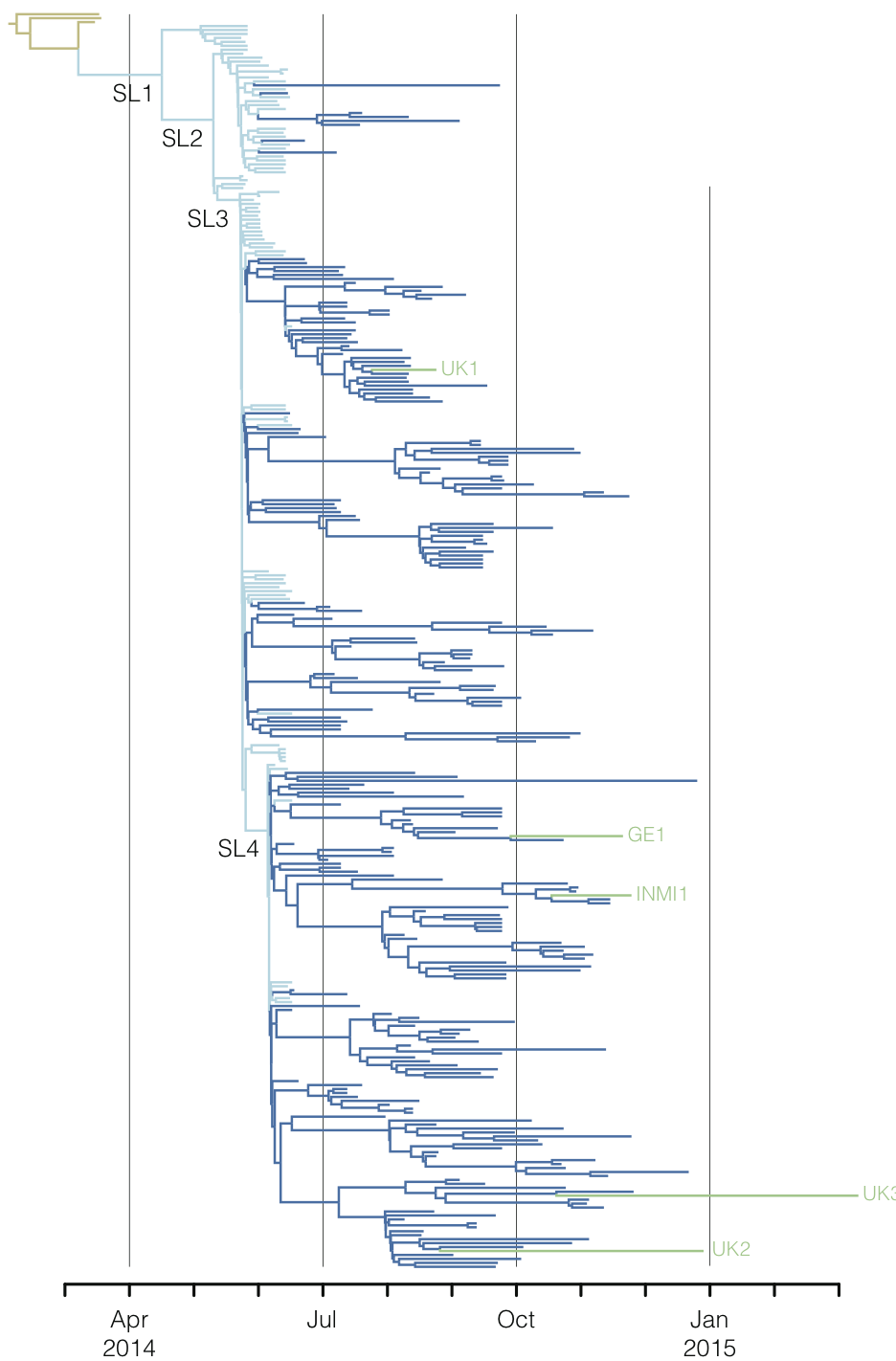
Regional West African Ebola phylogeny
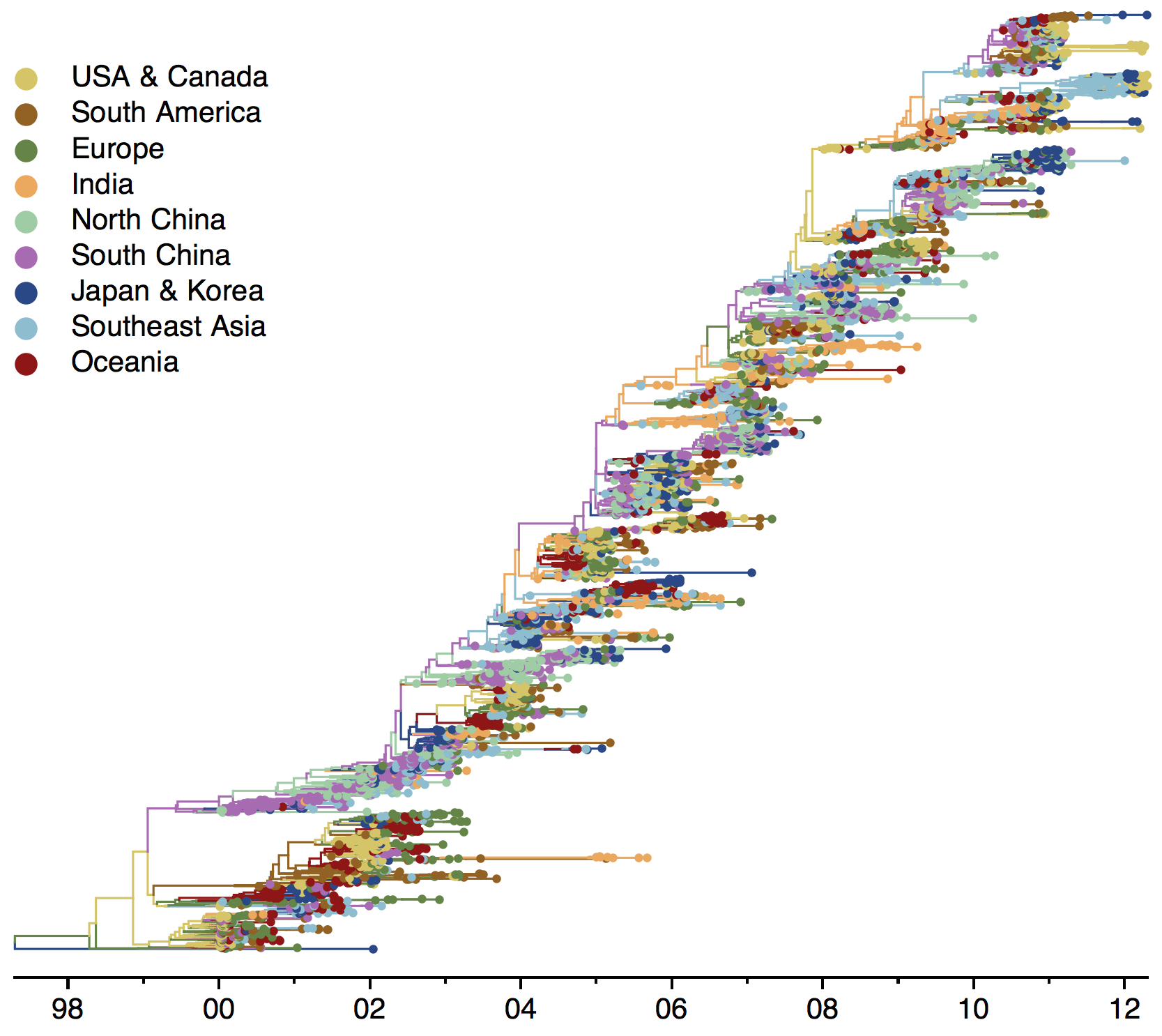
Global influenza phylogeny
Phylogenetic tracking has the capacity to revolutionize epidemiology
Outline
- Analysis of Ebola epidemic spread in West Africa
- Nextstrain platform for real-time phylodynamics
- Actionable genomic epidemiology for Ebola in the DRC
- Seasonal influenza evolution and vaccine strain selection
- Forecasting influenza strain turnover
Ebola
Ebola epidemic of 2014-2016 was unprecedented in scope
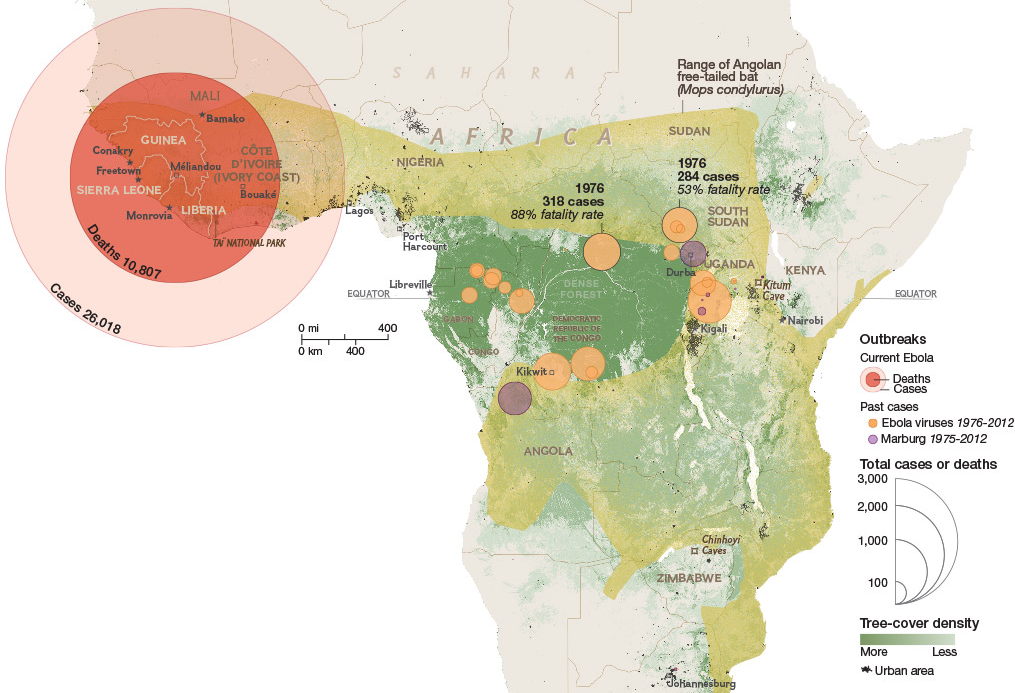
Ebola epidemic in West Africa
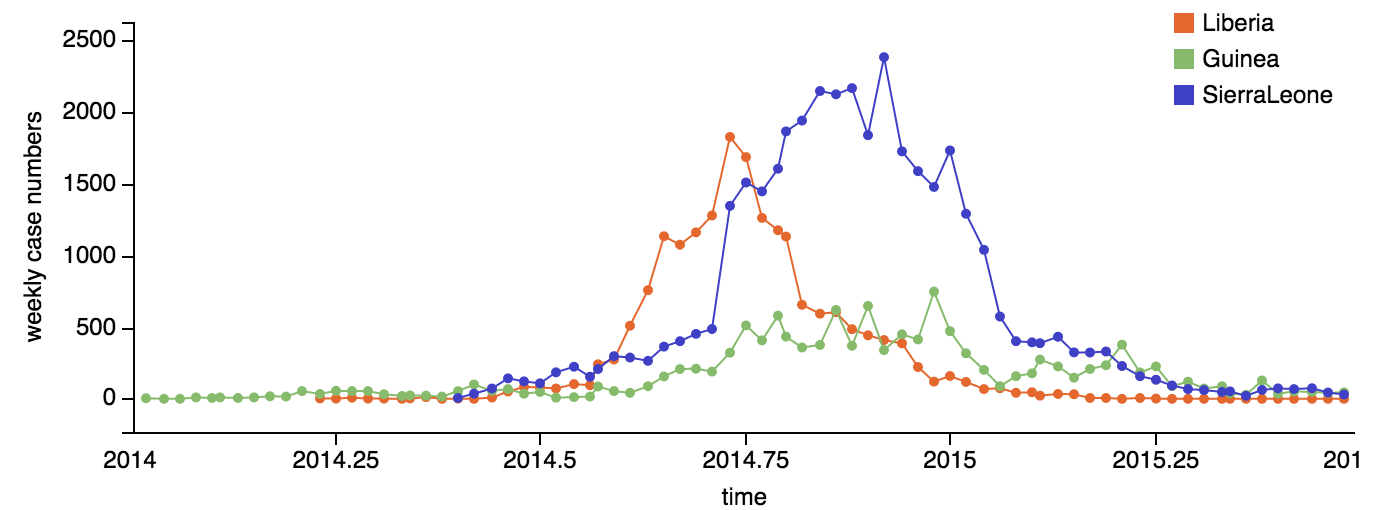
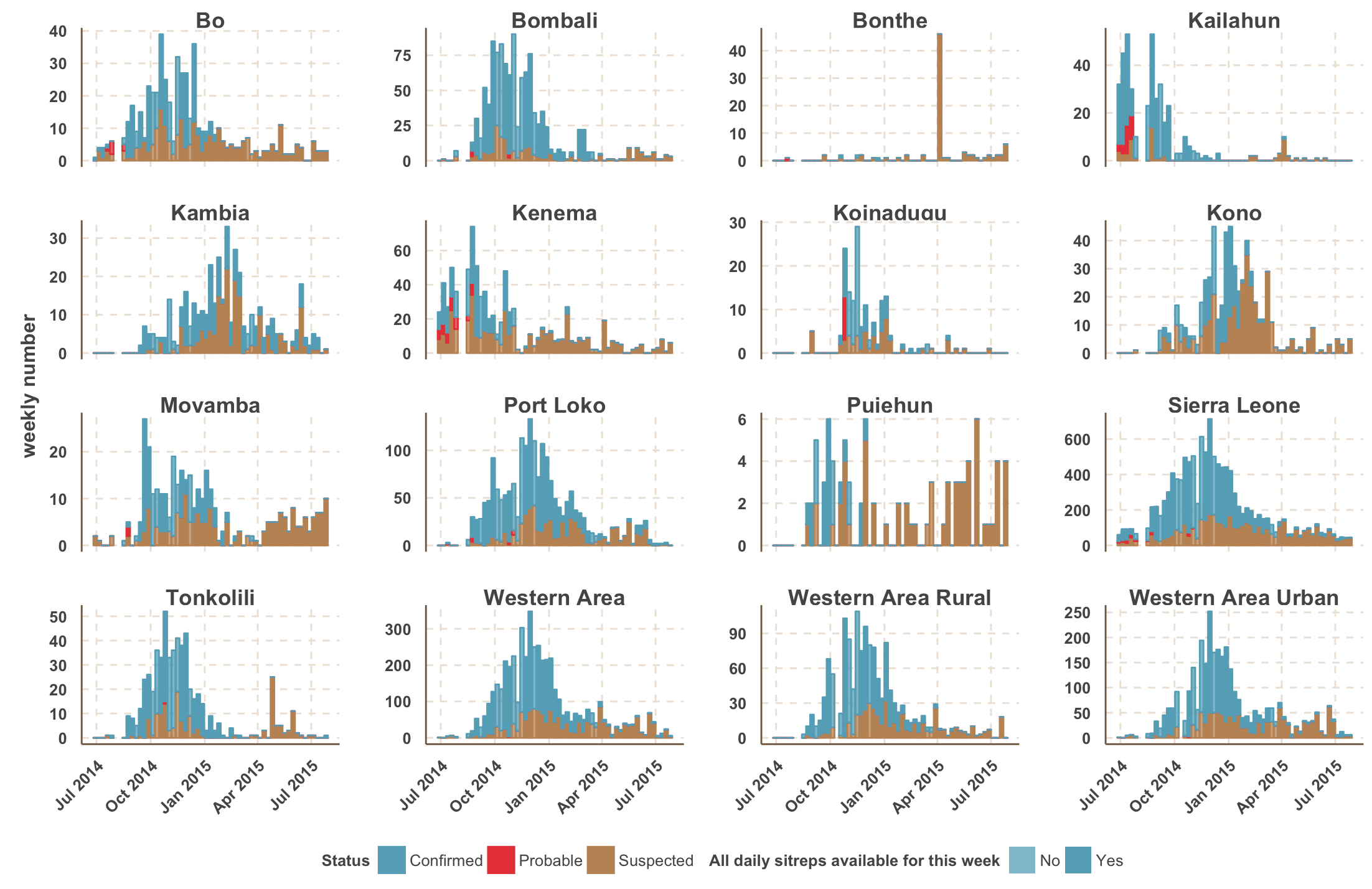
Ebola epidemic within Sierra Leone
Virus genomes reveal factors that spread and sustained the Ebola epidemic
with ![]() Gytis Dudas, Andrew Rambaut, Luiz Carvalho, Marc Suchard, Philippe Lemey,
Gytis Dudas, Andrew Rambaut, Luiz Carvalho, Marc Suchard, Philippe Lemey,
and many others
Sequencing of 1610 Ebola virus genomes collected during the 2013-2016 West African epidemic
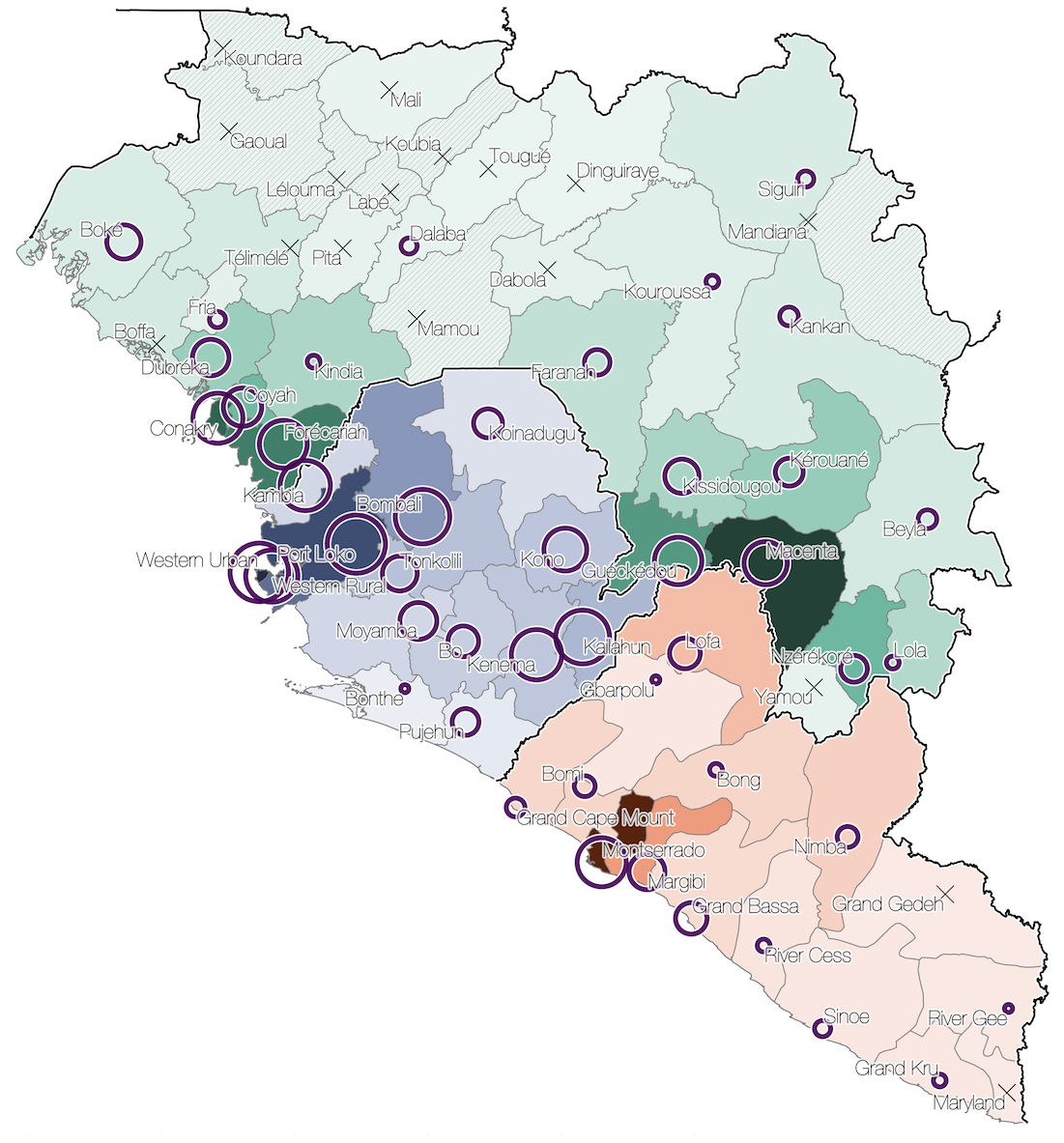
Sequenced genomes were representative of spatiotemporal diversity
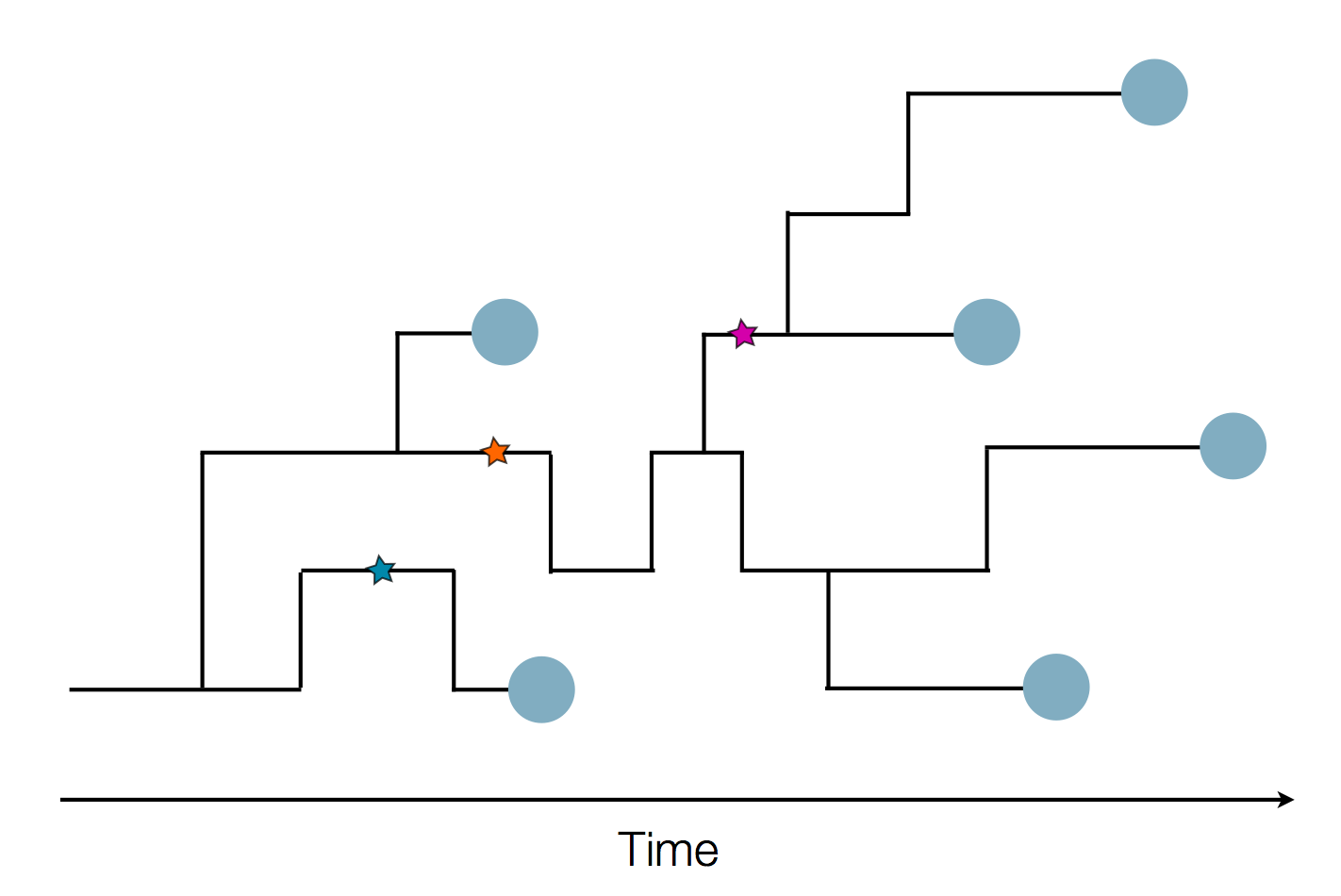
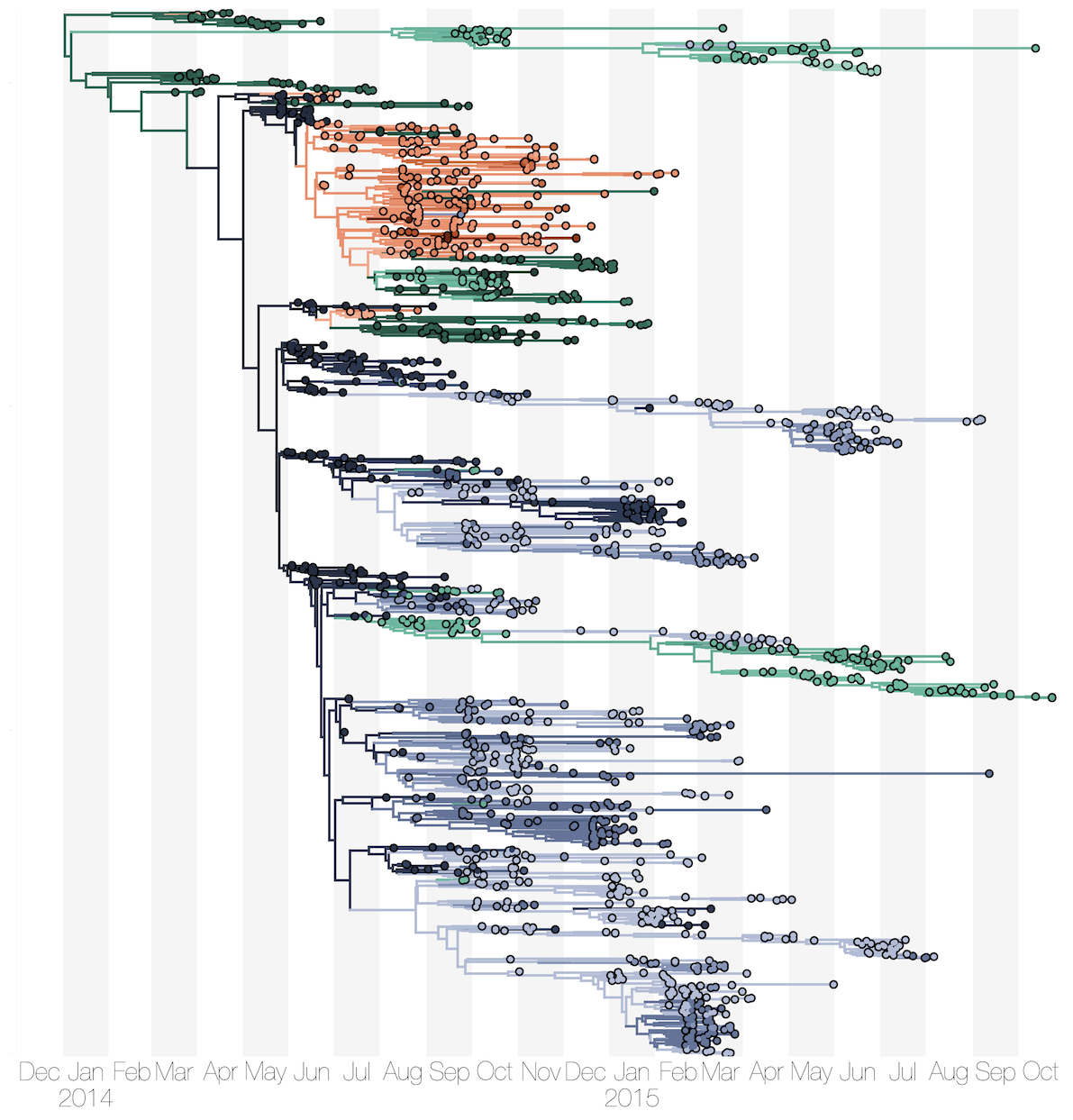
Phylogenetic reconstruction of epidemic
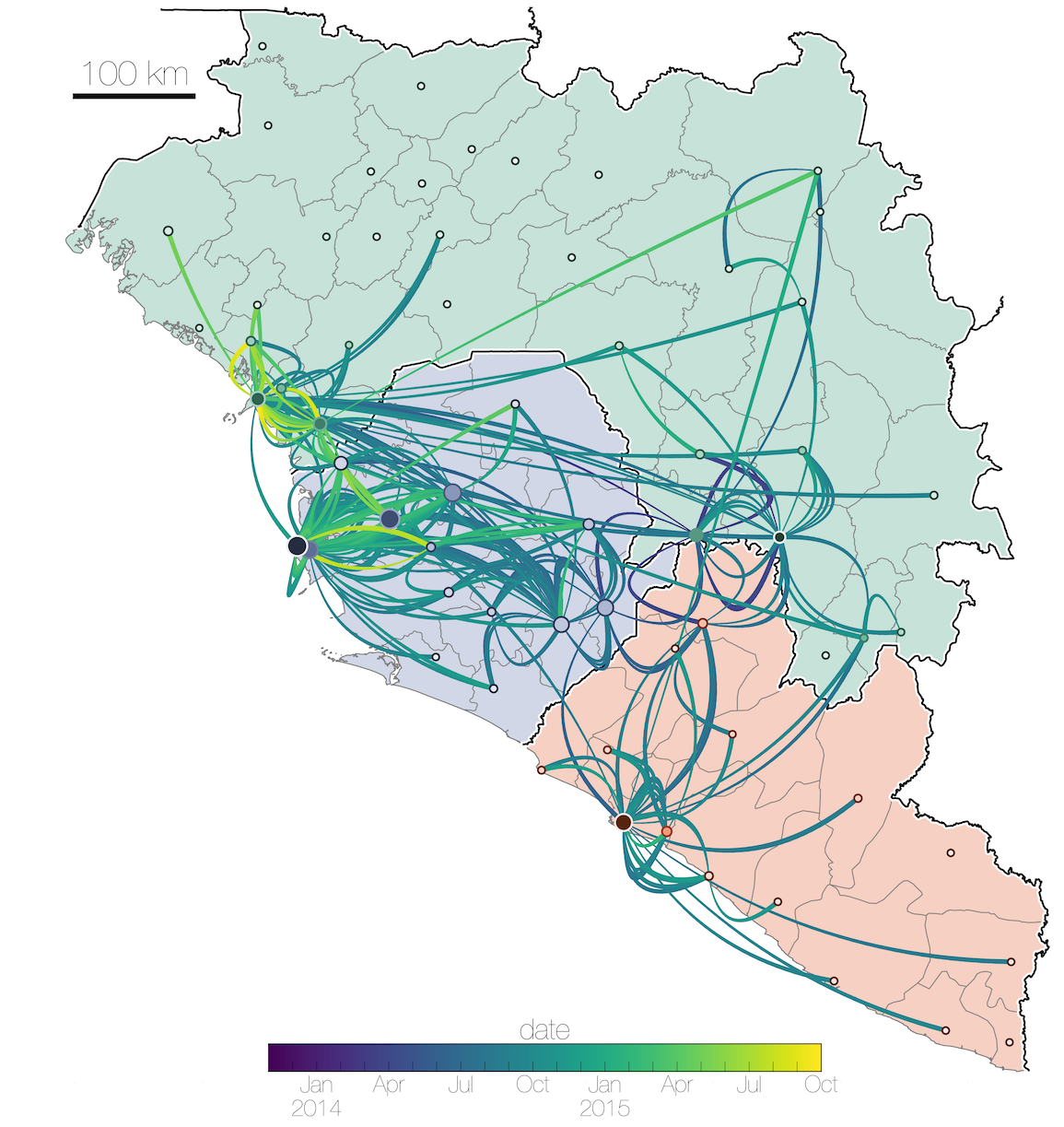
Tracking migration events
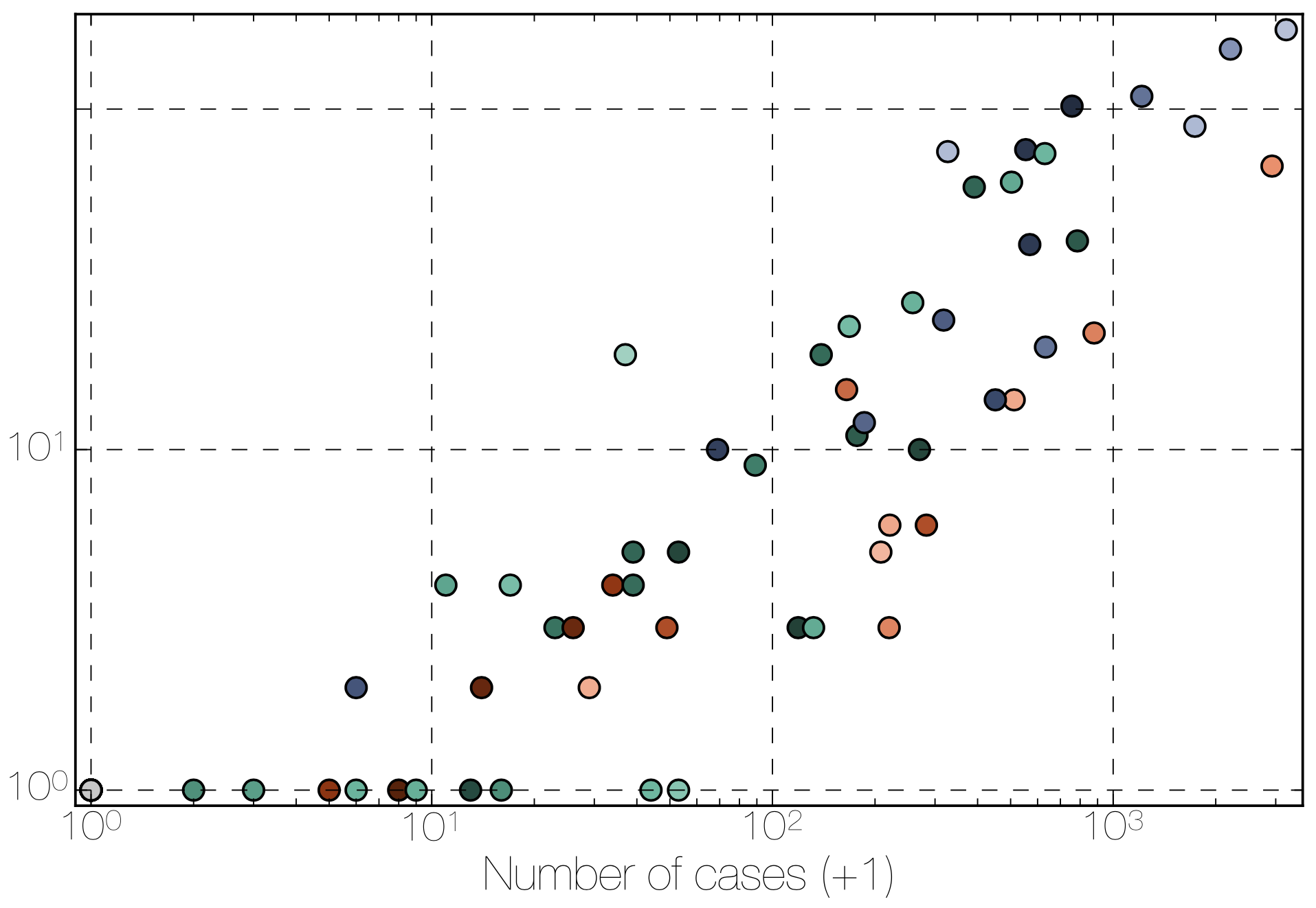
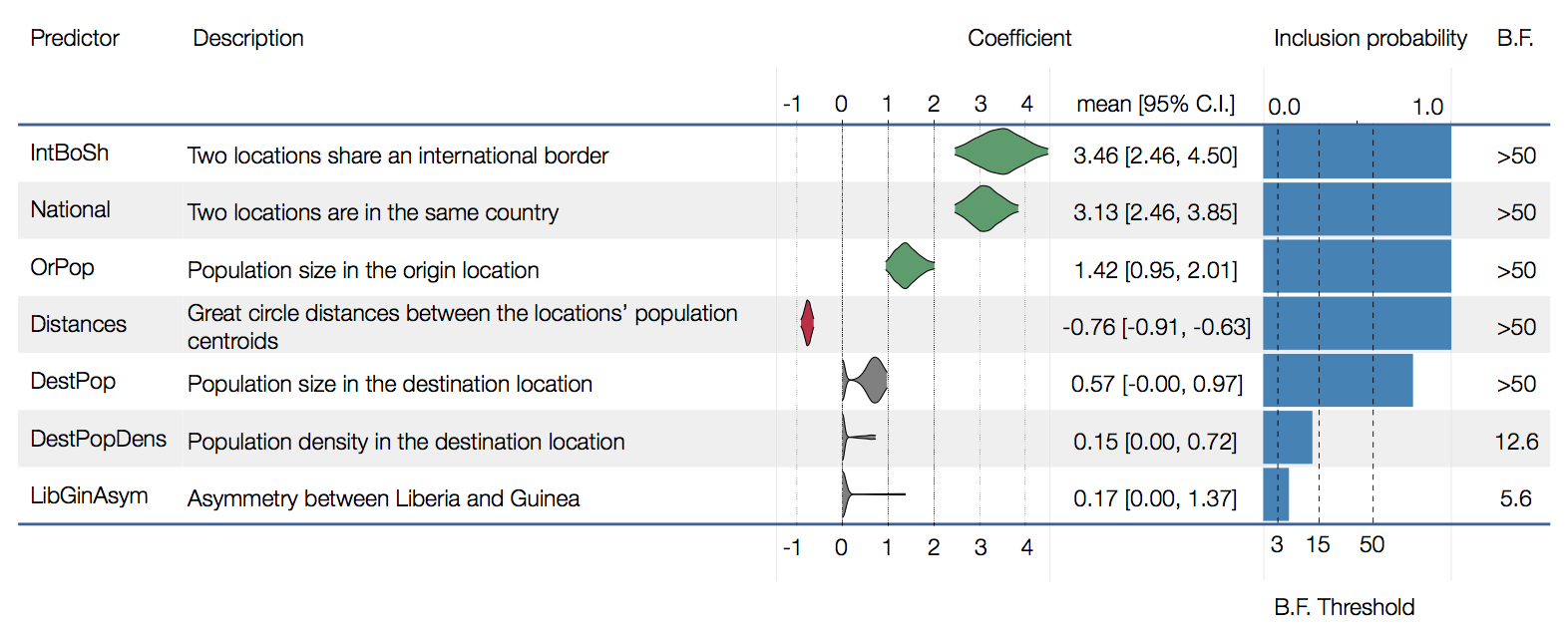
