SARS-CoV-2 evolutionary dynamics
Trevor Bedford (@trvrb)
Associate Professor, Fred Hutchinson Cancer Research Center
14 Sep 2021
VIDD Seminar
Fred Hutch
Slides at: bedford.io/talks
This talk
Non-COVID "lab-lead" papers since promotion on Jan 2018
 |
Dudas et al. 2018. MERS-CoV spillover at the camel-human interface. eLife. |  |
Hadfield et al. 2018. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. | |
 |
Lee et al. 2018. Deep mutational scanning of hemagglutinin helps predict evolutionary fates of human H3N2 influenza variants. PNAS. | 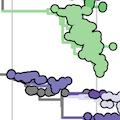 |
Bell et al. 2019. Dengue genetic divergence generates within-serotype antigenic variation, but serotypes dominate evolutionary dynamics. eLife. | |
 |
Hadfield et al. 2019. Twenty years of West Nile virus spread and evolution in the Americas visualized by Nextstrain. PLoS Pathog. |  |
Black et al. 2019. Genomic epidemiology supports multiple introductions and cryptic transmission of Zika virus in Colombia. BMC Infect Dis. | |
 |
Potter et al. 2019. Evolution and rapid spread of a reassortant A(H3N2) virus that predominated the 2017-2018 influenza season. Virus Evol. |  |
Dudas et al. 2019. The ability of single genes vs full genomes to resolve time and space in outbreak analysis. BMC Evol Biol. | |
 |
Moncla et al. 2020. Quantifying within-host evolution of H5N1 influenza in humans and poultry in Cambodia. PLoS Pathog. |  |
Black et al. 2020. Ten recommendations for supporting open pathogen genomic analysis in public health. Nat Med. | |
 |
Hilton et al. 2020. dms-view: Interactive visualization tool for deep mutational scanning data. JOSS. |  |
