Molecular evolution of SARS-CoV-2
Trevor Bedford (@trvrb)
Associate Professor, Fred Hutchinson Cancer Research Center
6 Feb 2021
CoVPN Meeting
NIH
Slides at: bedford.io/talks
1. Real-time tracking of SARS-CoV-2 evolution
2. Emergence of variants of concern
3. Expectations for antigenic evolution
4. Continued genomic surveillance
Real-time tracking of SARS-CoV-2 evolution
Over 480k SARS-CoV-2 genomes shared to GISAID and evolution tracked in real-time at nextstrain.org
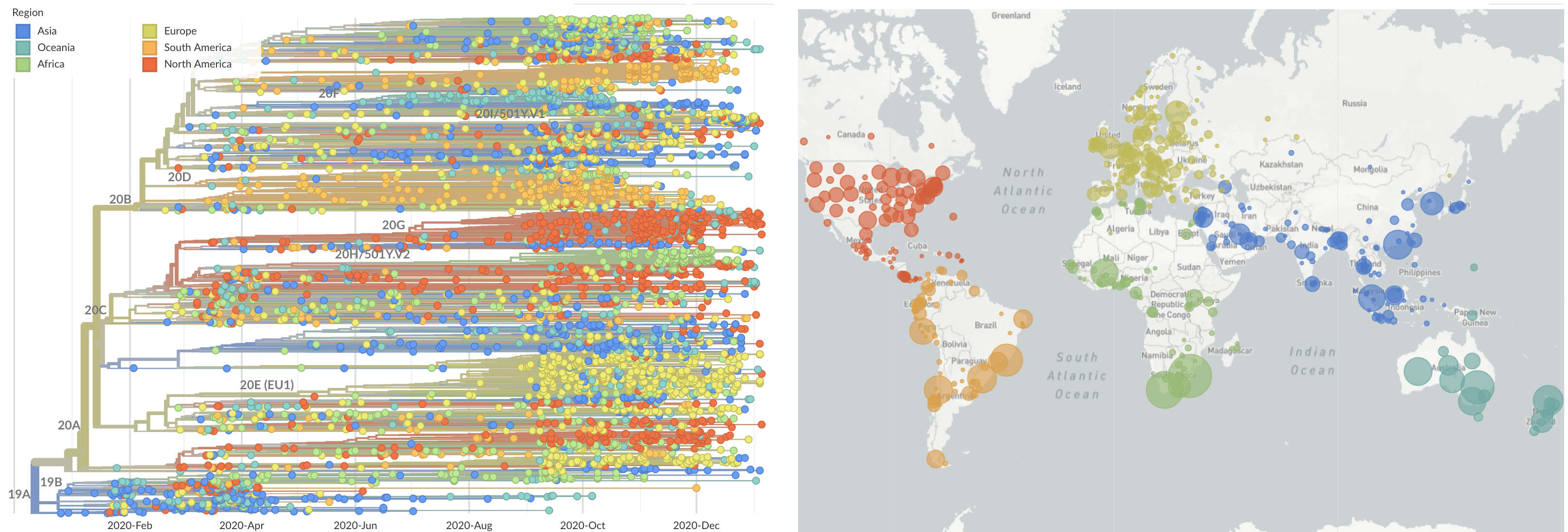
SARS-CoV-2 lineages establish globally in February and March
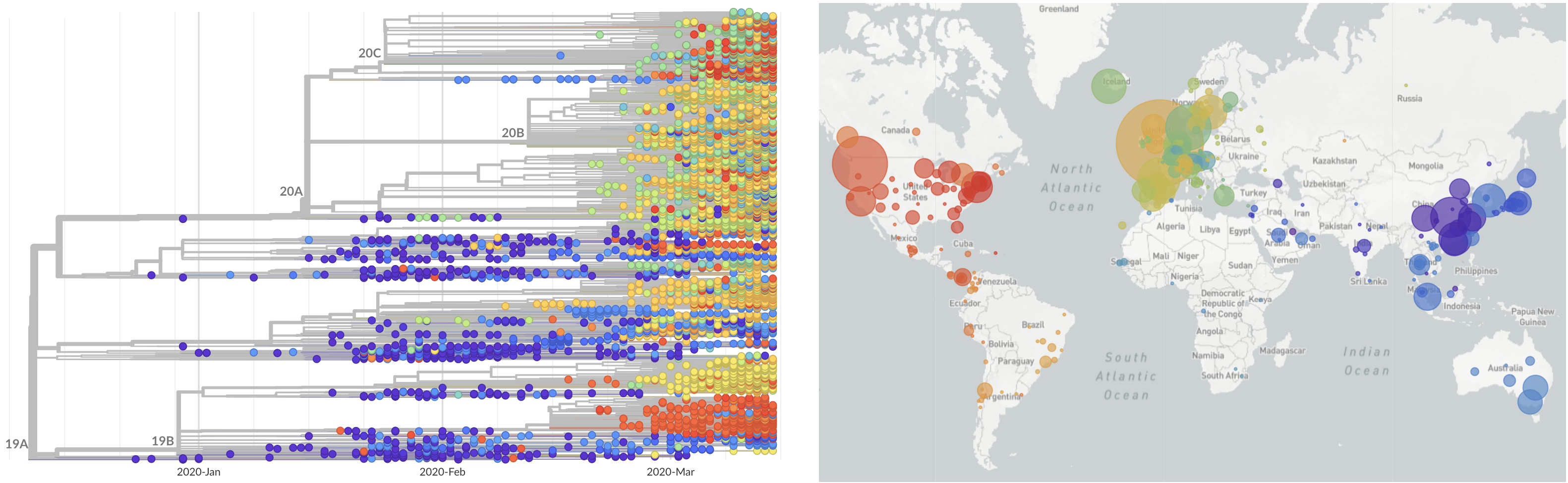