Factors influencing migration rates
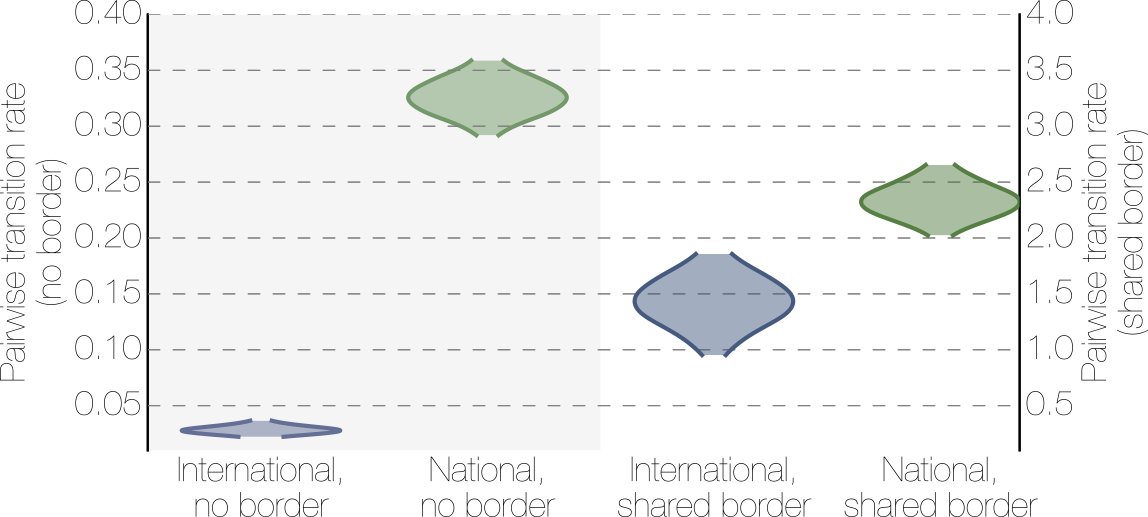
Effect of borders on migration rates
Spatial structure at the country level
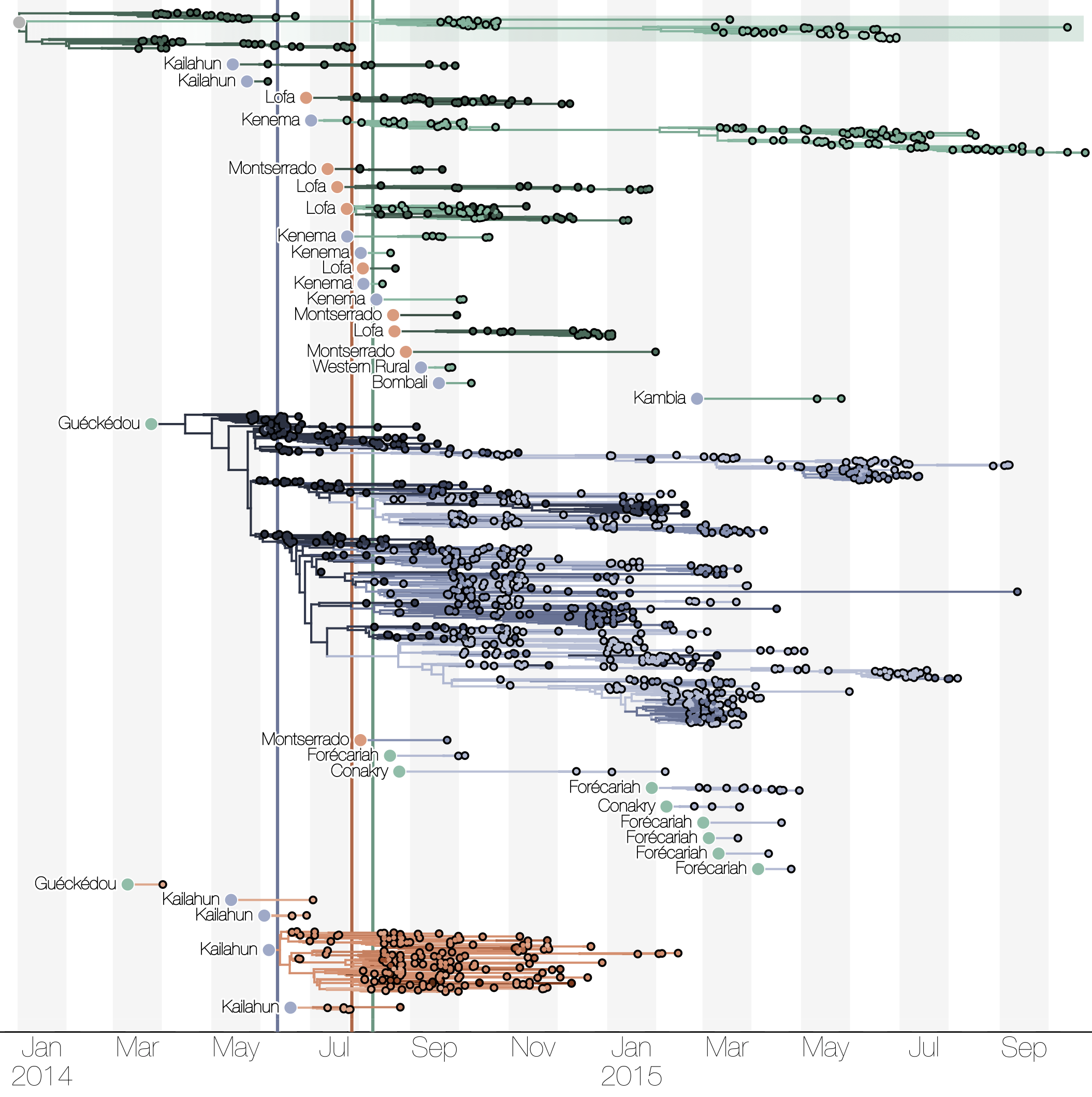
Substantial mixing at the regional level
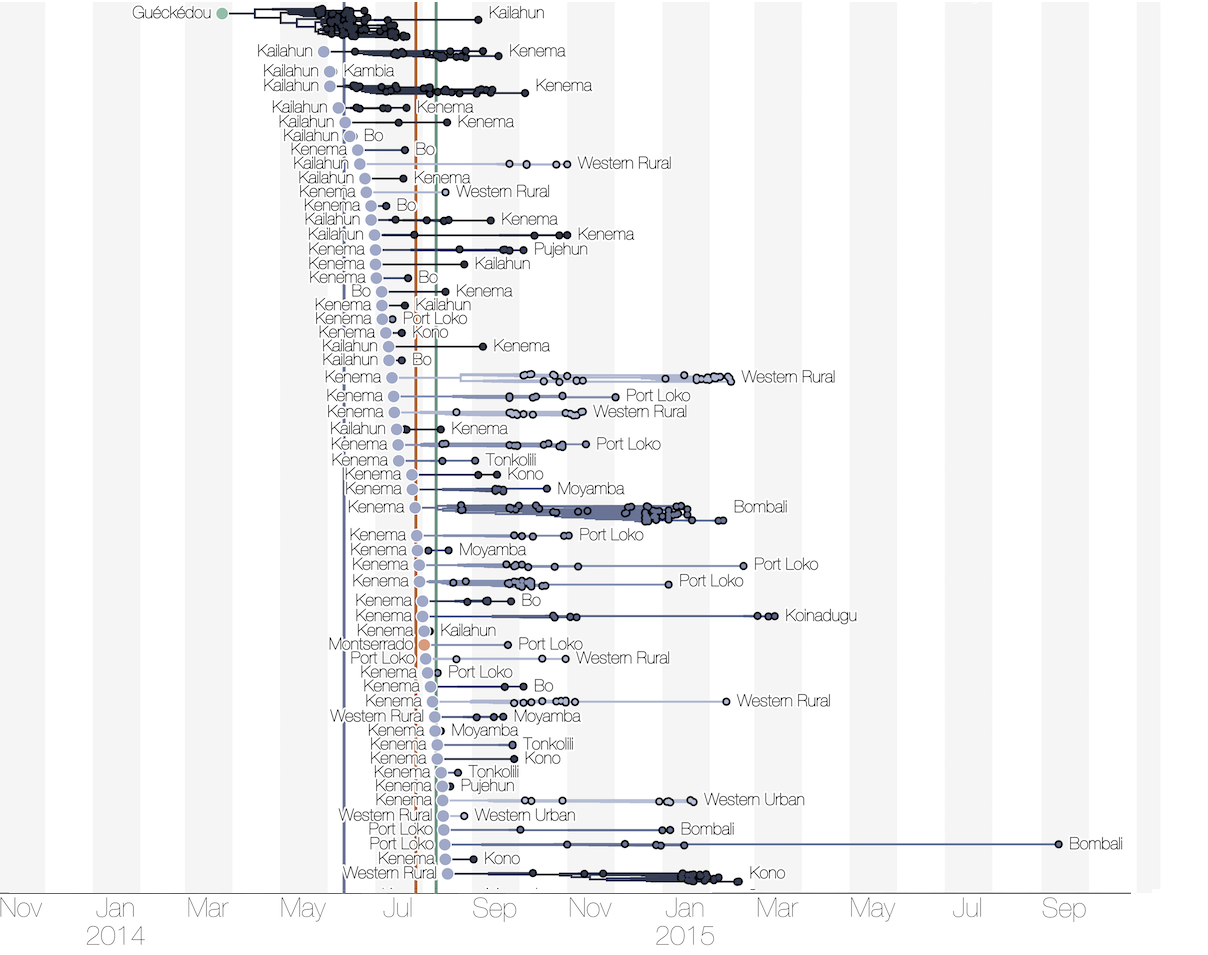
Each introduction results in a minor outbreak
Regional outbreaks due to multiple introductions
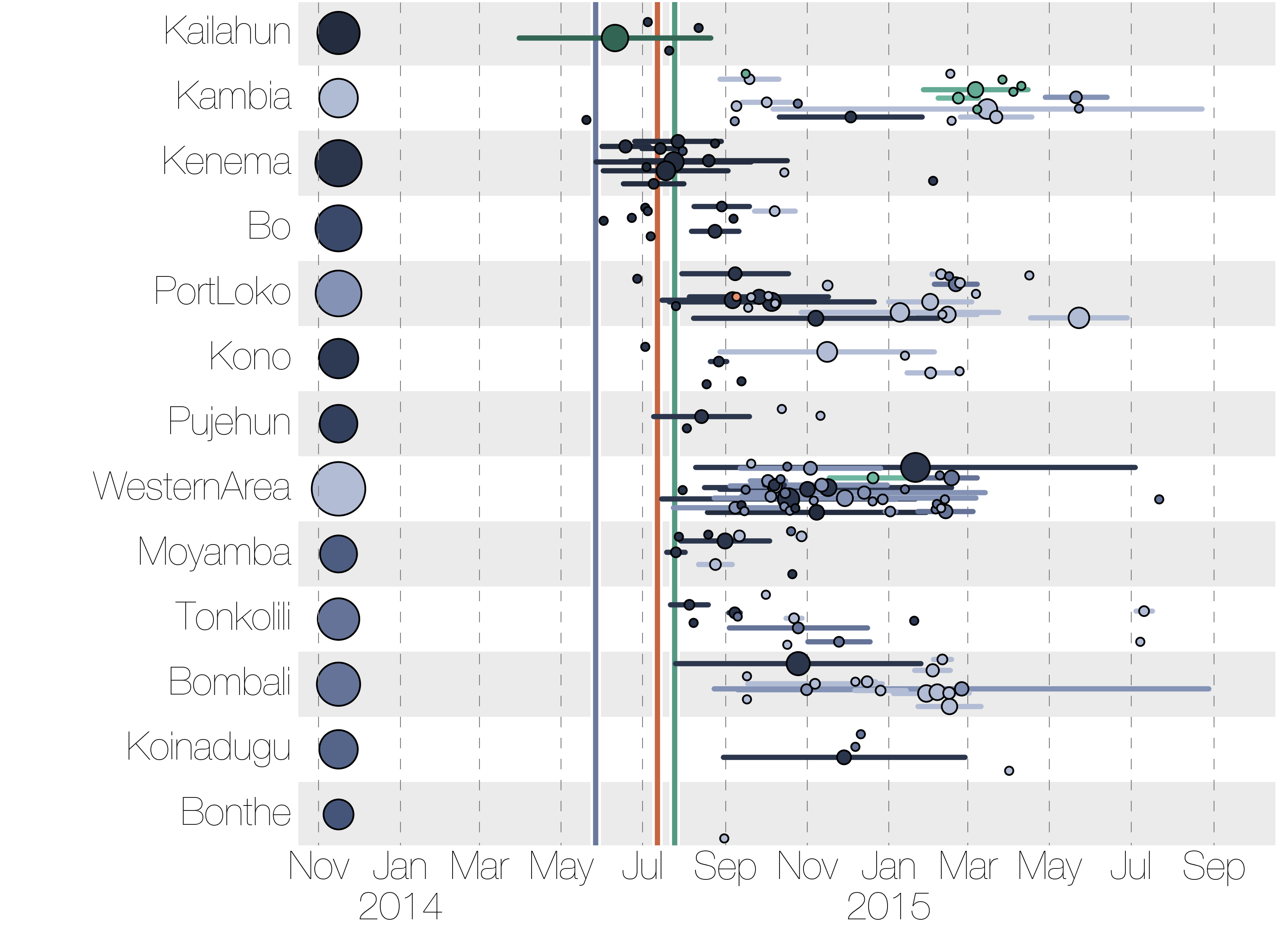
Regional outbreaks due to multiple introductions
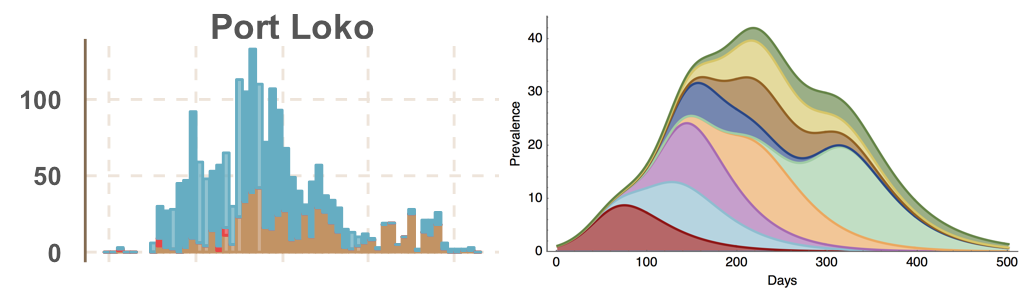
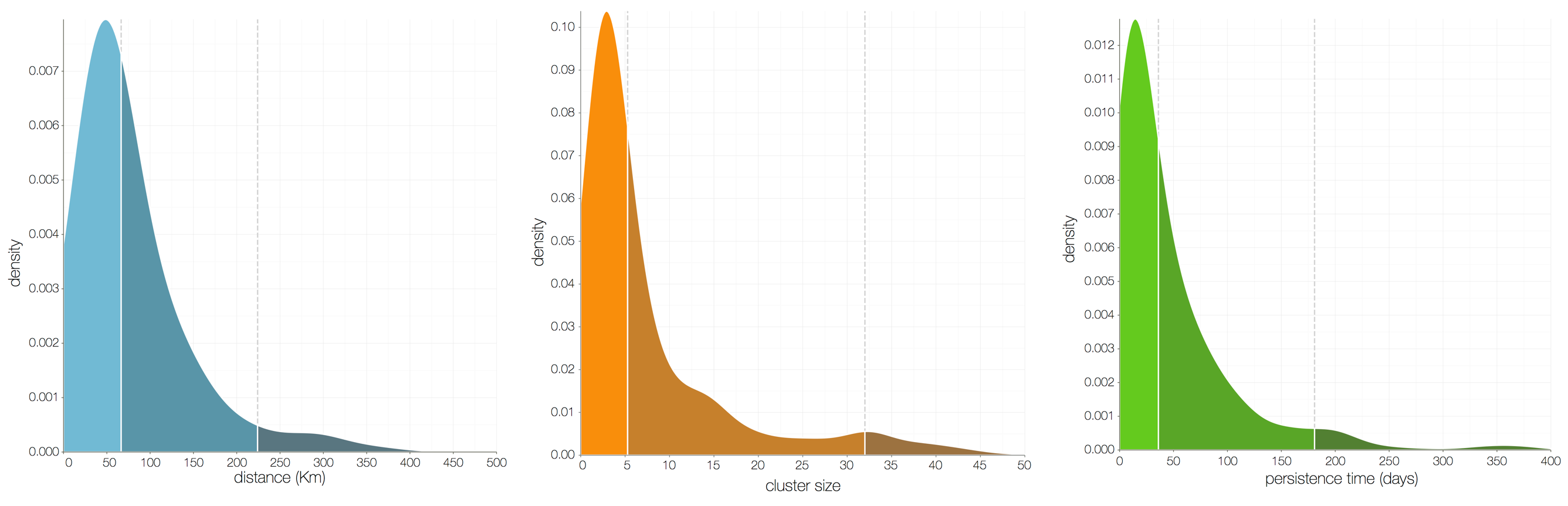
Ebola spread in West Africa followed a gravity model with moderate slowing by international borders, in which spread is driven by short-lived migratory clusters
Actionable inferences
Genomic analyses were mostly done in a retrospective manner
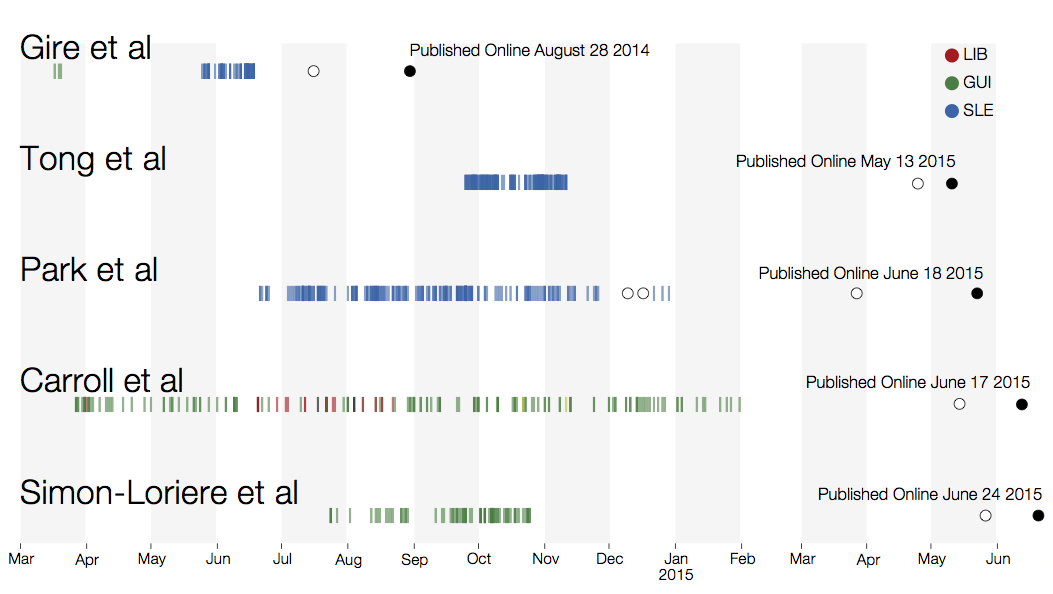
Key challenges to making genomic epidemiology actionable
- Timely analysis and sharing of results critical
- Dissemination must be scalable
- Integrate many data sources
- Results must be easily interpretable and queryable
Nextstrain
Project to conduct real-time molecular epidemiology and evolutionary analysis of emerging epidemics
with
![]() Richard Neher,
Richard Neher,
![]() James Hadfield,
James Hadfield,
![]() Emma Hodcroft,
Emma Hodcroft,
![]() Thomas Sibley,
Thomas Sibley,
![]() John Huddleston,
John Huddleston,
![]() Louise Moncla,
Louise Moncla,
![]() Misja Ilcisin,
Misja Ilcisin,