Huddleston et al. 2020. Integrating genotypes and phenotypes improves long-term forecasts of seasonal influenza A/H3N2 evolution. eLife. | |
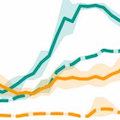 |
Kistler and Bedford. 2021. Evidence for adaptive evolution in the receptor-binding domain of seasonal coronaviruses OC43 and 229E. eLife. | 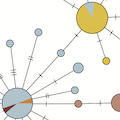 |
Mbala-Kingebeni et al. 2021. Ebola virus transmission initiated by systemic Ebola virus disease relapse. NEJM. | |
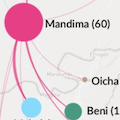 |
Kinganda-Lusamaki et al. 2021. Integration of genomic sequencing into the response to the Ebola virus outbreak in Nord Kivu, DRC. Nat Med. | 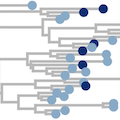 |
Moncla et al. 2021. Repeated introductions and intensive community transmission fueled a mumps virus outbreak in Washington State. eLife. |
Non-COVID "lab-lead" papers since promotion on Jan 2018
|
Dudas et al. 2018. MERS-CoV spillover at the camel-human interface. eLife. | |
Hadfield et al. 2018. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. | |
|
Lee et al. 2018. Deep mutational scanning of hemagglutinin helps predict evolutionary fates of human H3N2 influenza variants. PNAS. | |
Bell et al. 2019. Dengue genetic divergence generates within-serotype antigenic variation, but serotypes dominate evolutionary dynamics. eLife. | |
|
Hadfield et al. 2019. Twenty years of West Nile virus spread and evolution in the Americas visualized by Nextstrain. PLoS Pathog. | |
Black et al. 2019. Genomic epidemiology supports multiple introductions and cryptic transmission of Zika virus in Colombia. BMC Infect Dis. | |