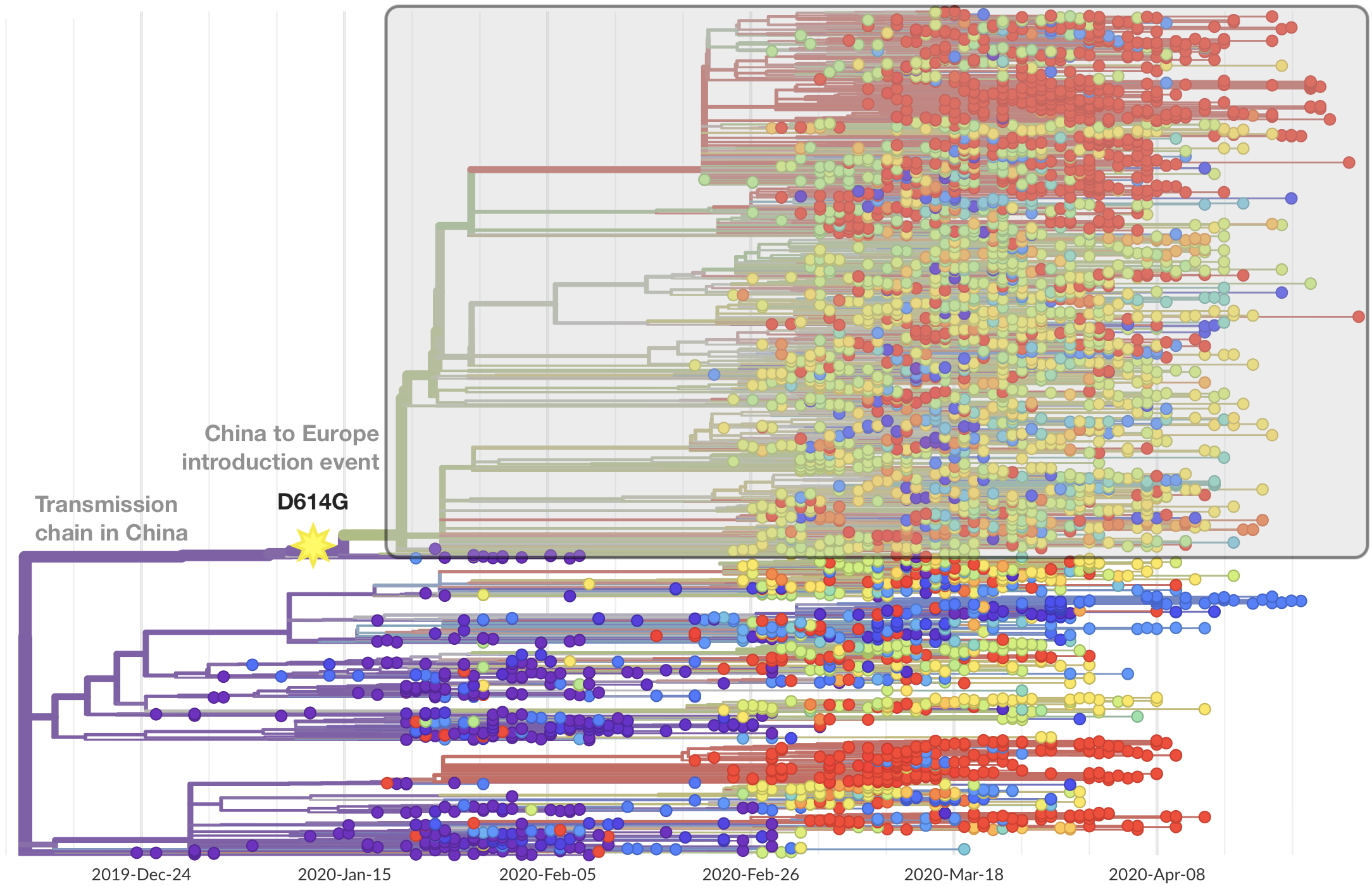
Limited early mutations like D614G spread globally during initial wave
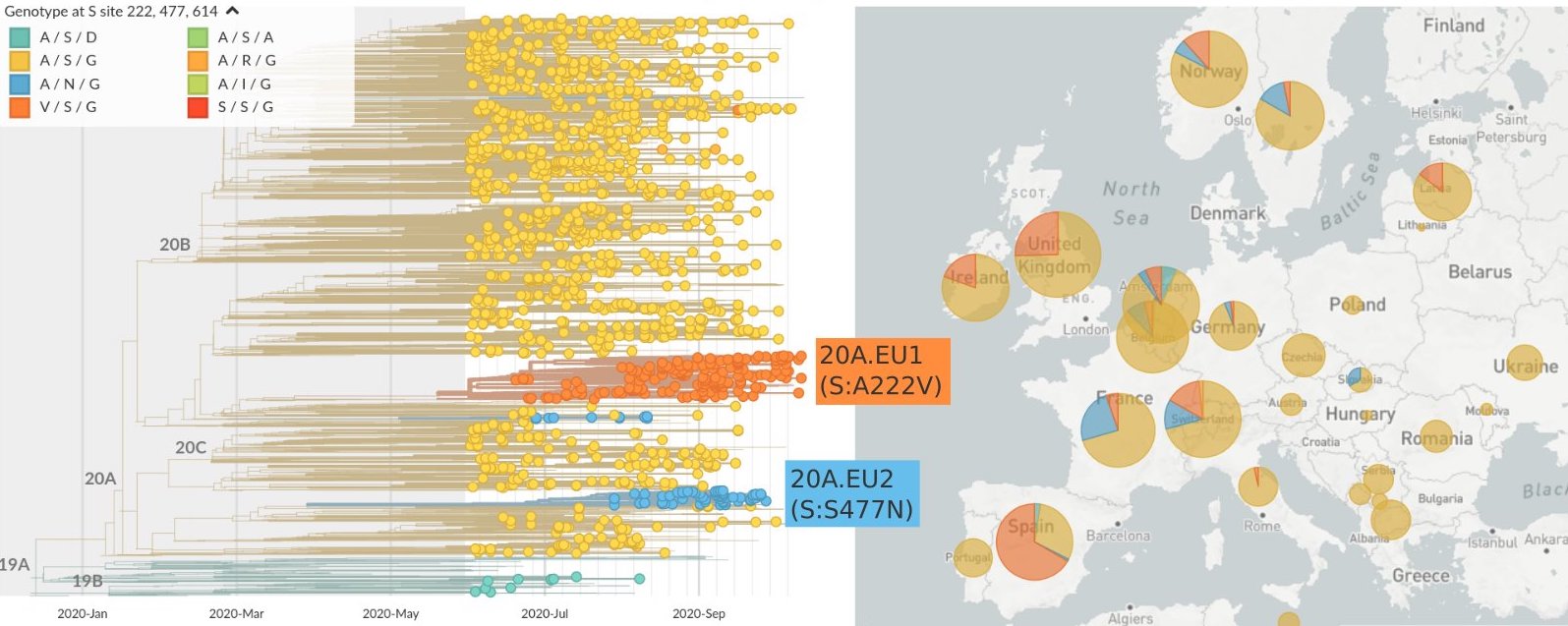
Summer and fall variants were confined to regional dominance
Emergence of variants of concern
484K and 501Y repeatedly emerging across the world

Emergence of 501Y.V1 (B.1.1.7) in the UK

Emergence of 501Y.V2 (B.1.351) in the South Africa

Emergence of 501Y.V3 (P.1) in the Brazil

Substantial convergent evolution

Three occurences makes a pattern. Working hypothesis of rare events occurring during chronic infection, driven by natural selection for immune escape.