![]() Kairsten Fay,
Kairsten Fay,
![]() Jover Lee,
Jover Lee,
![]() Allison Black,
Allison Black,
![]() Colin Megill,
Colin Megill,
![]() Sidney Bell,
Sidney Bell,
![]() Barney Potter,
Barney Potter,
![]() Charlton Callender
Charlton Callender
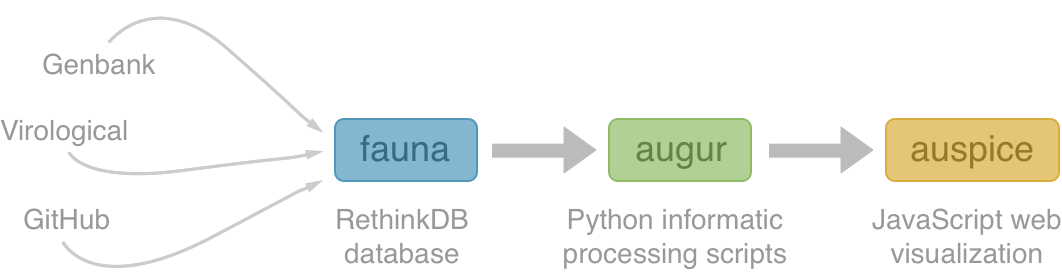
Nextstrain architecture
All code open source at github.com/nextstrain
Two central aims: (1) rapid and flexible phylodynamic analysis and
(2) interactive visualization
Rapid build pipeline for 1600 Ebola genomes
- Align with MAFFT (34 min)
- Build ML tree with RAxML (54 min)
- Temporally resolve tree and geographic ancestry with TreeTime (16 min)
- Total pipeline (1 hr 46 min)
Flexible pipelines constructed through command line modules
- Modules called via
augur filter,augur tree,augur traits, etc... - Designed to be composable across pathogen builds
- Defined pipeline, making steps obvious
- Provides dependency graph for fast recomputation
- Pathogen-specific repos give users an obvious foundation to build from
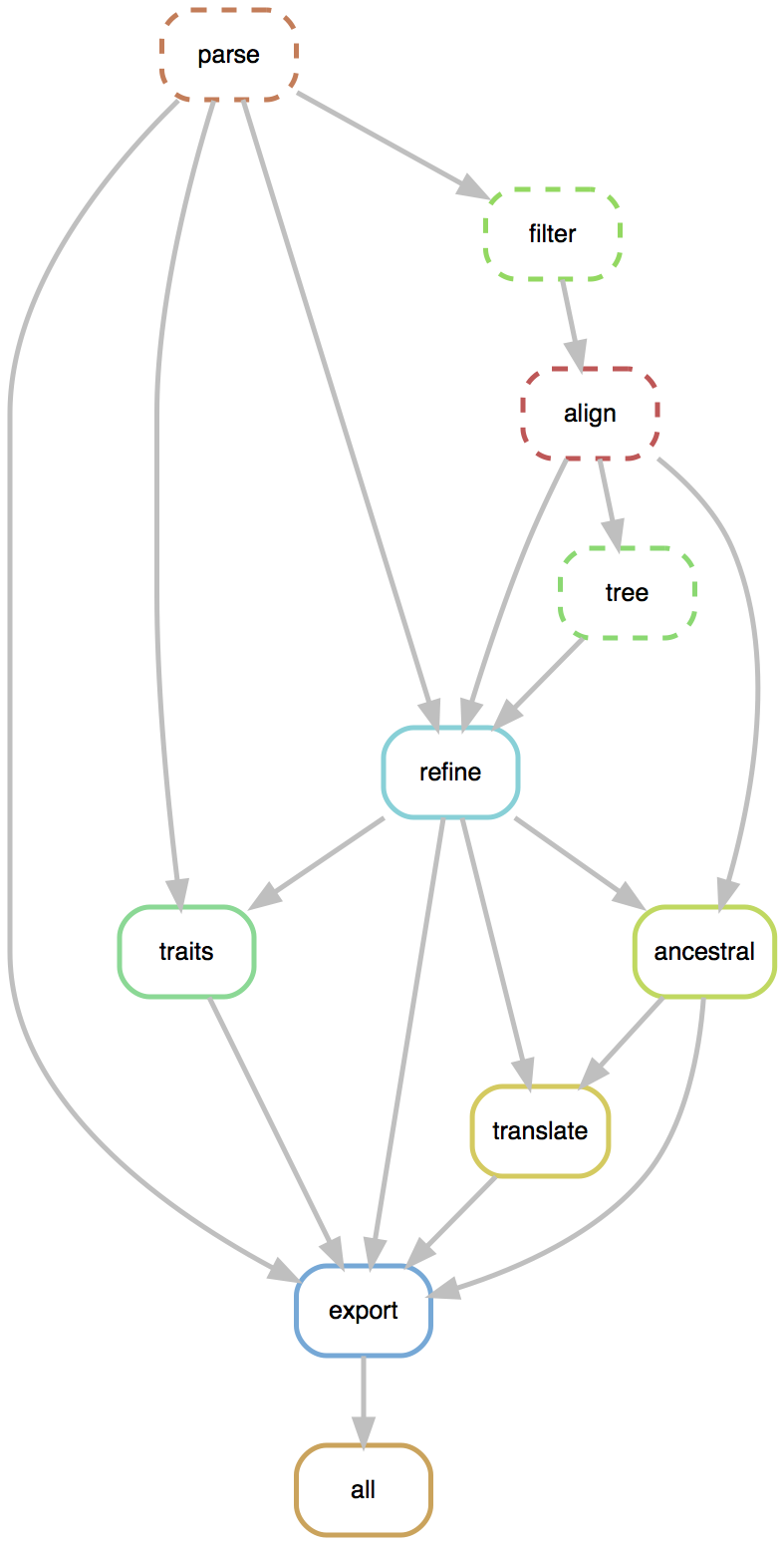
Nextstrain is two things
- a bioinformatics toolkit and visualization app, which can be used for a broad range of datasets
- a collection of real-time pathogen analyses kept up-to-date on the website nextstrain.org
nextstrain.org
Rapid on-the-ground sequencing in Makeni, Sierra Leone
"Community" builds to promote frictionless sharing of results
- Attempting to write us out of the picture