|
Potter et al. 2019. Evolution and rapid spread of a reassortant A(H3N2) virus that predominated the 2017-2018 influenza season. Virus Evol. | |
Dudas et al. 2019. The ability of single genes vs full genomes to resolve time and space in outbreak analysis of Ebola. BMC Evol Biol. | |
|
Moncla et al. 2020. Quantifying within-host evolution of H5N1 influenza in humans and poultry in Cambodia. PLoS Pathog. | |
Black et al. 2020. Ten recommendations for supporting open pathogen genomic analysis in public health. Nat Med. | |
|
Hilton et al. 2020. dms-view: Interactive visualization tool for deep mutational scanning data. J Open Source Softw. | |
Huddleston et al. 2020. Integrating genotypes and phenotypes improves long-term forecasts of seasonal influenza A/H3N2 evolution. eLife. | |
|
Kistler and Bedford. 2021. Evidence for adaptive evolution in the receptor-binding domain of seasonal coronaviruses OC43 and 229E. eLife. | |
Mbala-Kingebeni et al. 2021. Ebola virus transmission initiated by systemic Ebola virus disease relapse. NEJM. | |
|
Kinganda-Lusamaki et al. 2021. Integration of genomic sequencing into the response to the Ebola virus outbreak in Nord Kivu, DRC. Nat Med. | |
Moncla et al. 2021. Repeated introductions and intensive community transmission fueled a mumps virus outbreak in Washington State. eLife. |
COVID-19
1. Real-time tracking of SARS-CoV-2 evolution
2. Emergence of variants of concern
3. Current circulation patterns
4. Assessing adaptive evolution
5. Variant transmission dynamics
6. Genomic surveillance
7. Future perspectives
Real-time tracking of SARS-CoV-2 evolution