Rapid within-host evolution during persistent infection
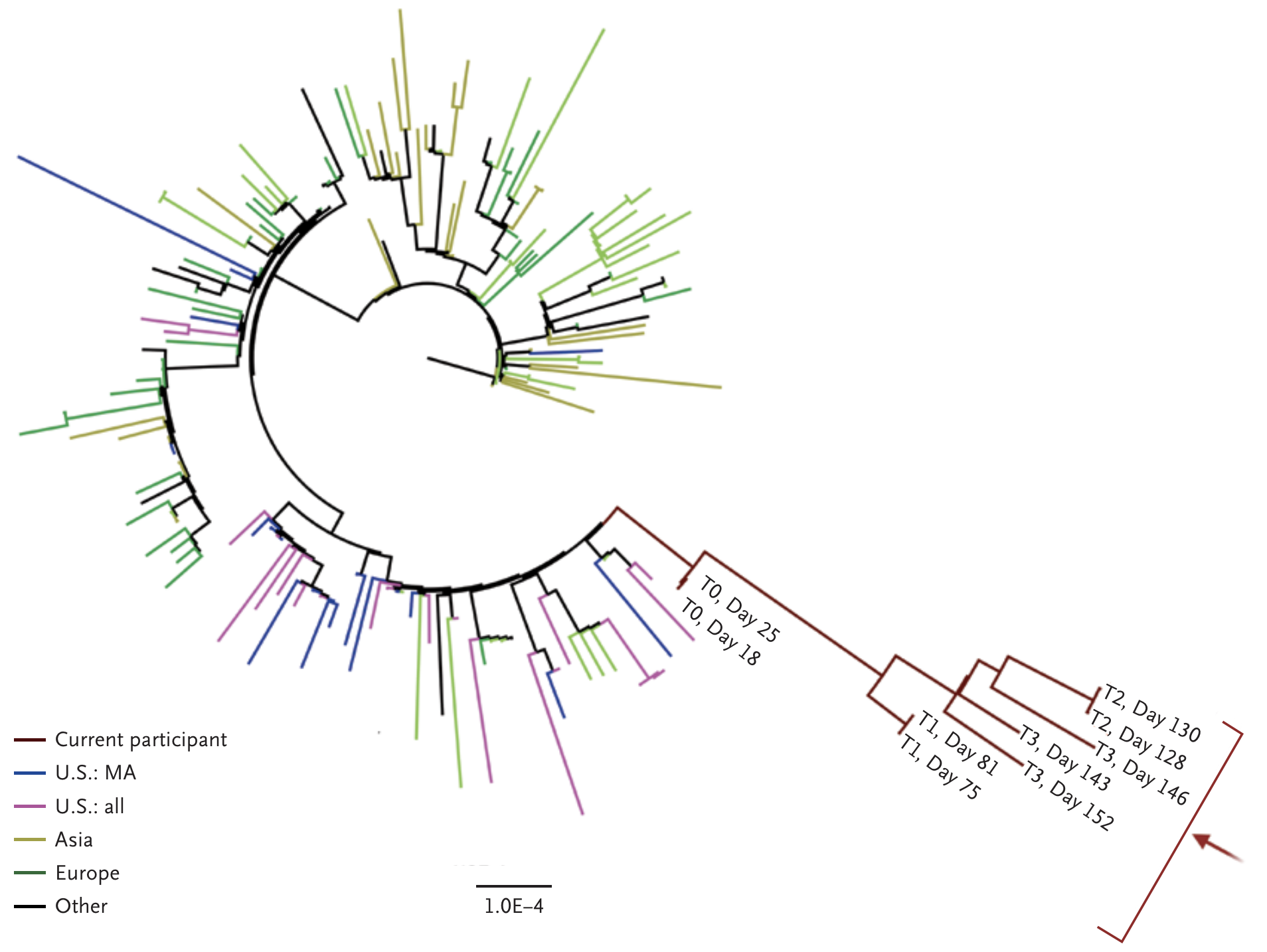
484K and 501Y observed during this evolution
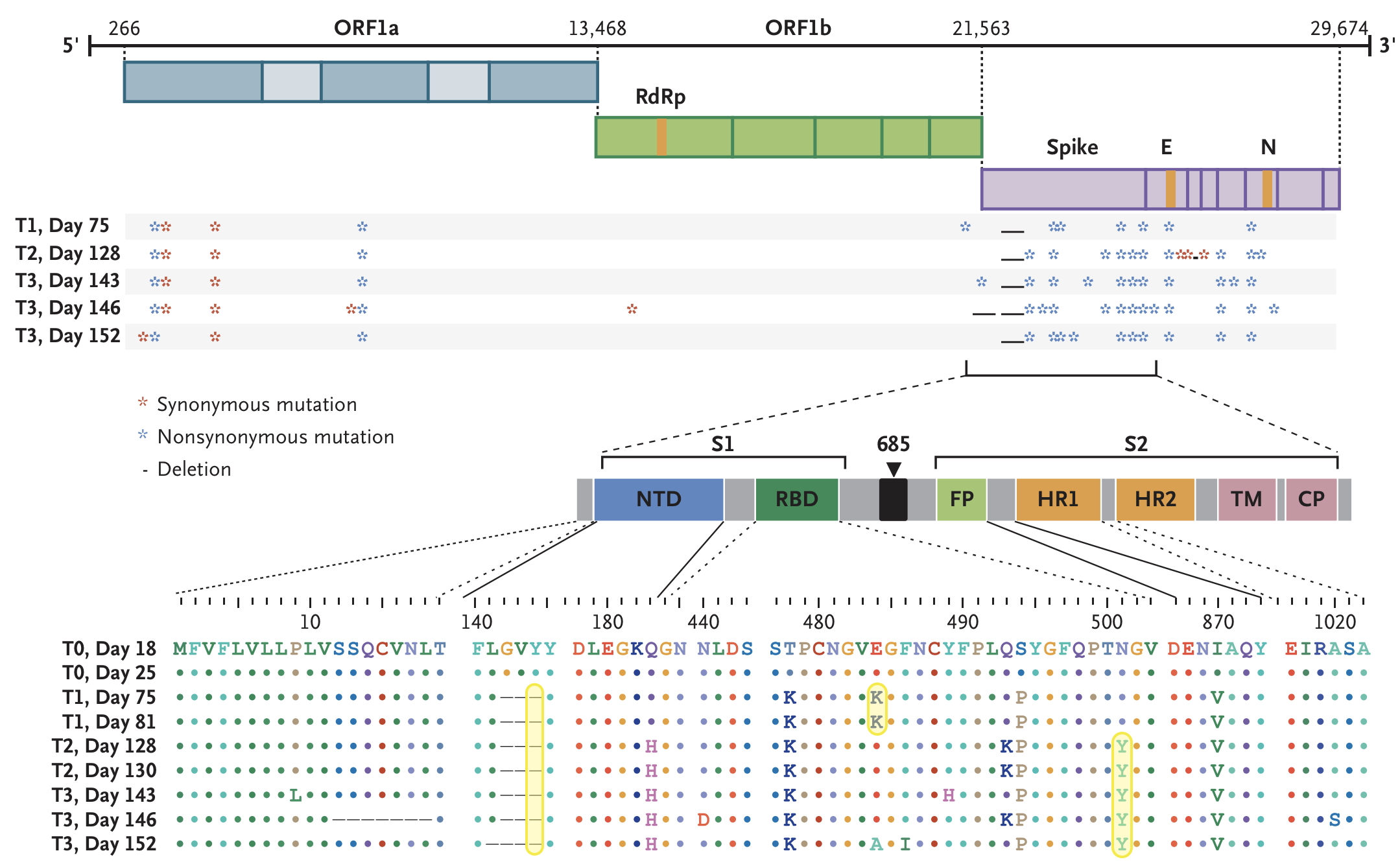
Expectations for antigenic evolution
Up to December, my expectation was evolution as seen in seasonal coronaviruses