- JSON outputs uploaded to
github.com/czbiohub/dengue, would be available atnextstrain.org/community/czbiohub/dengue - Used now for a variety of pathogens including Lassa in Nigeria, global RSV and cassava virus
Ongoing DRC outbreak
Genomic epidemiology applied to North Kivu Ebola outbreak
with Placide Mbala-Kingebeni, Eddy Kinganda Lusamaki, Catherine Pratt, Mike Wiley, James Hadfield, Allison Black, Jean-Jacques Muyembe Tamfum, Steve Ahuka-Mundeke, Daniel Mukadi, Gustavo Palacios, Amadou Sall, Ousmane Faye, Eric Delaporte, Martine Peeters and many others
Nextstrain is being used to track North Kivu outbreak
- Institut National de Recherche Biomédicale (INRB) is sequencing samples in Kinshasa
- Data and pipeline housed at github.com/inrb-drc/ebola-nord-kivu
- Resulting analyses are being deployed live to nextstrain.org/community/inrb-drc/ebola-nord-kivu
Allison Black (PhD student) and James Hadfield (postdoc) working with scientists at the INRB. Goal is to provide training in bioinformatics, Nextstrain and genomic epidemiology.

View of current genomic data
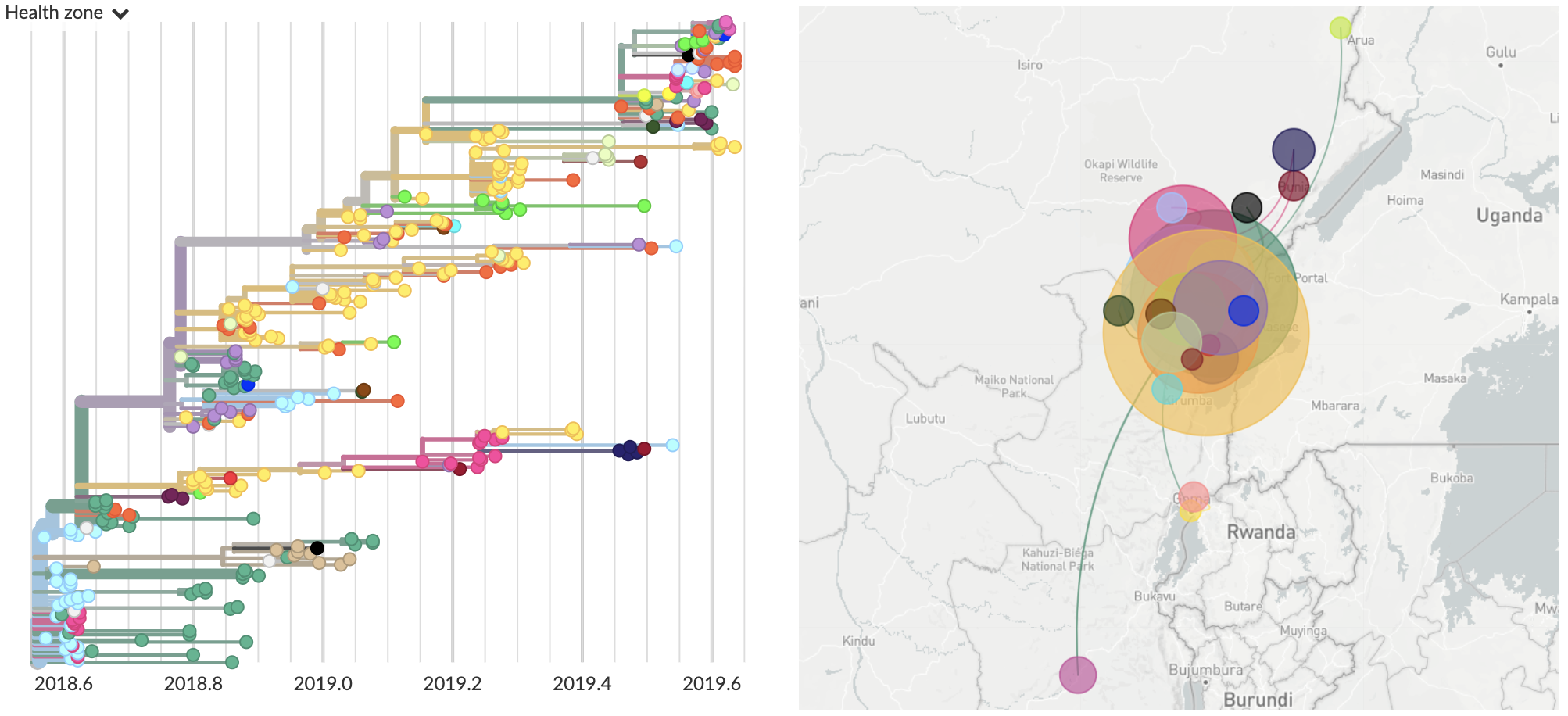
Current dataset:
- 376 full genomes sequenced (15% of confirmed cases)
- Most recent sequenced virus collected Sep 12 (4 weeks ago)
Often (but not always) need specific actionable pieces of information rather than large-scale understanding. For Ebola outbreak response, I believe this needs to revolve around contact tracing.