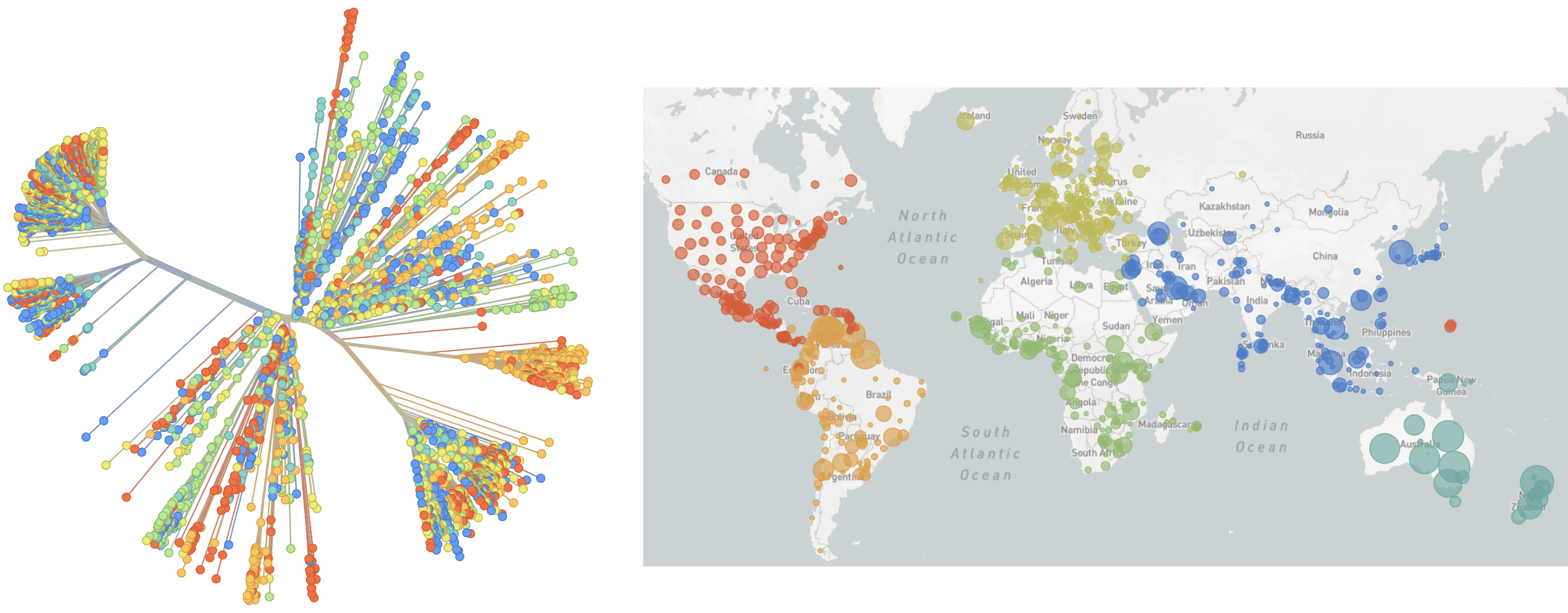
Over 3.4M SARS-CoV-2 genomes shared to GISAID and evolution tracked in real-time at nextstrain.org
![]() Richard Neher,
Richard Neher,
![]() Emma Hodcroft,
Emma Hodcroft,
![]() James Hadfield,
James Hadfield,
![]() Thomas Sibley,
Thomas Sibley,
![]() John Huddleston,
John Huddleston,
![]() Ivan Aksamentov,
Ivan Aksamentov,
![]() Moira Zuber,
Moira Zuber,
![]() Eli Harkins,
Eli Harkins,
![]() Jover Lee,
Jover Lee,
![]() Cassia Wagner,
Cassia Wagner,
![]() Louise Moncla,
Louise Moncla,
![]() Misja Ilcisin,
Misja Ilcisin,
![]() Kairsten Fay,
Kairsten Fay,
![]() Allison Black,
Allison Black,
![]() Miguel Paredes,
Miguel Paredes,
![]() Sidney Bell,
Sidney Bell,
![]() Denisse Sequeira
Denisse Sequeira
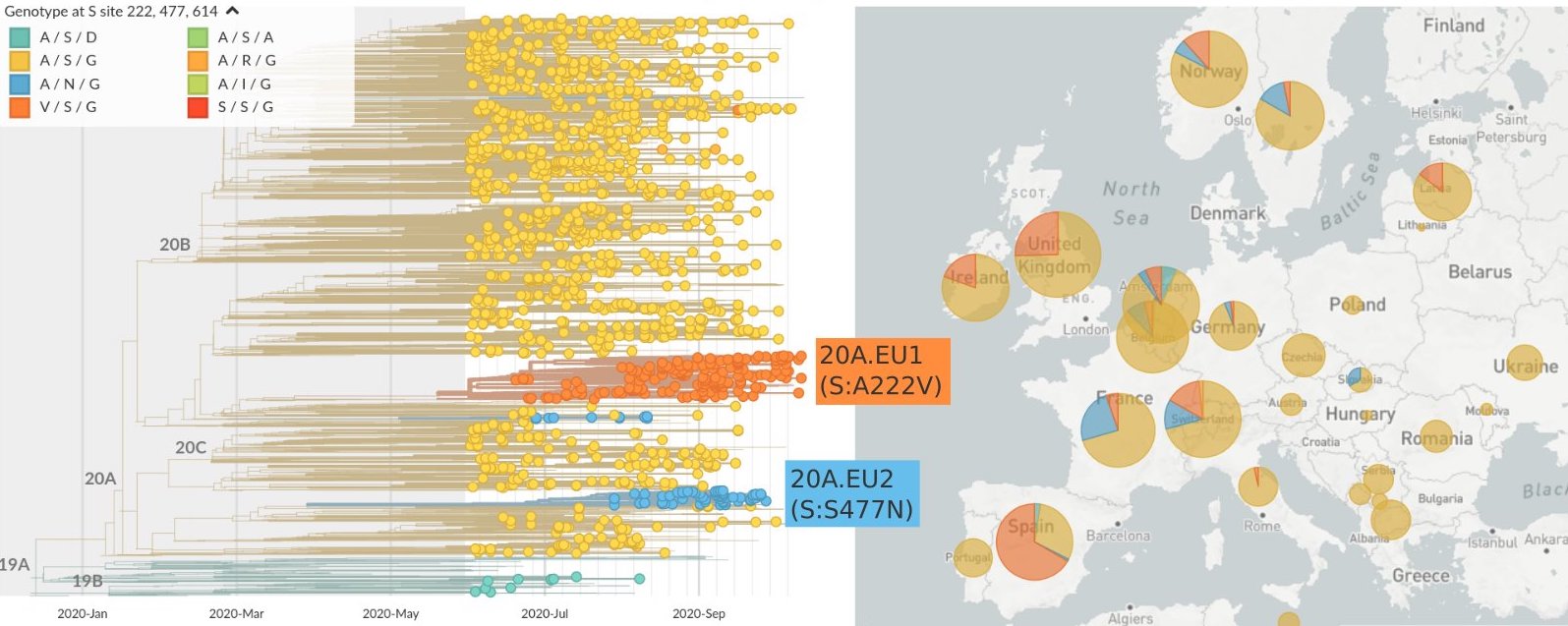