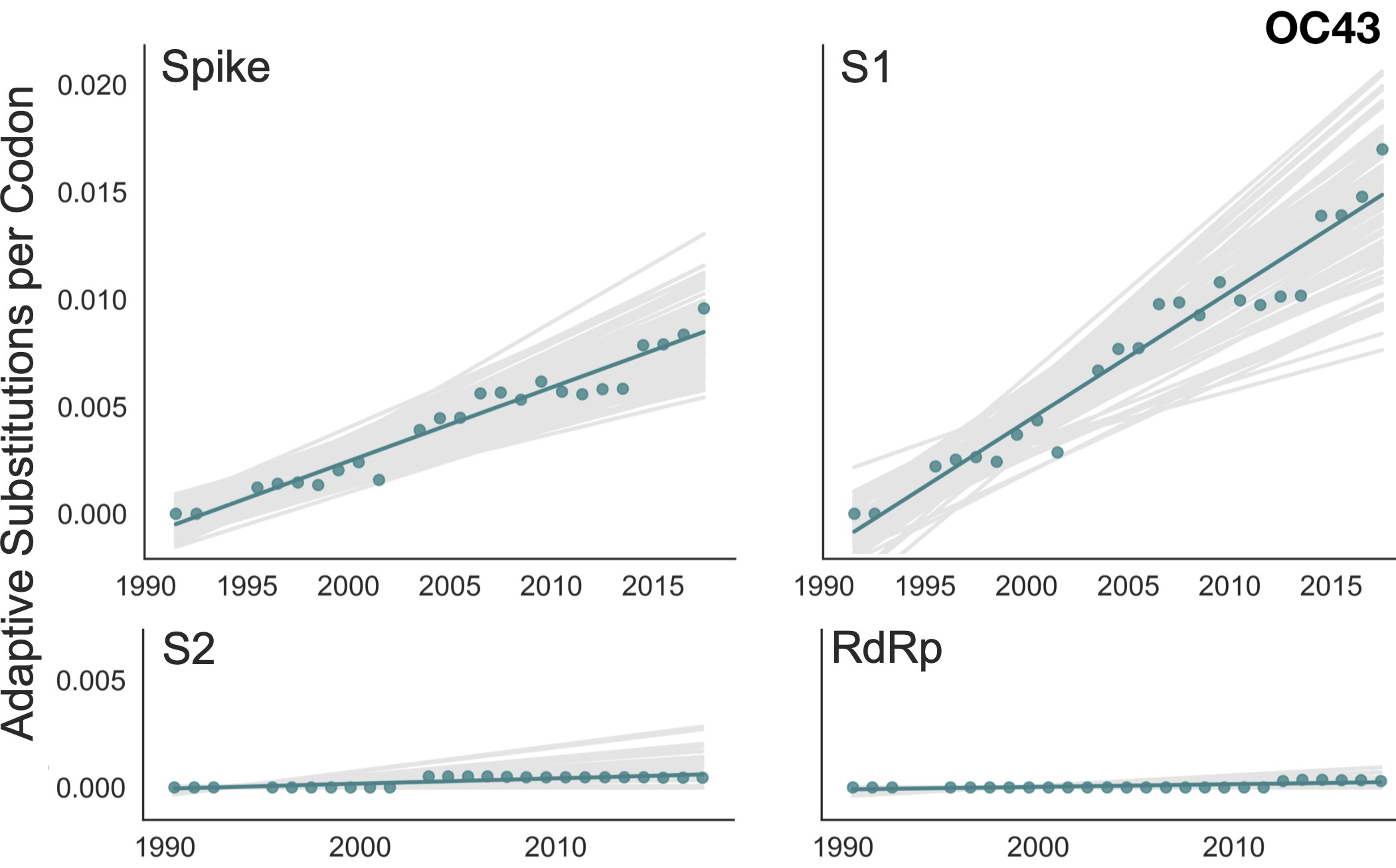
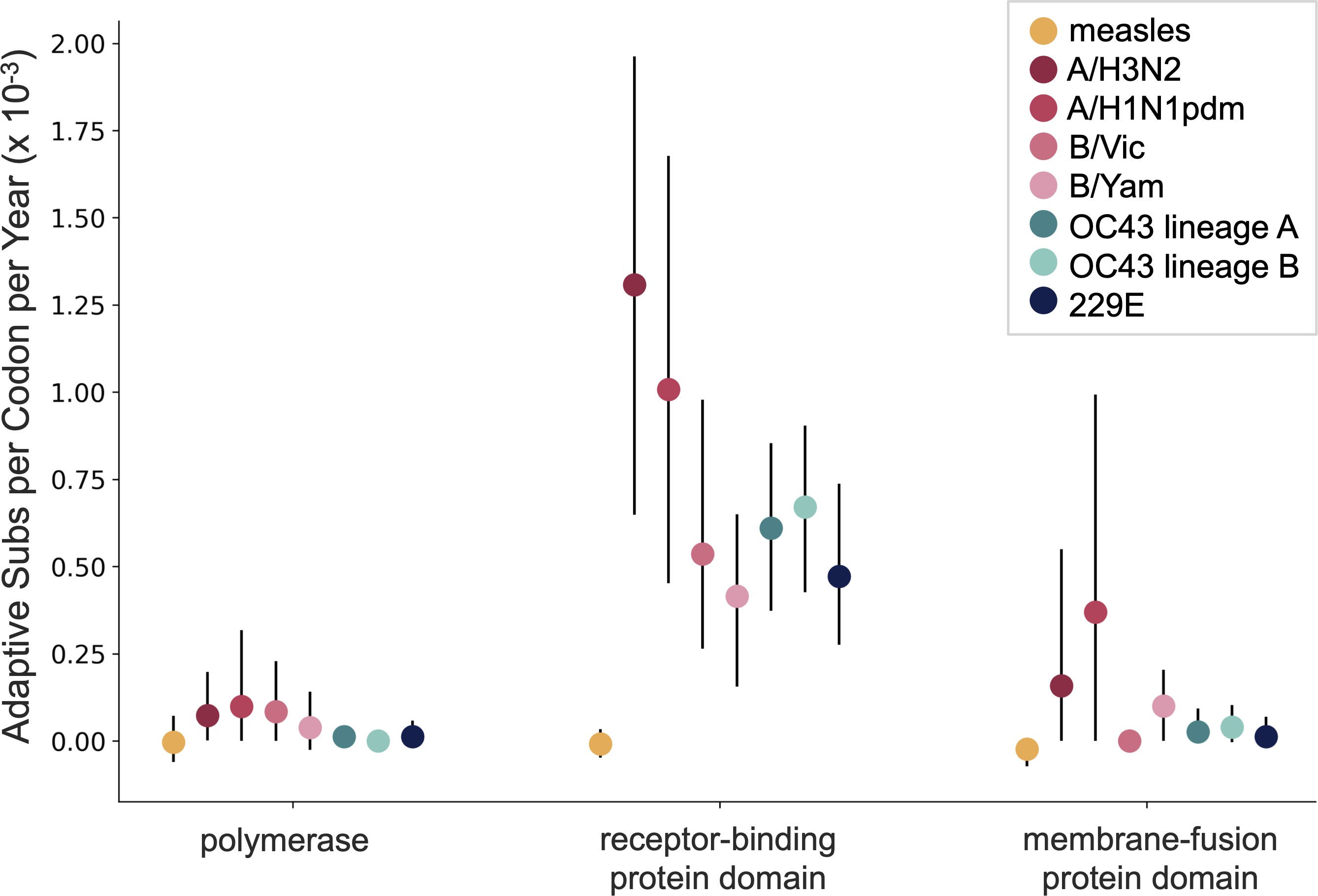
OC43 and 229E show flu B-like rates of adaptive evolution in S1
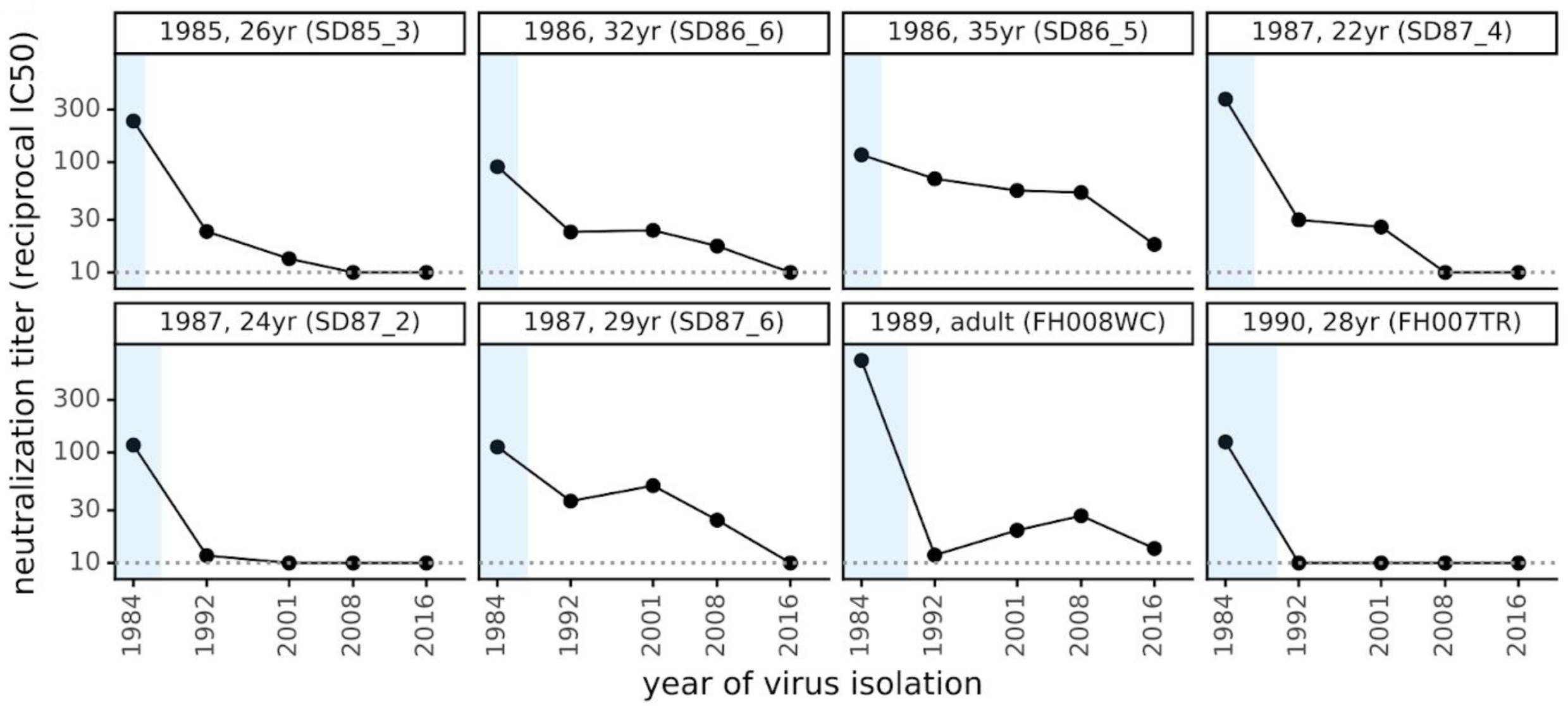
229E shows gradual antigenic drift as measured by neutralization assays