Superspreader event in June
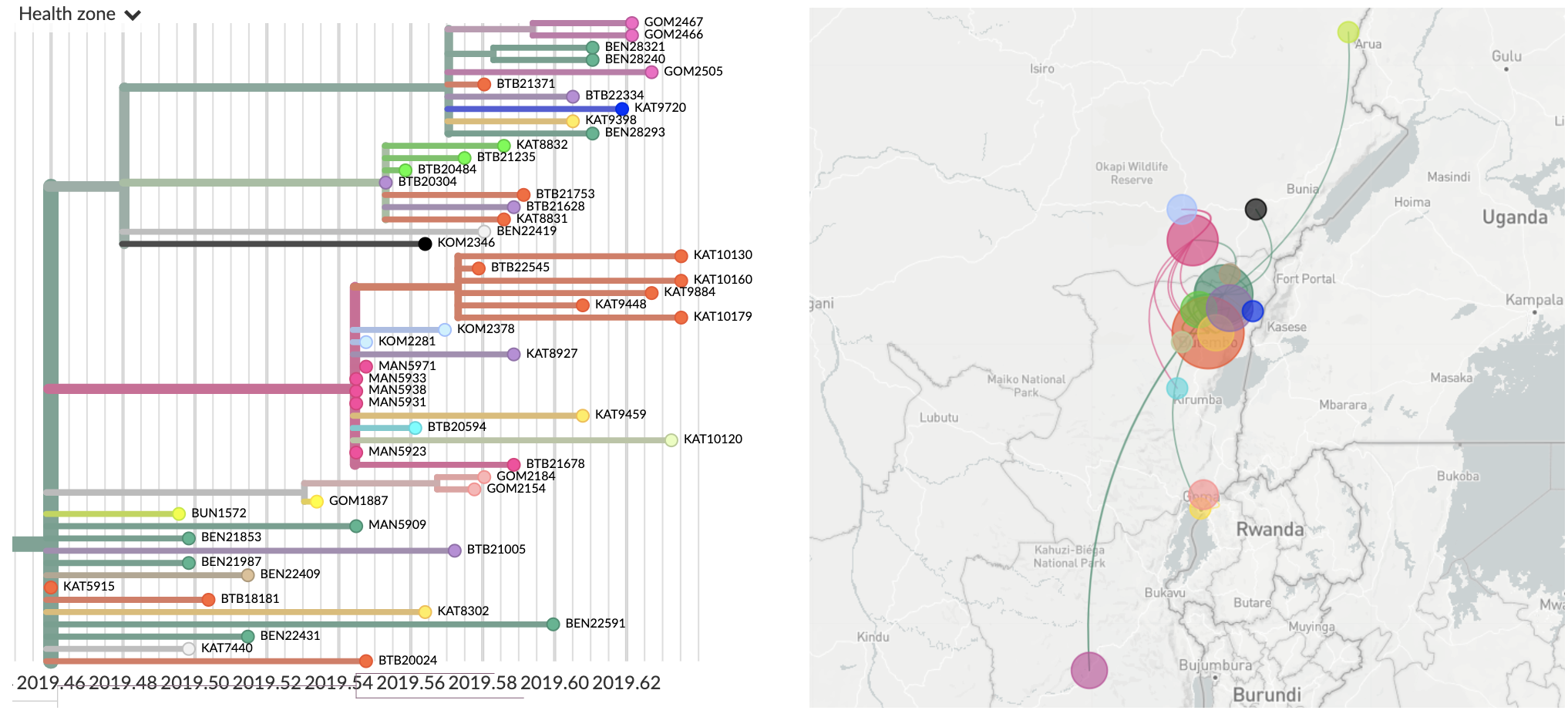
Using narratives to walk through specific transmission inferences
- We're rolling out new narratives functionality in Nextstrain
- These are Markdown posts that allow you to pair narrative text to visualization state
- Made possible through an early decision to embed visualization state in URL
- Example narrative for Ebola in the DRC here: nextstrain.org/narratives/inrb-ebola-example-sit-rep
Tracking seasonal influenza virus evolution
Population turnover of A/H3N2 influenza is extremely rapid
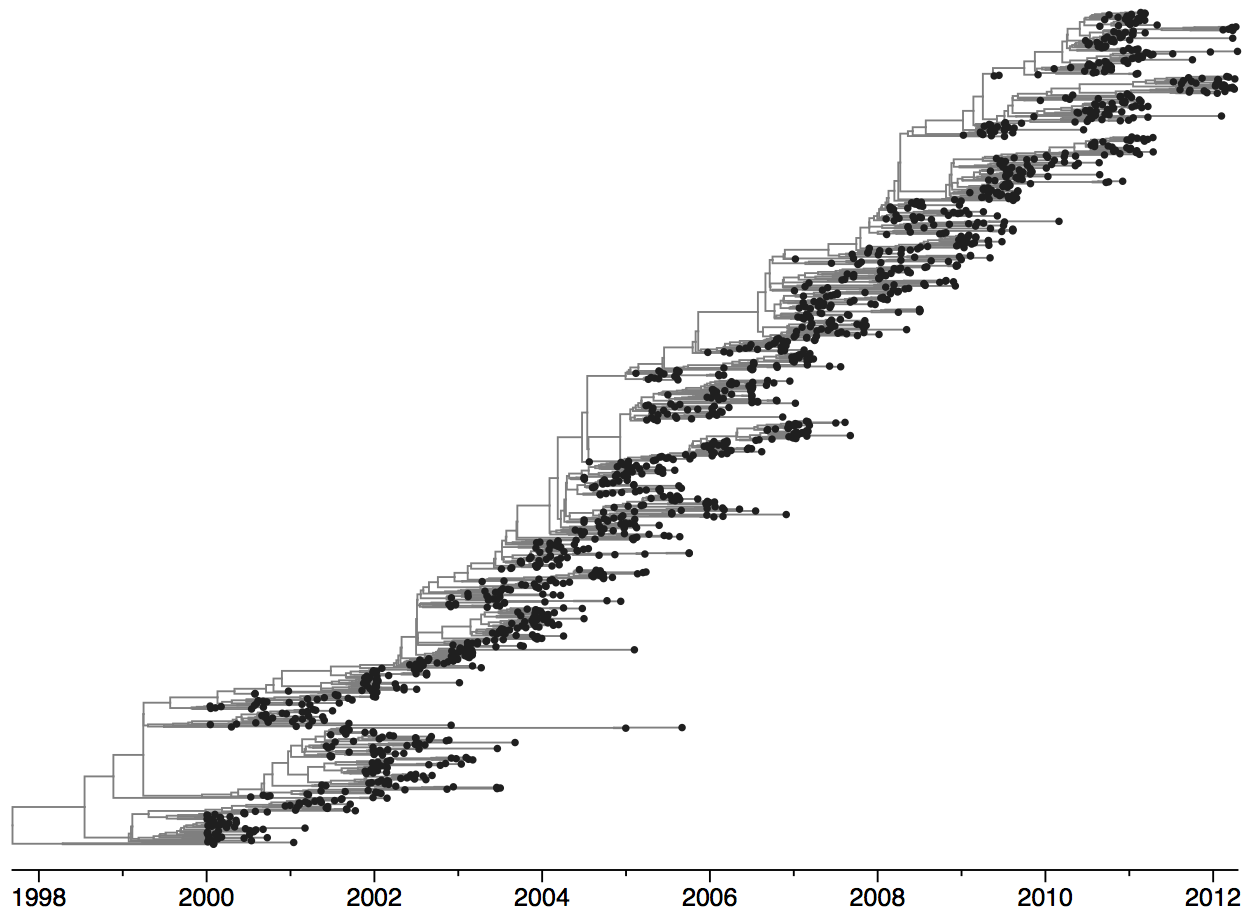
Clades emerge, die out and take over
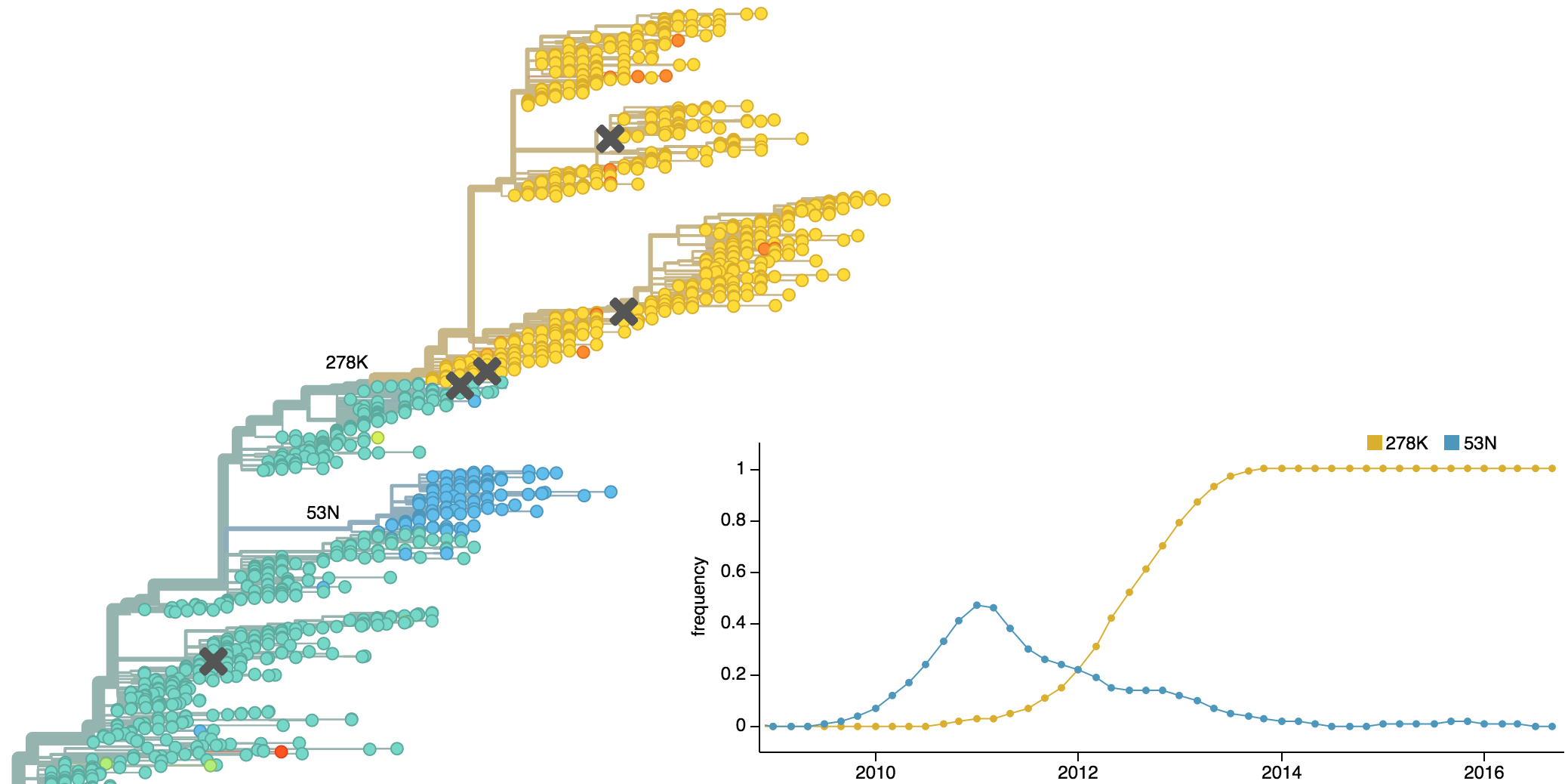
Clades show rapid turnover

Dynamics driven by antigenic drift
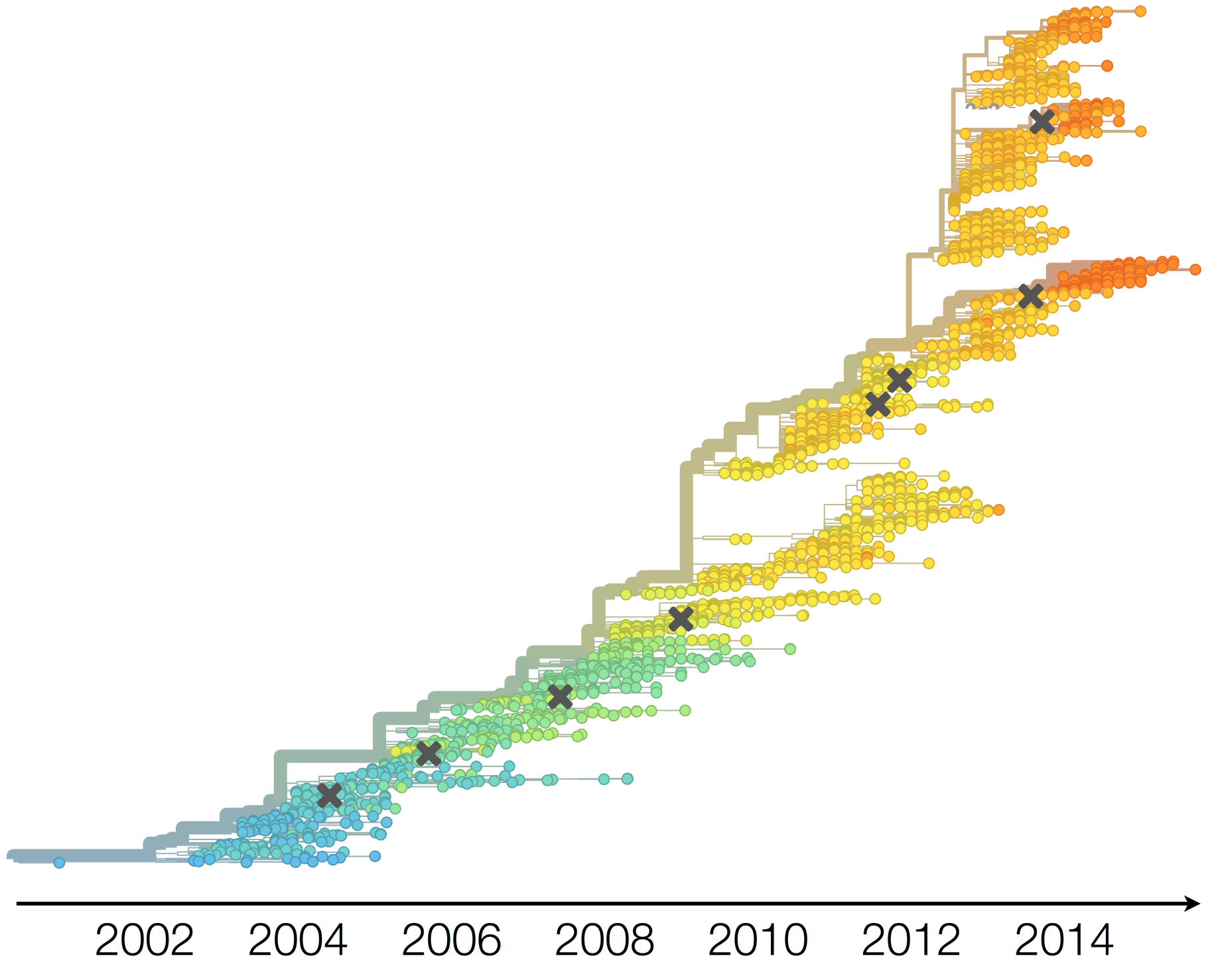
Drift necessitates vaccine updates
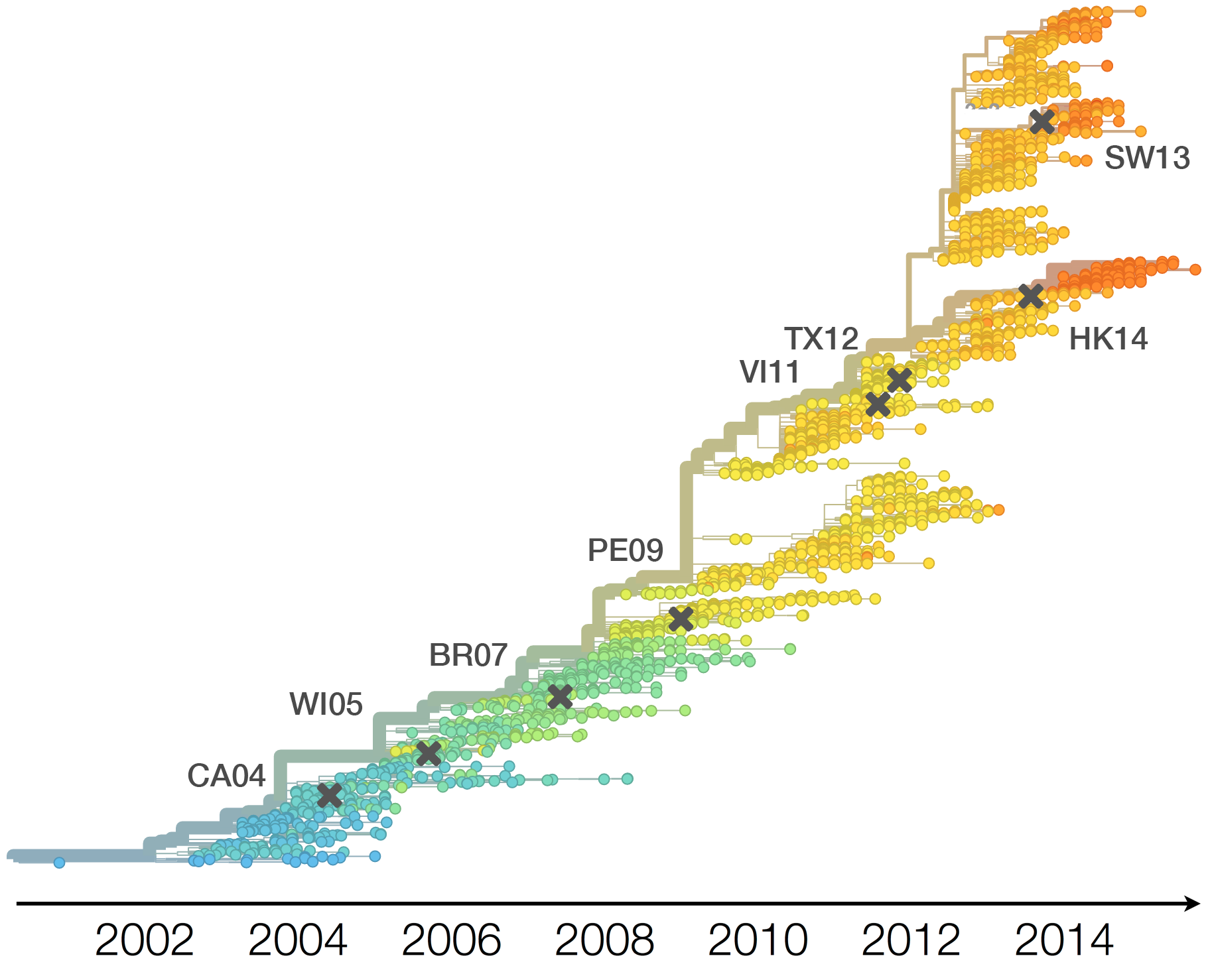
H3N2 vaccine updates occur every ~2 years
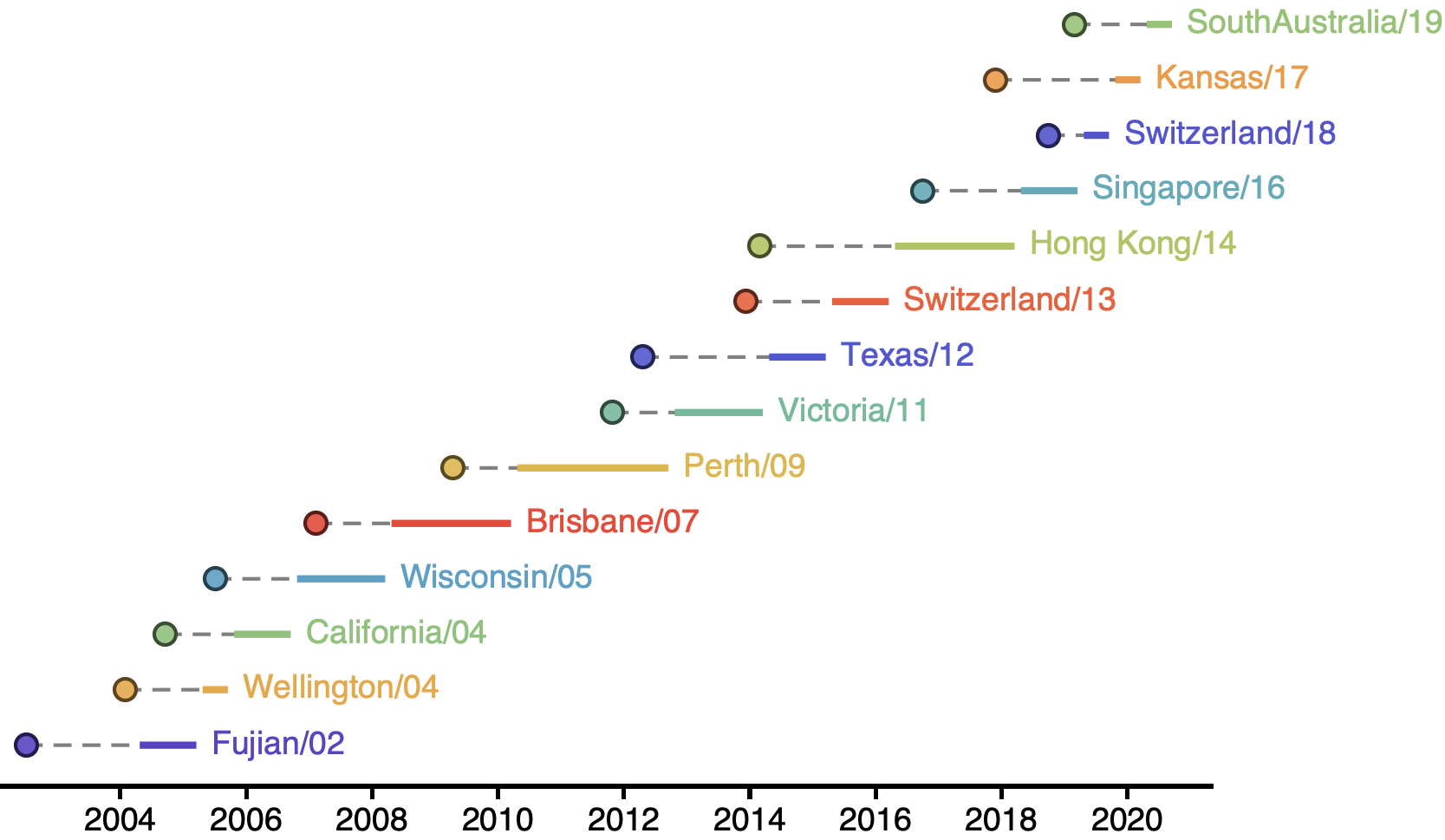
Vaccine strain selection by WHO
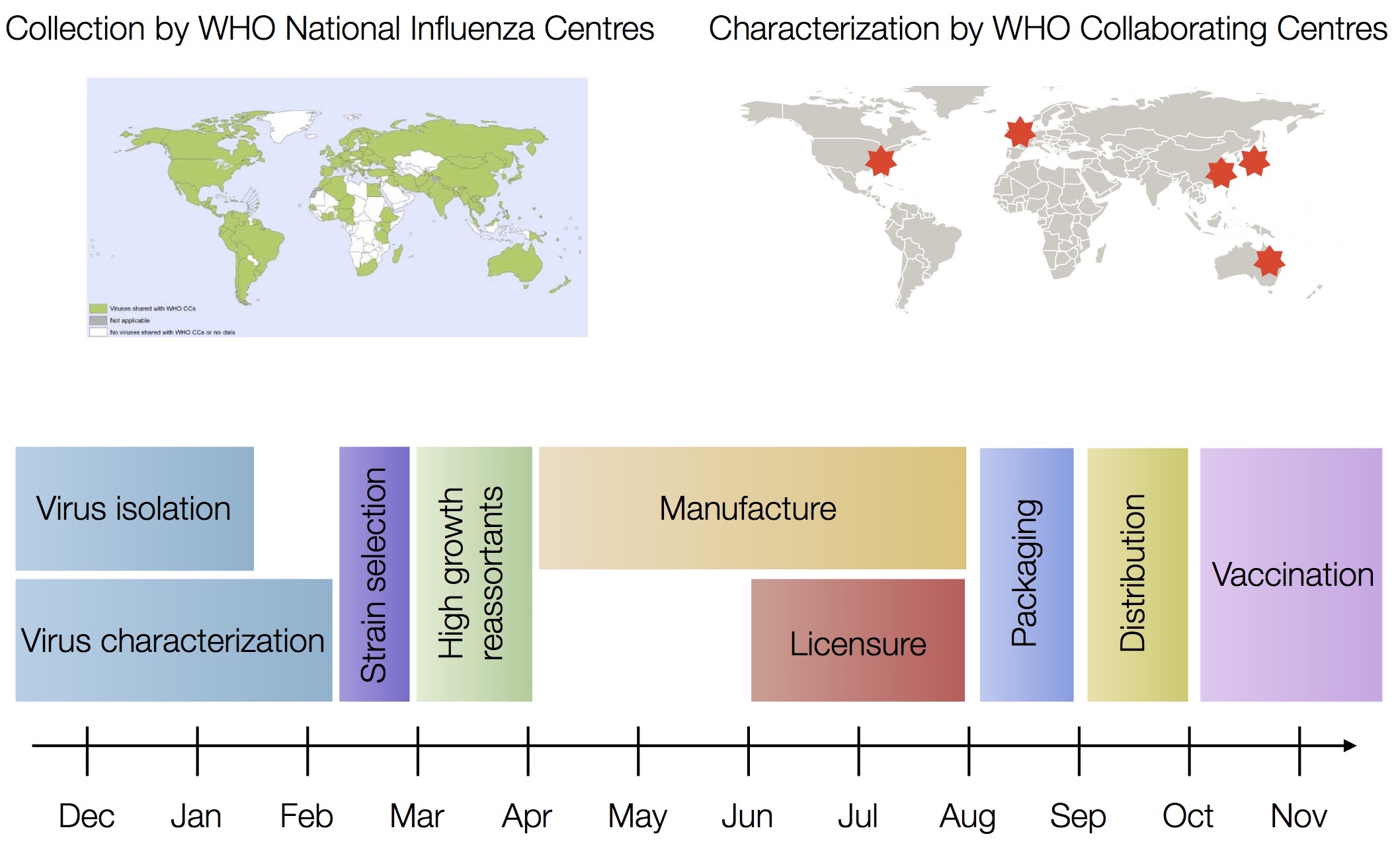
Working with Richard Neher, we decided to tackle this head on and build something that:
- Charts behavior of specific strains
- Can be kept continually up to date