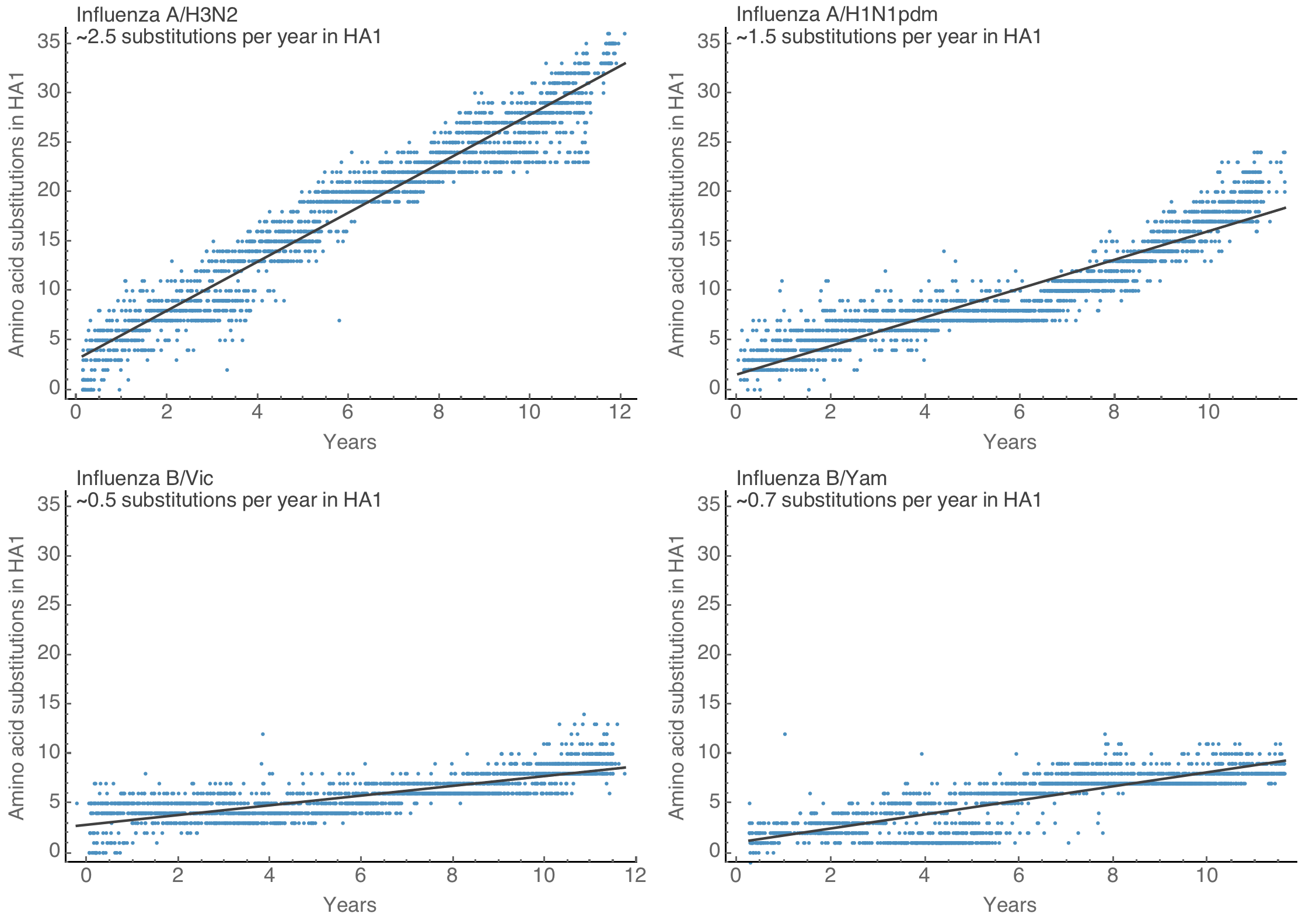
~23 mutations per year across SARS-CoV-2 genome

Substantial increase in spike S1 amino acid substitutions in VOCs

Spike S1 rate of 2.9 subs per year similar to rate in HA1 of flu A
Influenza H3N2 drifts at ~1 two-fold HI dilution per year
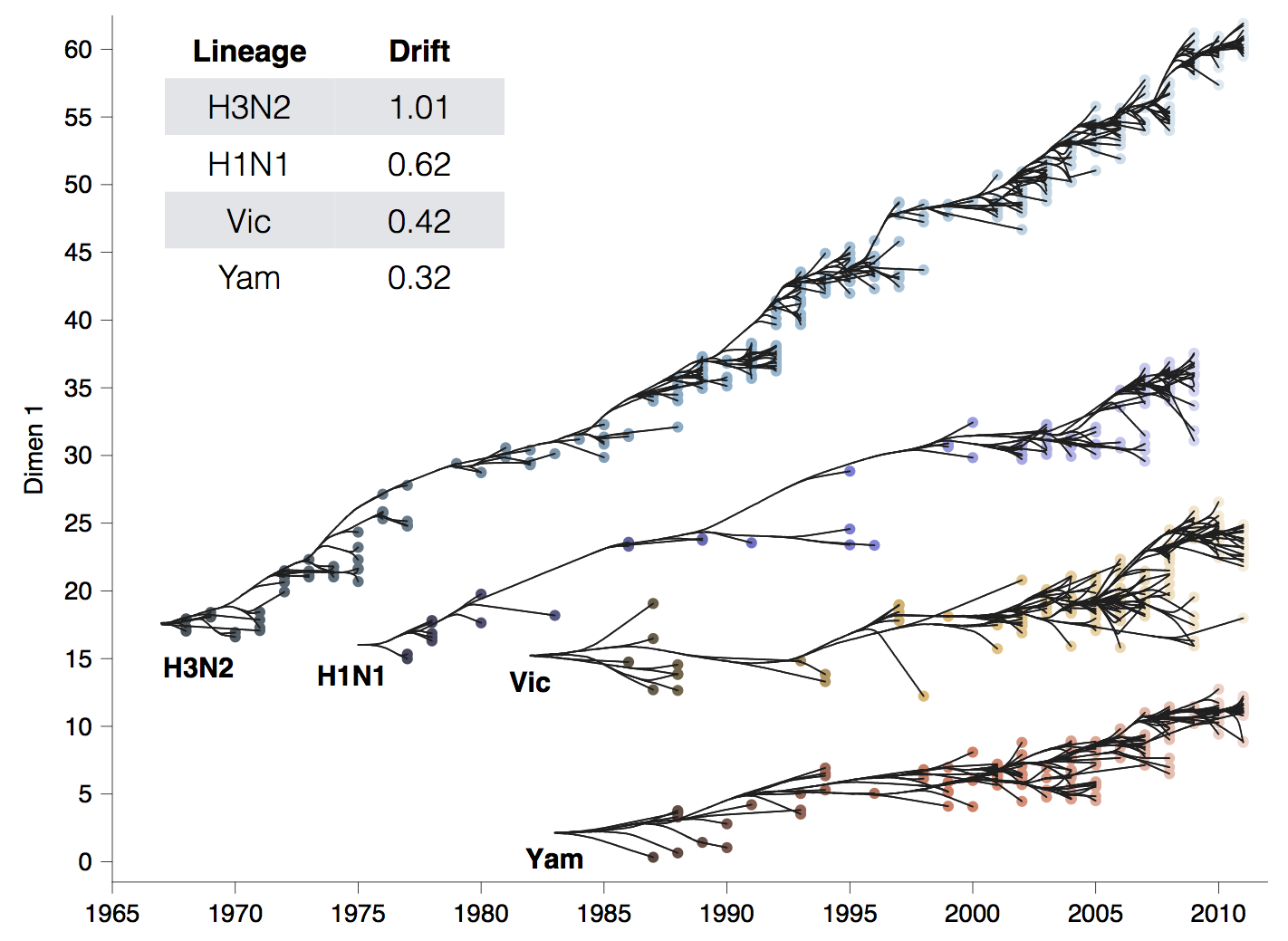
SARS-CoV-2 VOCs have evolved ~10 amino acid sites in S1 in over one year. This is rapid even for influenza A. 501Y.V2 shows an ~8-fold drop in neutralization titer, equivalent to ~3 years of H3N2 evolution.