Nextflu
Project to provide a real-time view of the evolving influenza population
Made possible by rapid and open sharing of WHO GISRS data through GISAID database
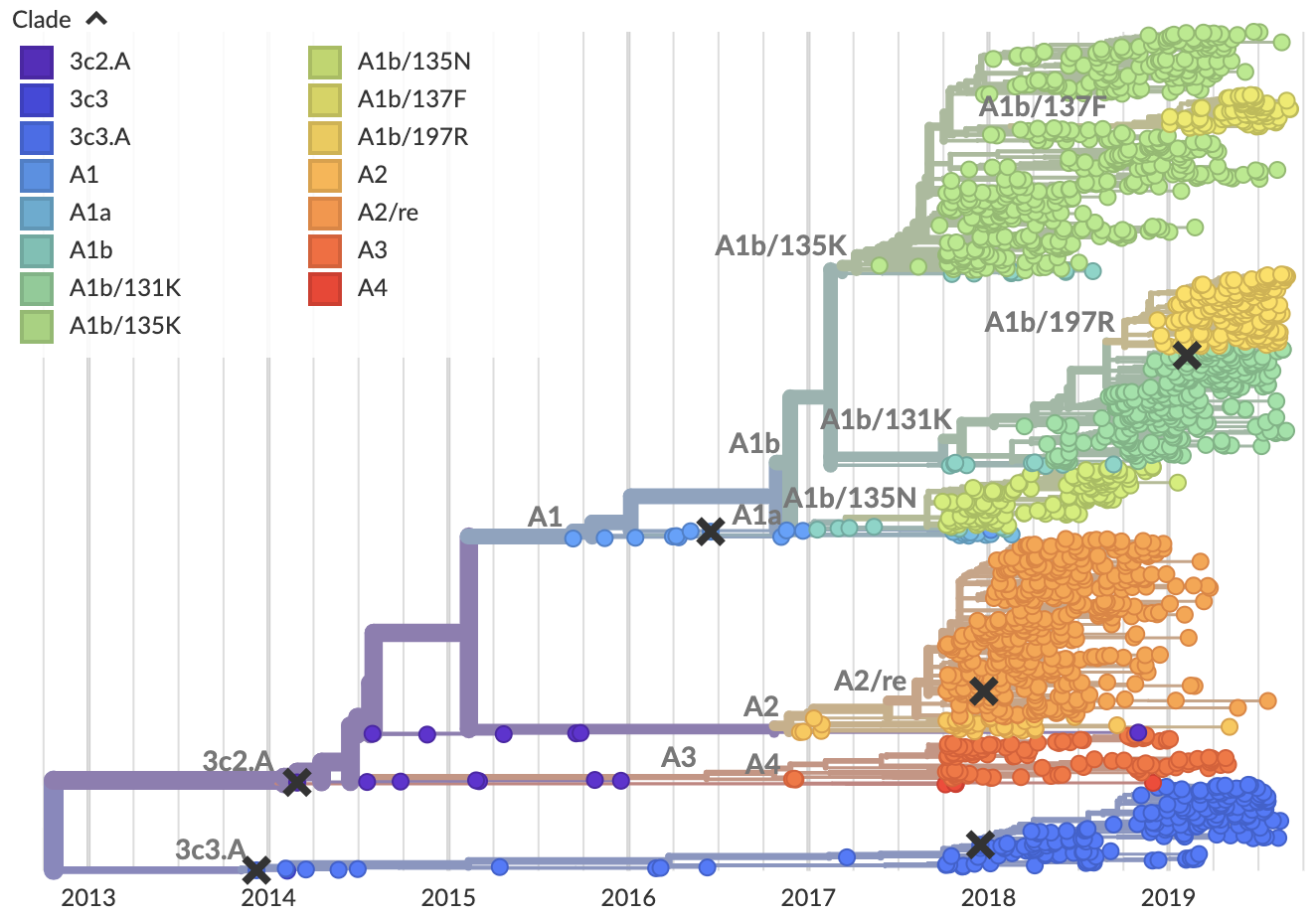
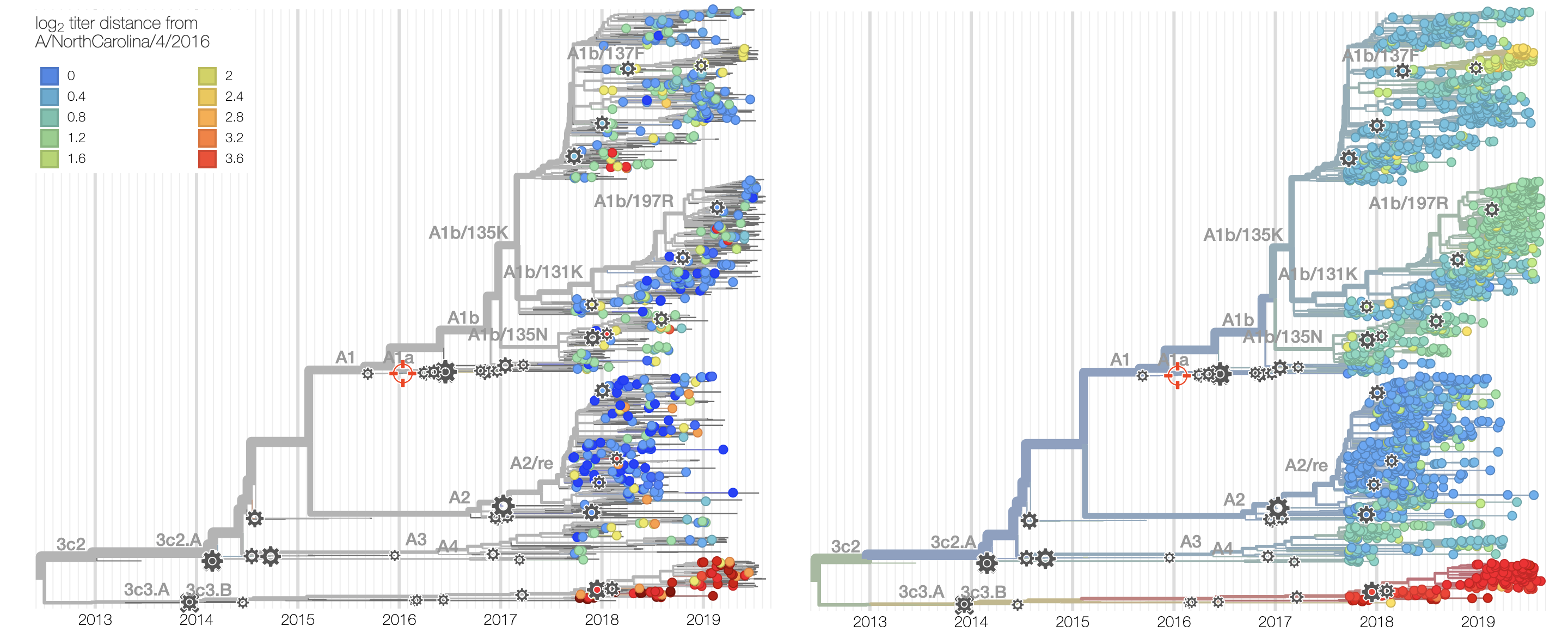
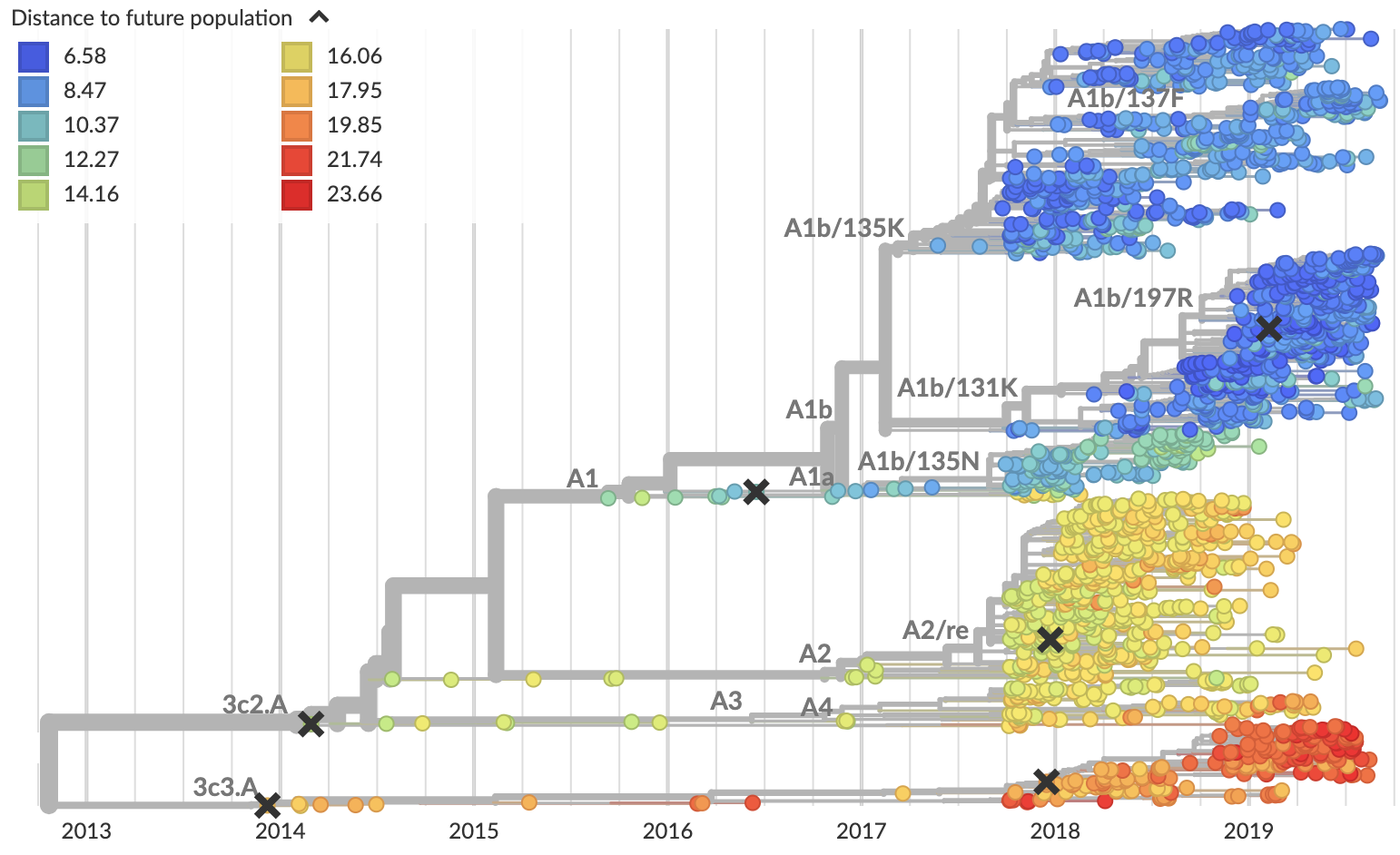
Current view of H3N2 from nextstrain.org/flu
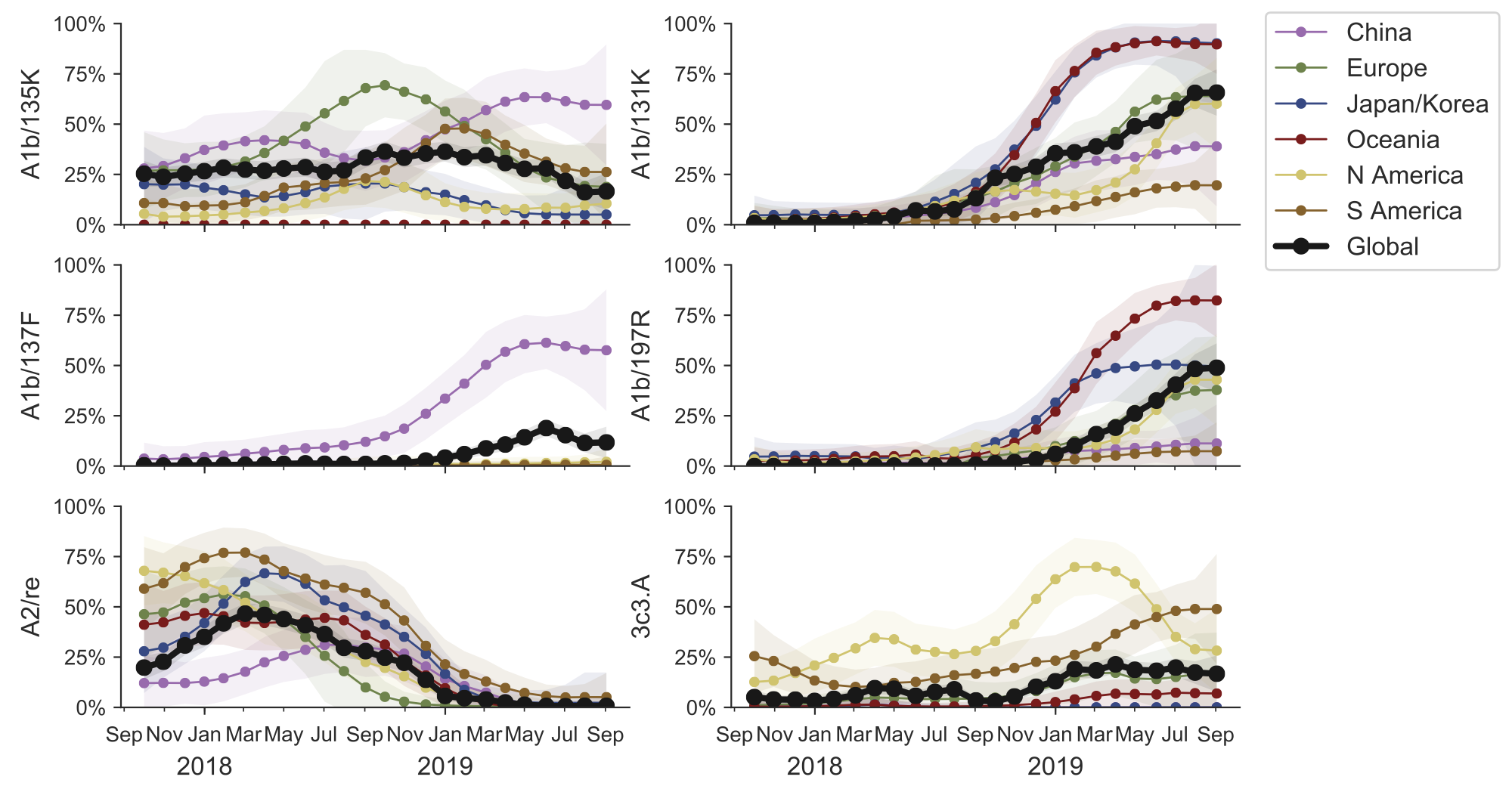
Clade frequencies show recent rise of A1b/197R viruses
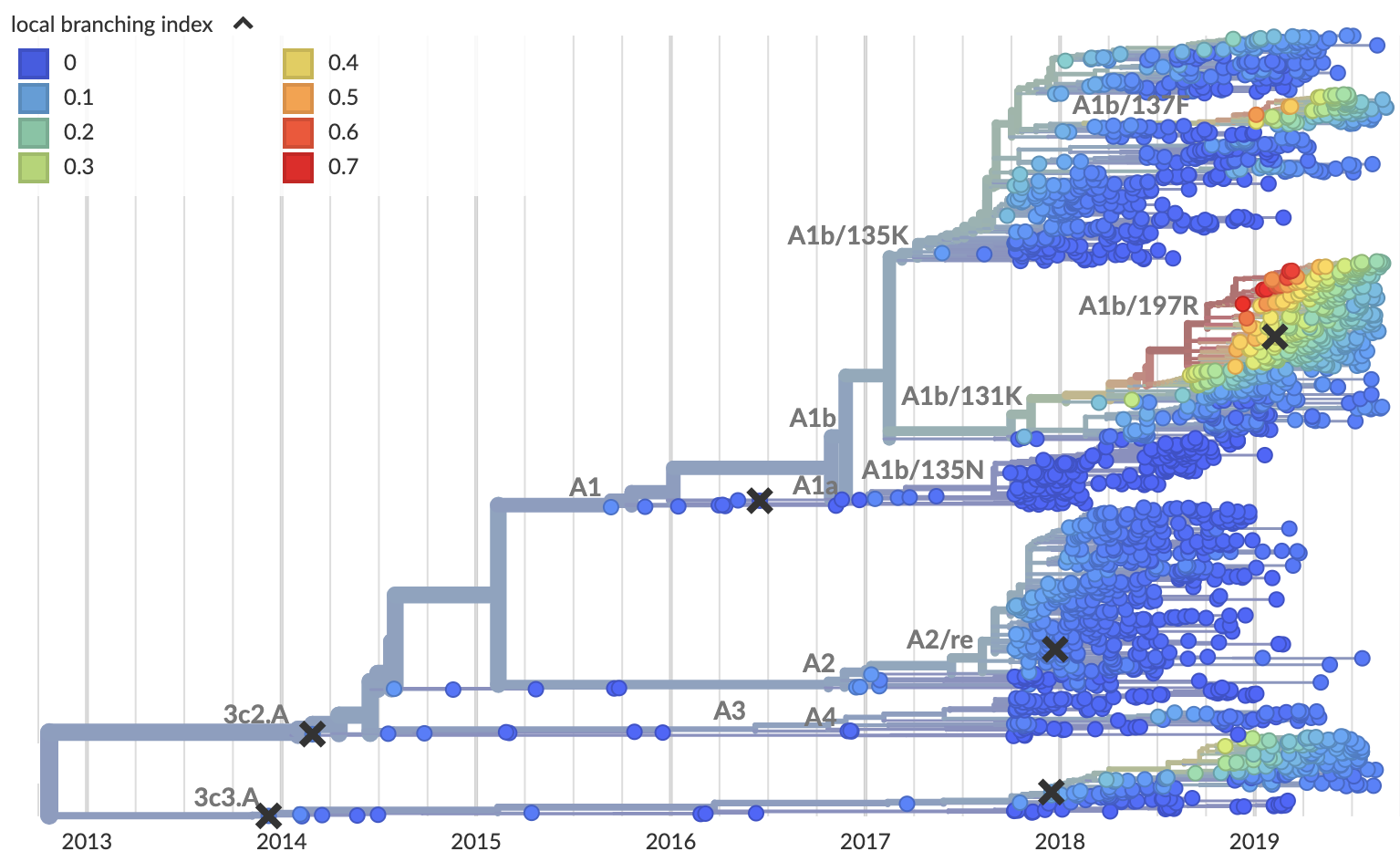
Local branching index (LBI) also points to A1b/197R as the most rapidly spreading clade
Serological assay data indicates largely similar antigenic phenotypes in A1b viruses
Forecasting seasonal influenza A/H3N2 evolution
with ![]() John Huddleston
and
John Huddleston
and ![]() Richard Neher
Richard Neher
Fitness models project strain frequencies
Future frequency $x_i(t+\Delta t)$ of strain $i$ derives from strain fitness $f_i$ and present day frequency $x_i(t)$, such that
$$\hat{x}_i(t+\Delta t) = x_i(t) \, \mathrm{exp}(f_i \, \Delta t)$$
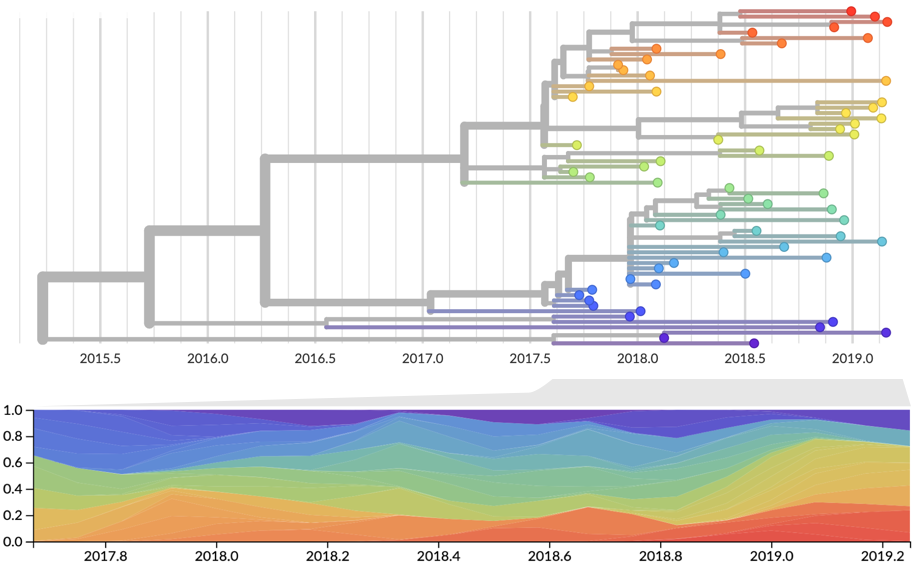
Total strain frequencies at each timepoint are normalized. This captures clonal interference between competing lineages.