Continued genomic surveillance
Substantial improvements in the both the volume and cadence of US genomic surveillance
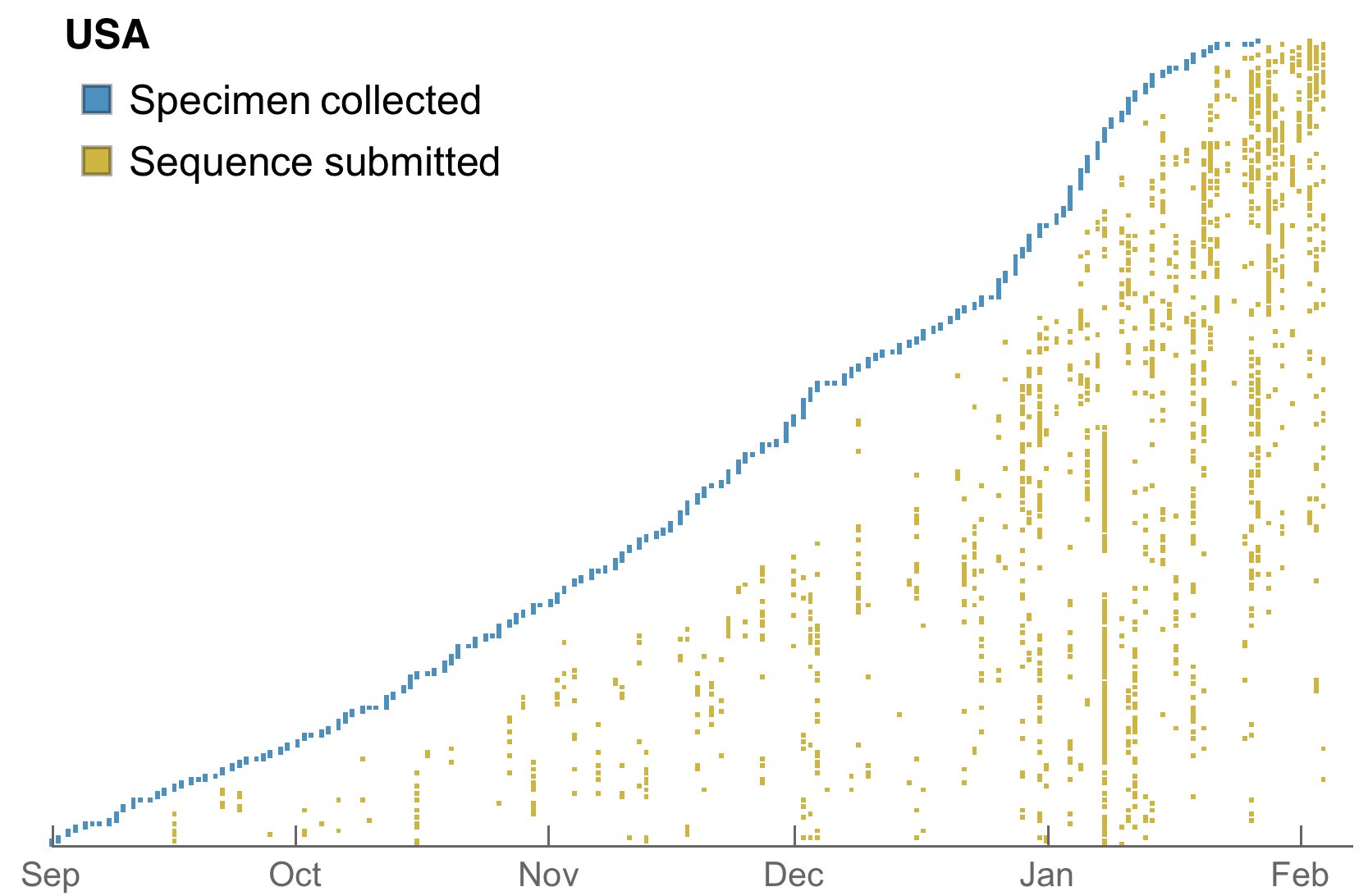
My standard metric is number of sequences available from samples collected in the past 30 days
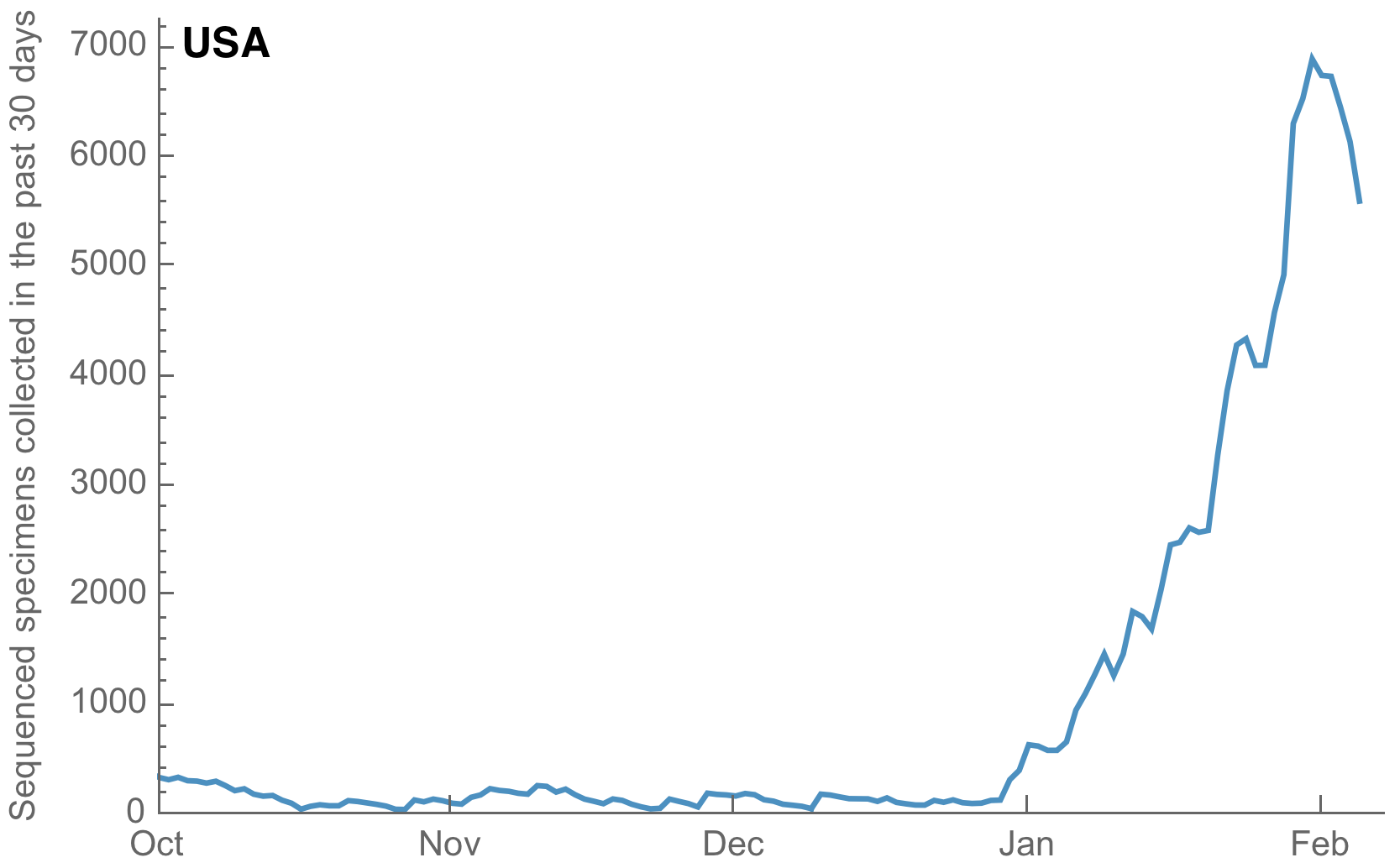
Substantial improvements though still behind front runners
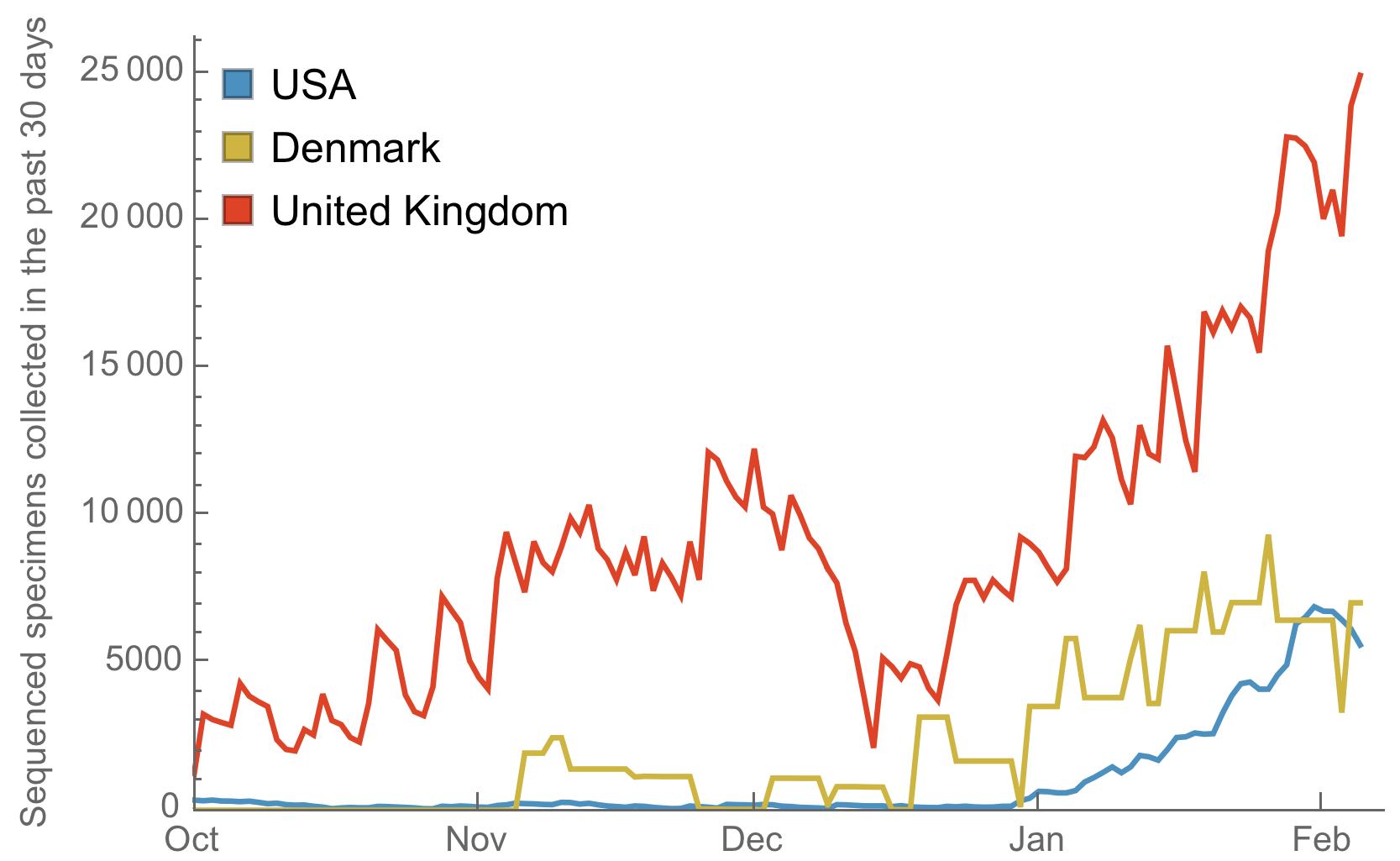
Generally, I think the process for influenza strain selection is a solid model
- mRNA vaccines allow faster turnarounds and may push for more regional strategies
- Work directly from human serological data rather than naive animals
- Multiple valencies possible
Acknowledgements
SARS-CoV-2 genomic epi: Data producers from all over the world, GISAID and the Nextstrain team
Bedford Lab:
![]() Alli Black,
Alli Black,
![]() John Huddleston,
John Huddleston,
![]() James Hadfield,
James Hadfield,
![]() Katie Kistler,
Katie Kistler,
![]() Louise Moncla,
Louise Moncla,
![]() Maya Lewinsohn,
Maya Lewinsohn,
![]() Thomas Sibley,
Thomas Sibley,
![]() Jover Lee,
Jover Lee,
![]() Kairsten Fay,
Kairsten Fay,
![]() Misja Ilcisin,
Misja Ilcisin,
![]() Cassia Wagner,
Cassia Wagner,
![]() Miguel Paredes,
Miguel Paredes,
![]() Nicola Müller,
Nicola Müller,
![]() Marlin Figgins,
Marlin Figgins,
![]() Eli Harkins
Eli Harkins