Two inputs
- Estimate of present-day strain frequencies $x(t)$
- Estimate of present-day strain fitnesses $f$
Strain frequency estimated via region-weighted KDE
Strain fitness estimated from viral attributes
The fitness $f$ of strain $i$ is estimated as
$$\hat{f}_i = \beta^\mathrm{A} \, f_i^\mathrm{A} + \beta^\mathrm{B} \, f_i^\mathrm{B} + \ldots$$
where $f^A$, $f^B$, etc... are different standardized viral attributes and $\beta^A$, $\beta^B$, etc... coefficients are trained based on historical evolution
| Antigenic drift | Intrinsic fitness | Recent growth |
|---|---|---|
| epitope mutations | non-epitope mutations | local branching index |
| HI titers | DMS data (via Bloom lab) | delta frequency |
Future population depends on frequency and fitness
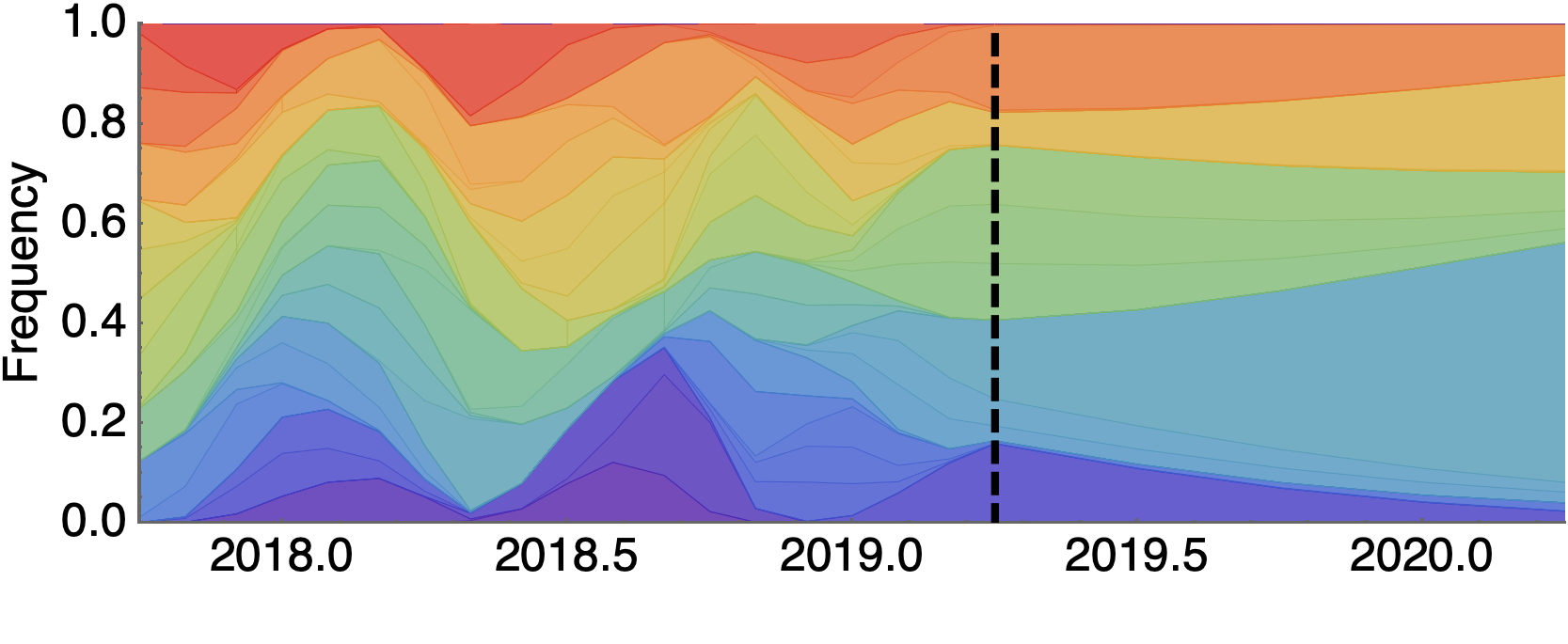
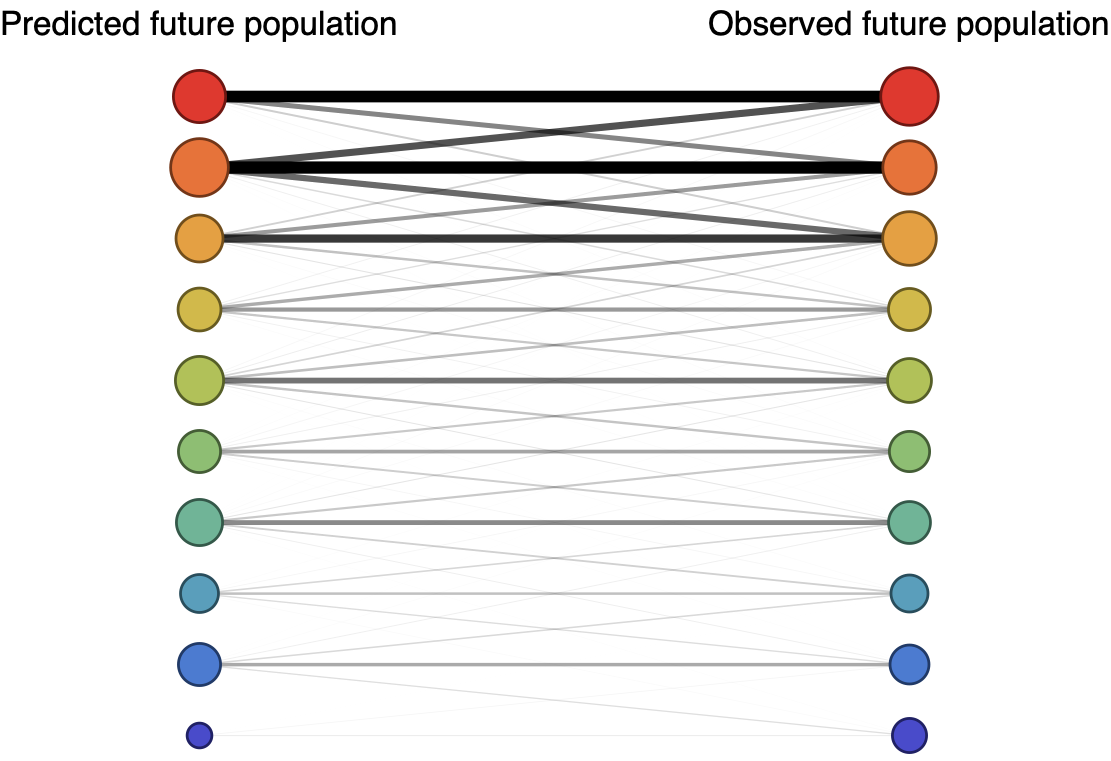
Forecast assessed based on weighted distance match to observed future population
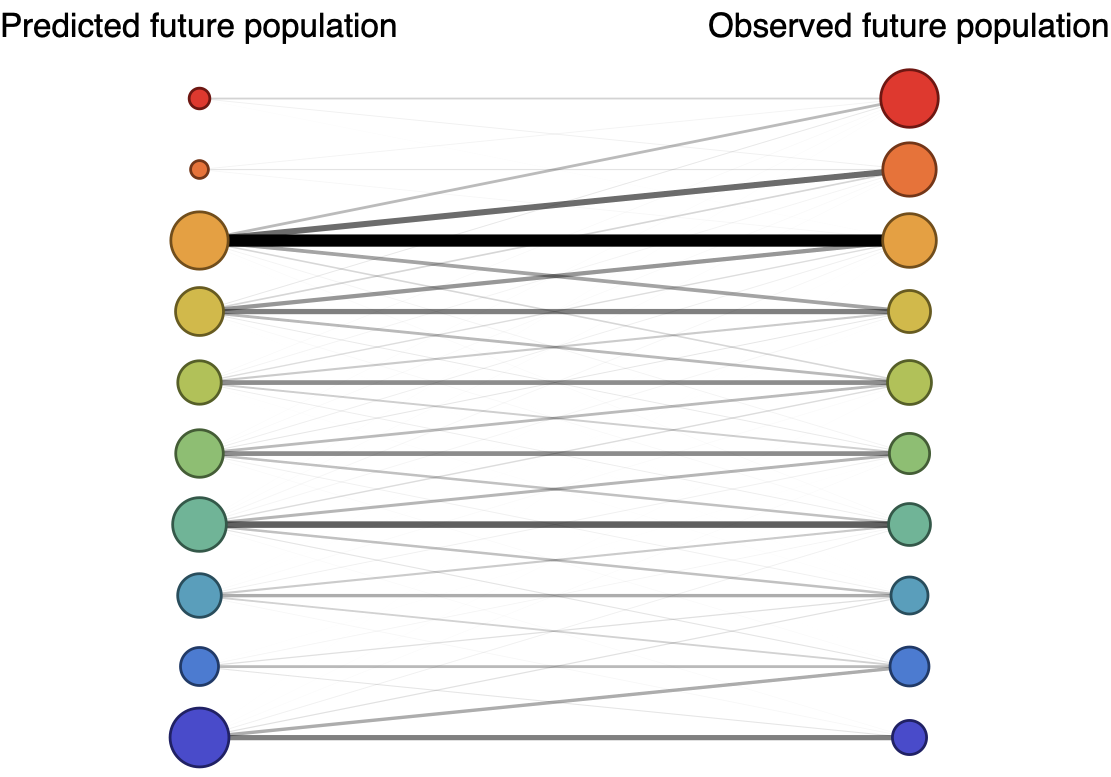
Forecast assessed based on weighted distance match to observed future population
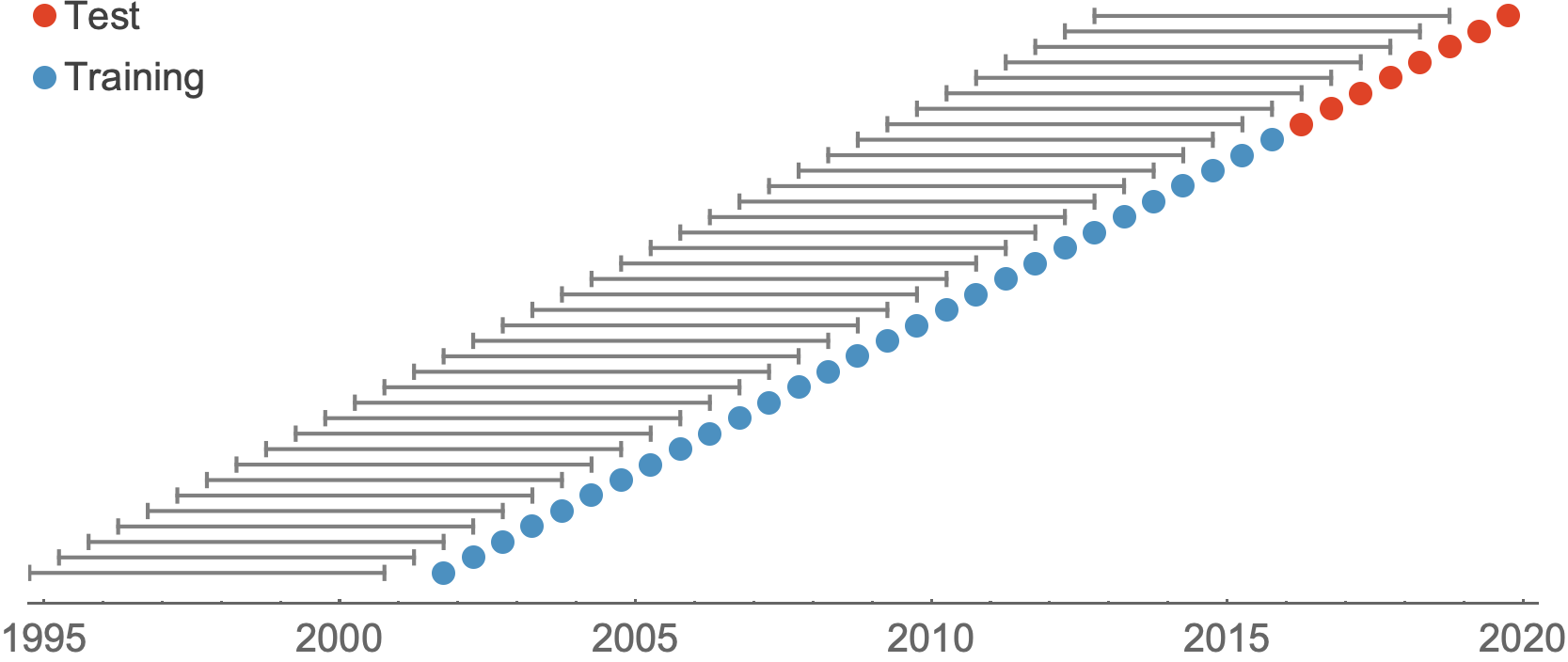
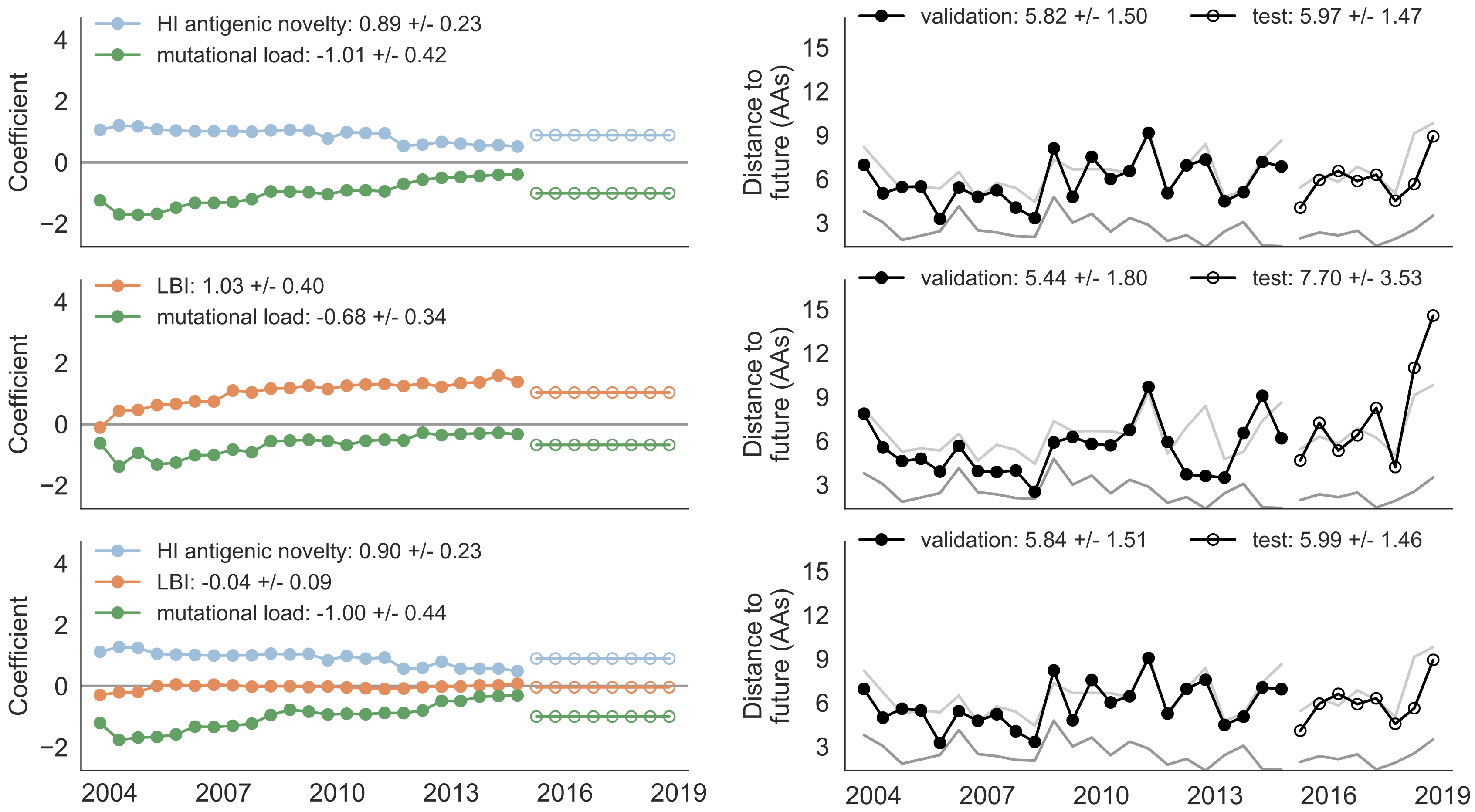
Train in 6-year sliding windows from 1995 to 2015 with most recent years held out as test
Single predictors favor HI drift, non-epitope fitness and local branching index
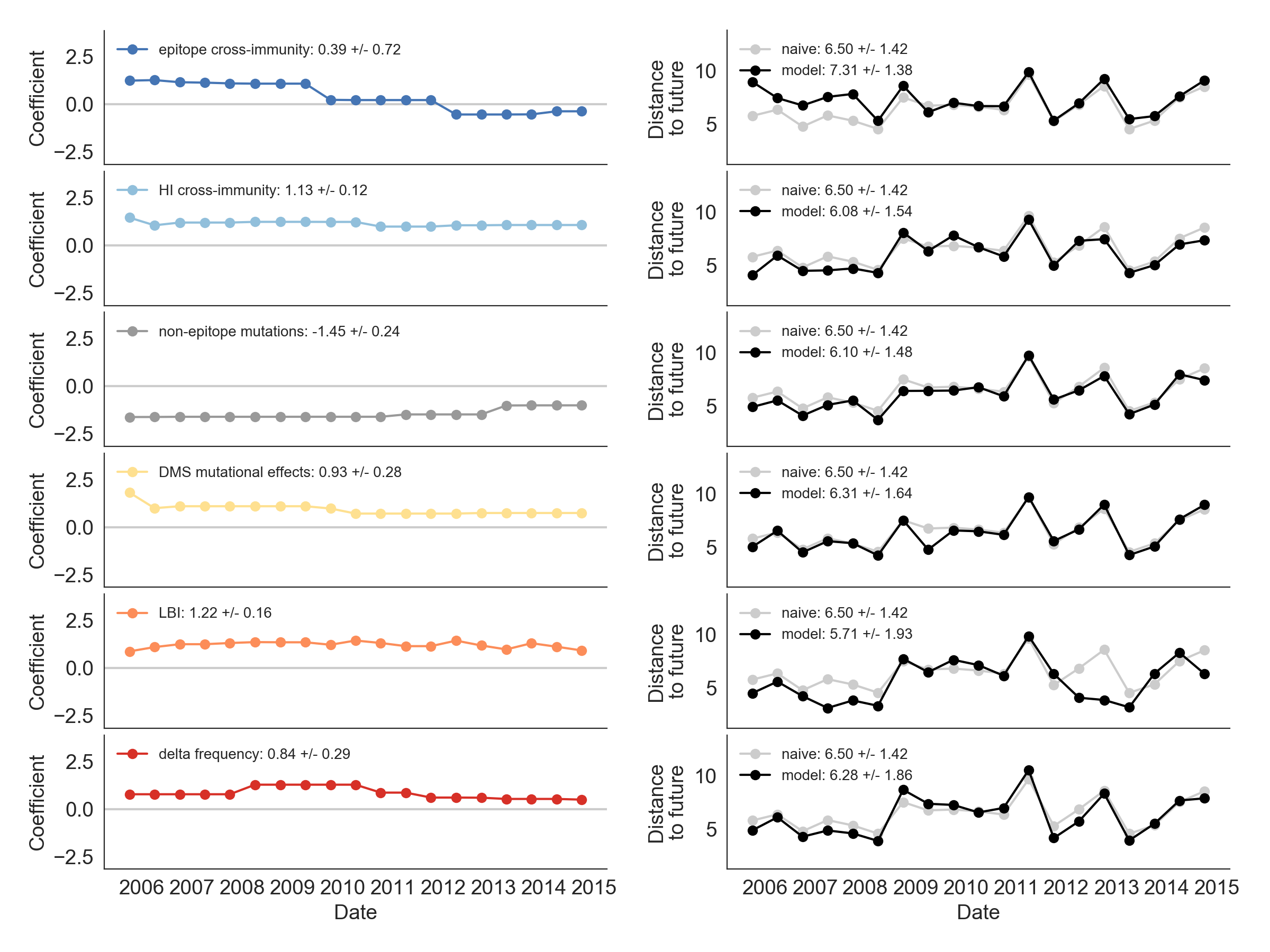
Composite models suggest a combination of local branching index and non-epitope fitness
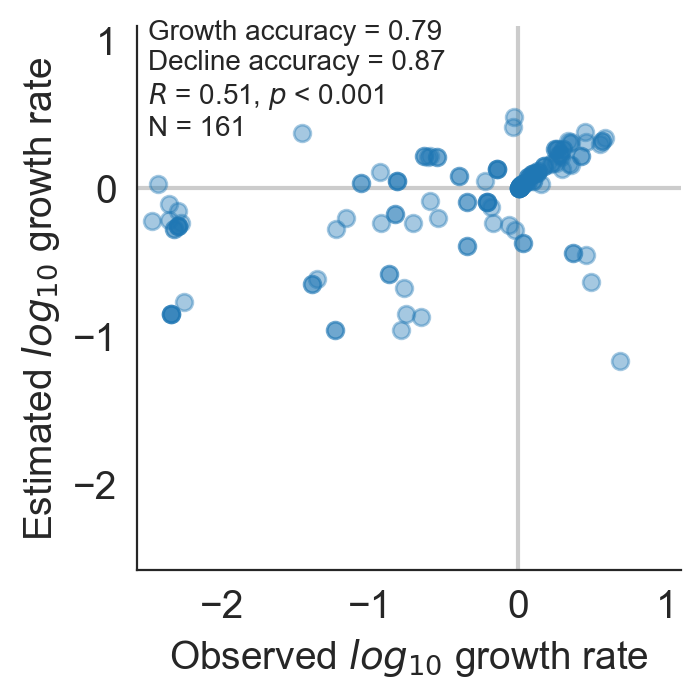
Model successfully predicts clade growth
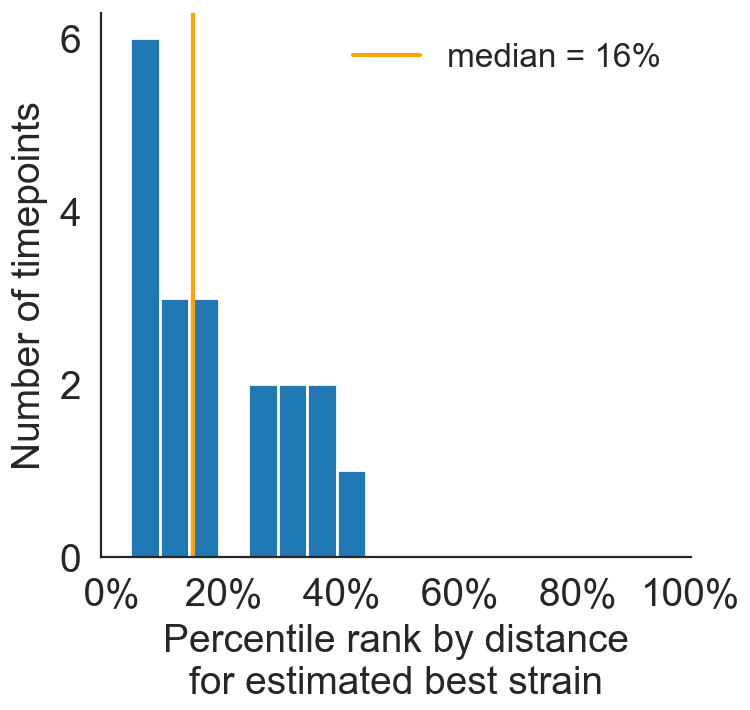
Best pick from model is generally close to best possible retrospective pick
Forecast from current virus population
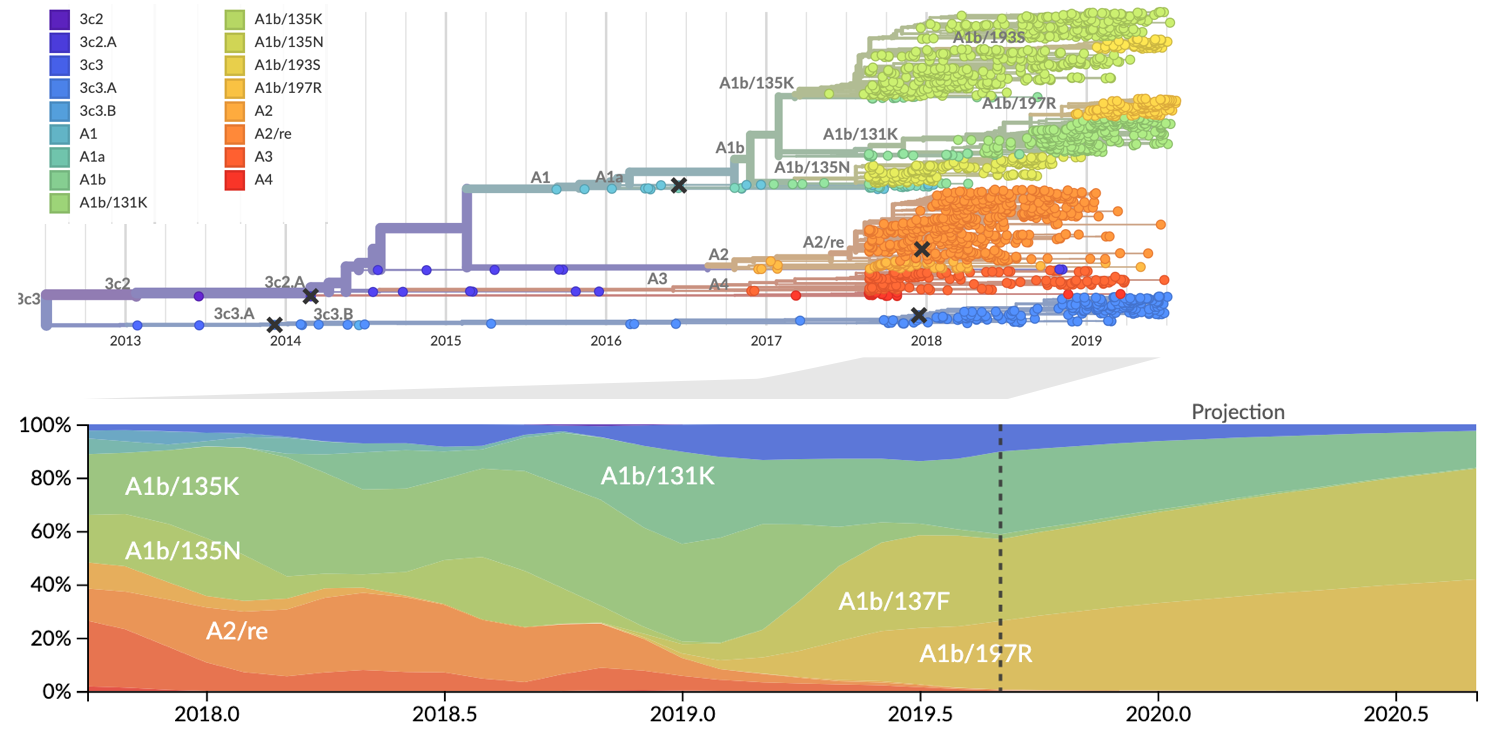
Predicted sequence match of circulating strains to future population
These forecasts are now rolled out live to nextstrain.org/flu
The same machinery can be applied to other viruses, like Sidney's work here on dengue
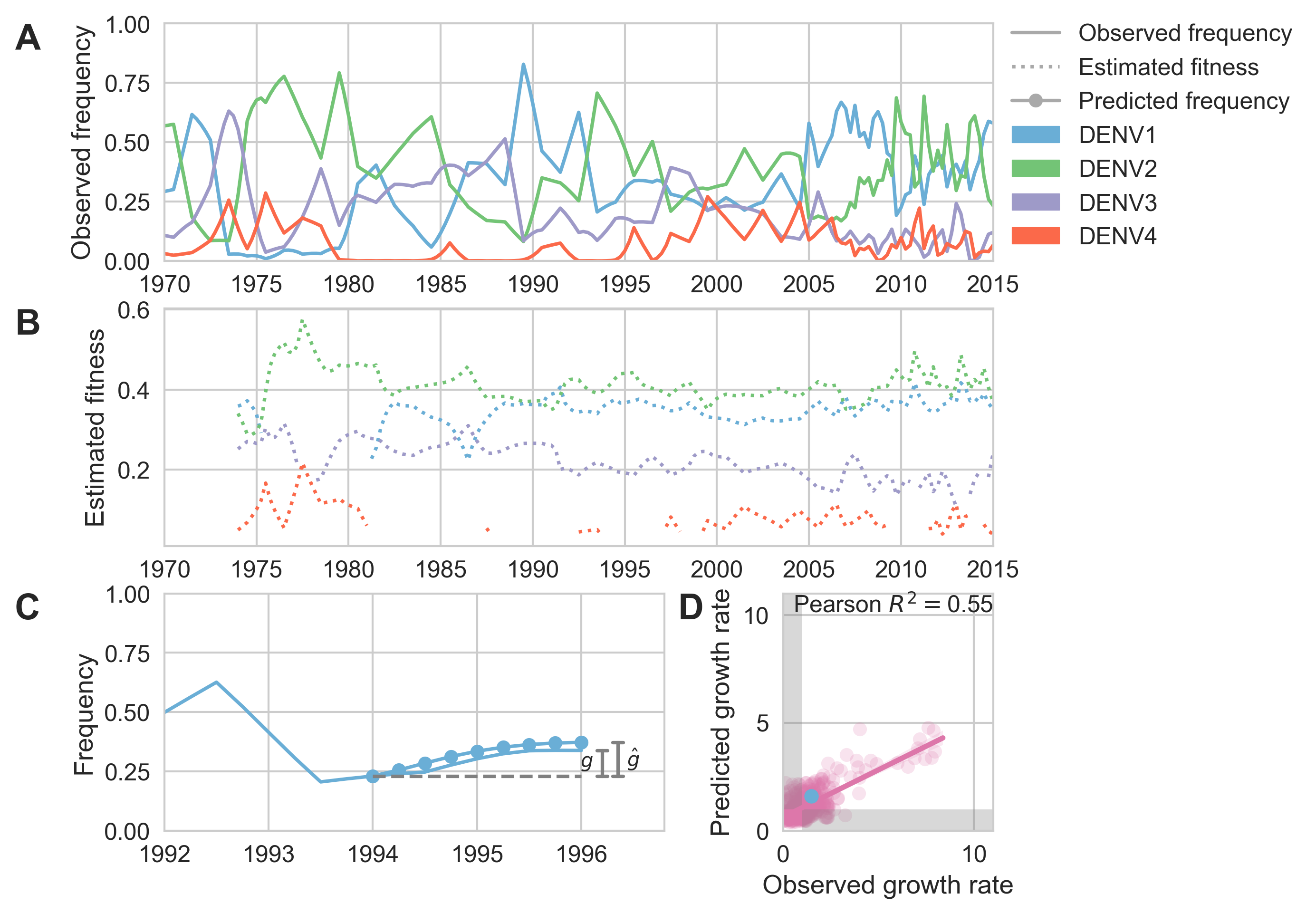
This work relies on rapid and open sharing of pathogen genomic data.
All Nextstrain code is entirely open source and intended to be used by the community. We've been working hard on improving documentation at nextstrain.org/docs. You're most welcome to kick the tires.
Acknowledgements
Bedford Lab:
![]() Alli Black,
Alli Black,
![]() John Huddleston,
John Huddleston,
![]() James Hadfield,
James Hadfield,
![]() Katie Kistler,
Katie Kistler,
![]() Louise Moncla,
Louise Moncla,
![]() Maya Lewinsohn,
Maya Lewinsohn,
![]() Thomas Sibley,
Thomas Sibley,
![]() Jover Lee,
Jover Lee,
![]() Kairsten Fay,
Kairsten Fay,
![]() Misja Ilcisin
Misja Ilcisin
Ebola in West Africa: Gytis Dudas, Andrew Rambaut, Luiz Carvalho, Philippe Lemey, Marc Suchard, Andrew Tatem Nextstrain: Richard Neher, James Hadfield, Emma Hodcroft, Tom Sibley, John Huddleston, Sidney Bell, Barney Potter, Colin Megill, Charlton Callender Ebola in DRC: James Hadfield, Allison Black, Eddy Kinganda Lusamaki, Placide Mbala-Kingebeni, Catherine Pratt, Mike Wiley, Jean-Jacques Muyembe Tamfum, Steve Ahuka-Mundeke, Daniel Mukadi, Gustavo Palacios, Amadou Sall, Ousmane Faye, Eric Delaporte, Martine Peeters, David Blazes, Cecile Viboud, David Spiro Seasonal flu: WHO Global Influenza Surveillance Network, GISAID, John Huddleston, Richard Neher, Barney Potter, Dave Wentworth, Becky Garten