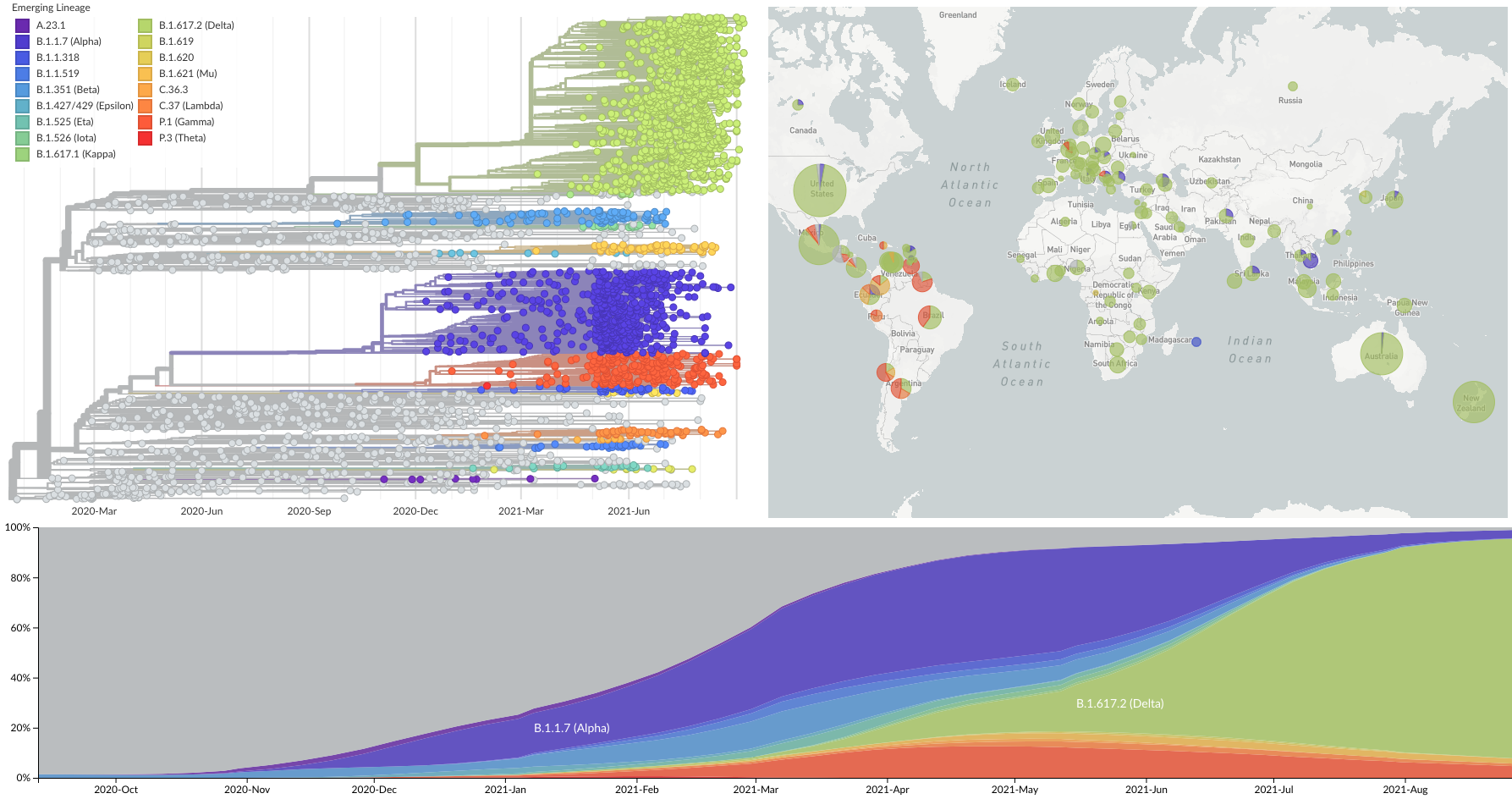
SARS-CoV-2 lineages establish globally in February and March
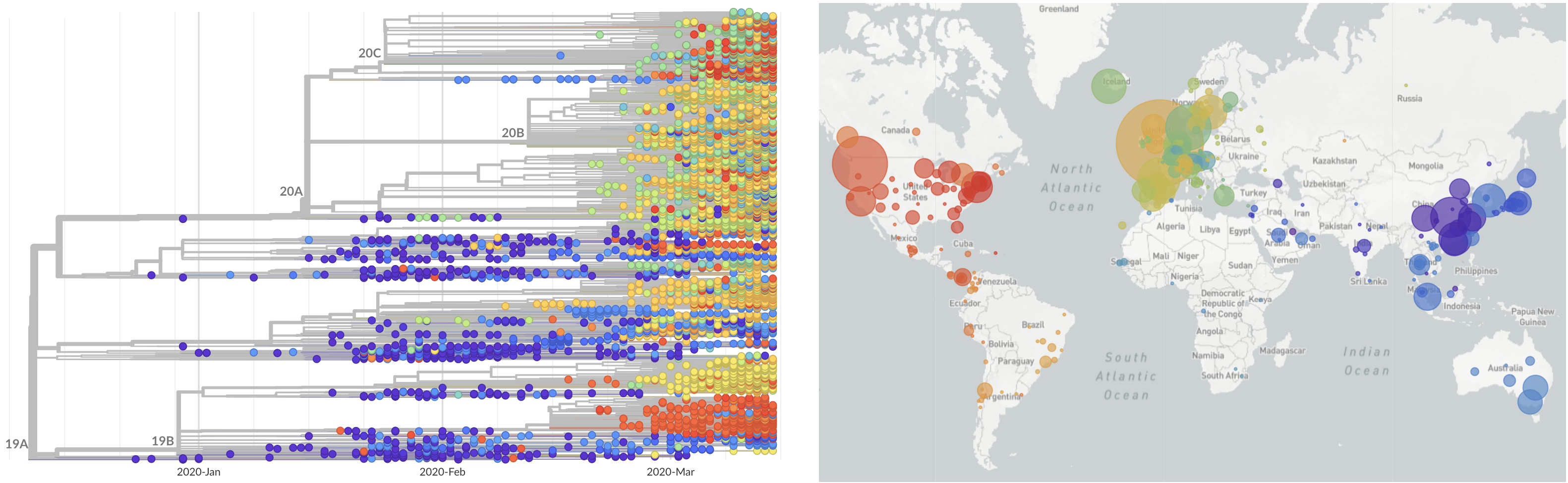
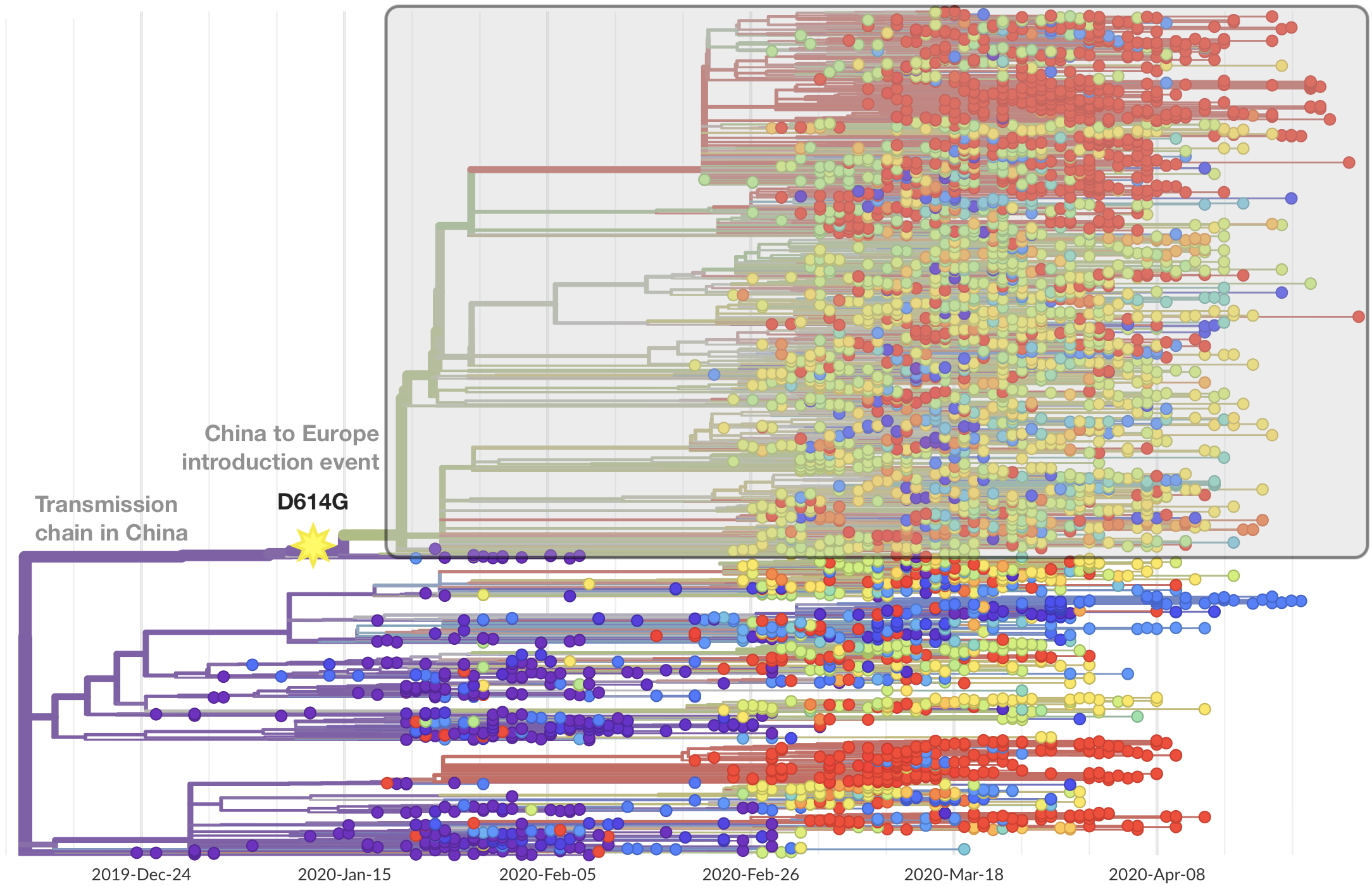
Limited early mutations like spike D614G spread globally during initial wave
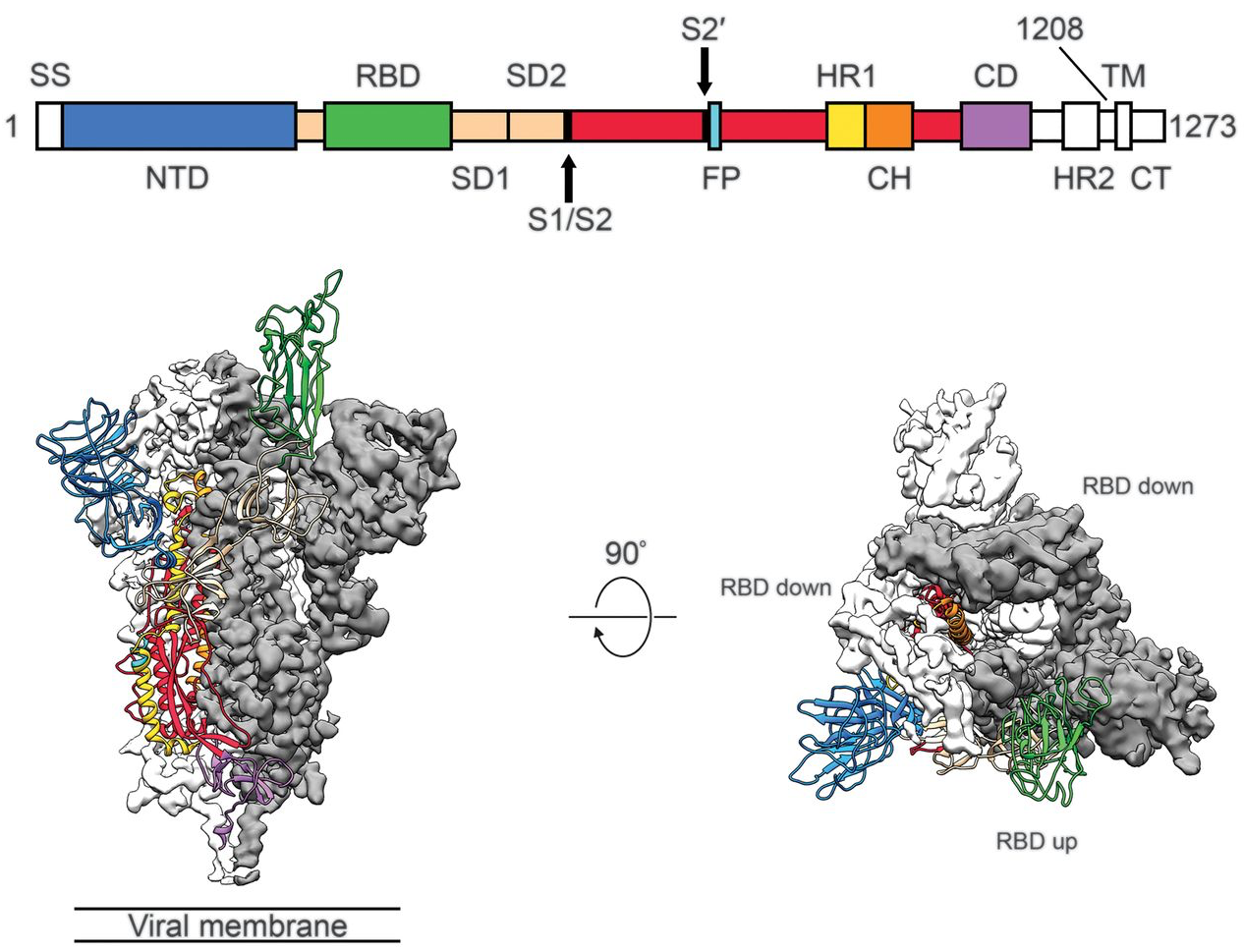
Spike protein is critical for cell invasion by the virus and is the primary target for human immune response
After initial wave, with mitigation
efforts and decreased travel,
regional clades emerge
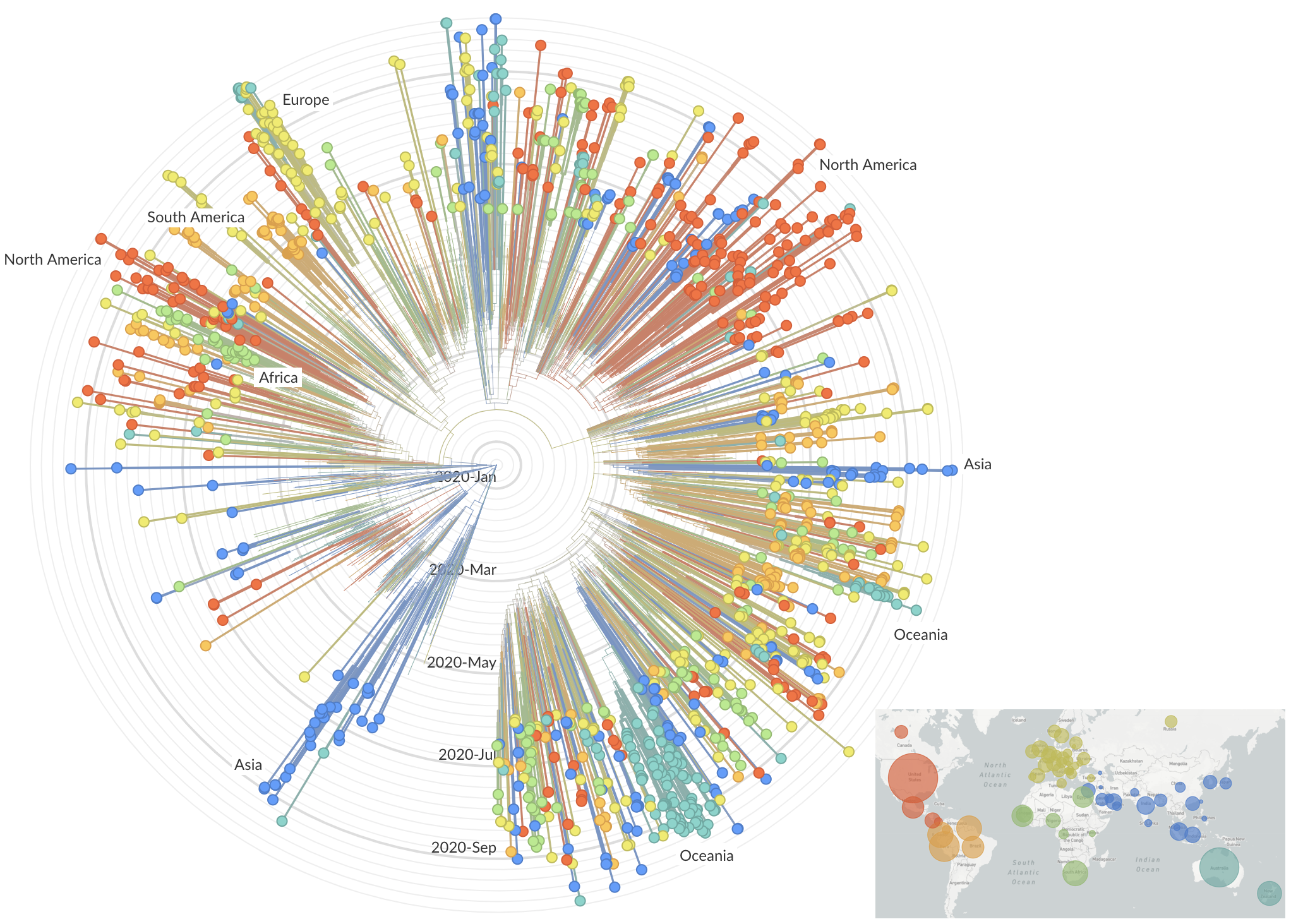
Mutations in summer and fall 2020 were confined to regional dominance
Emergence of variants of concern
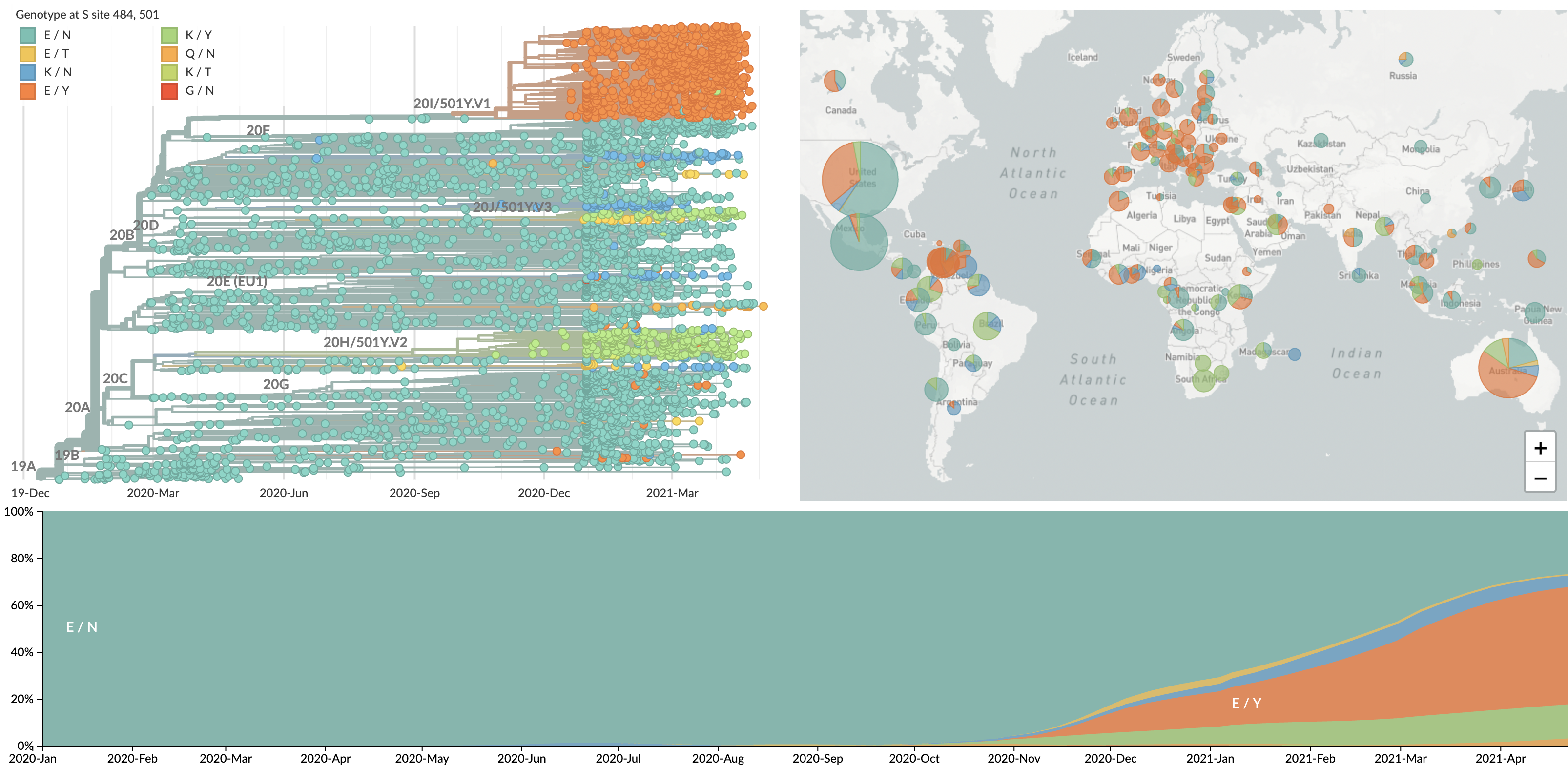
Repeated emergence of 484K and 501Y across the world
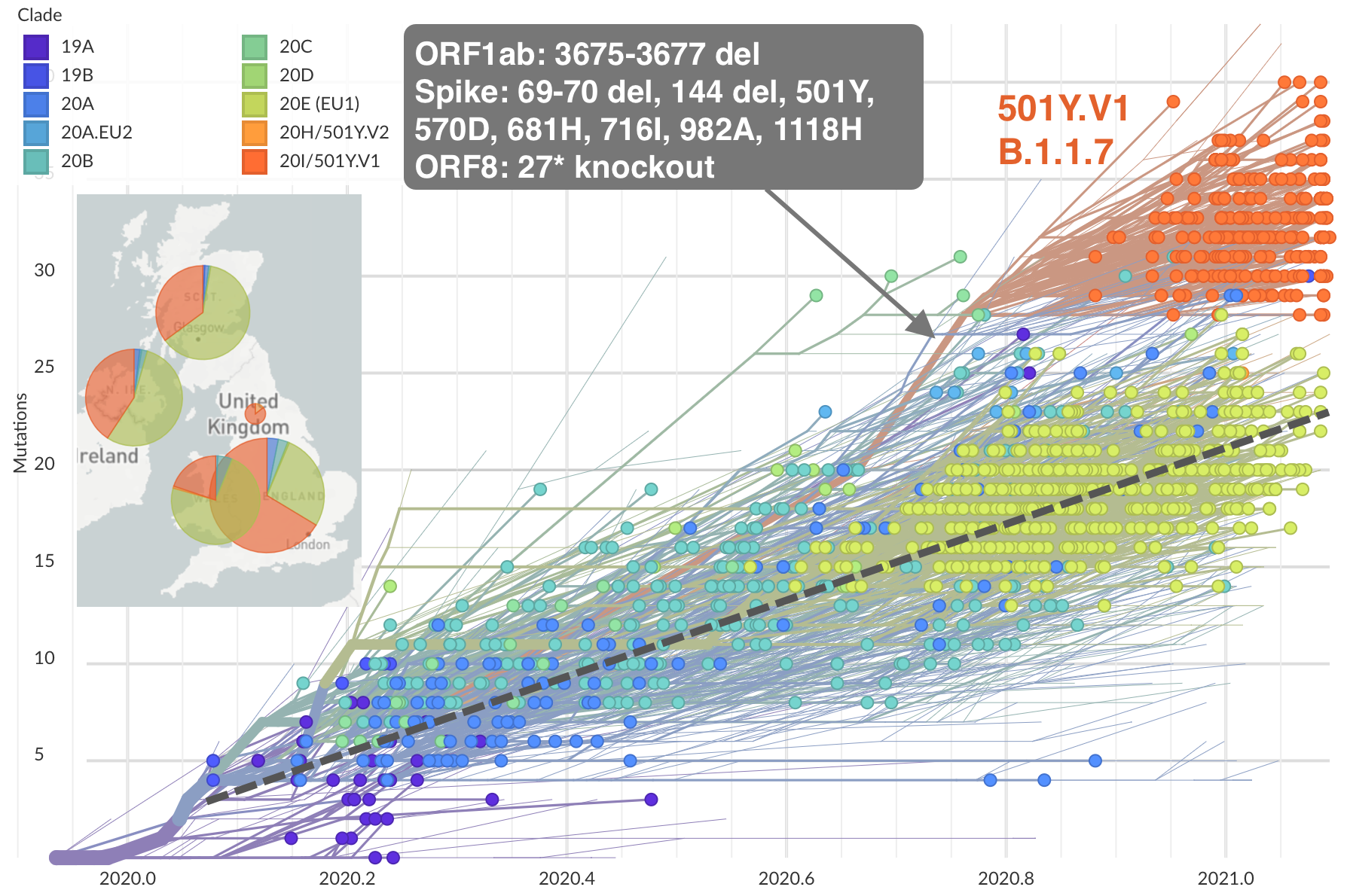
Emergence of Alpha (B.1.1.7) in the UK
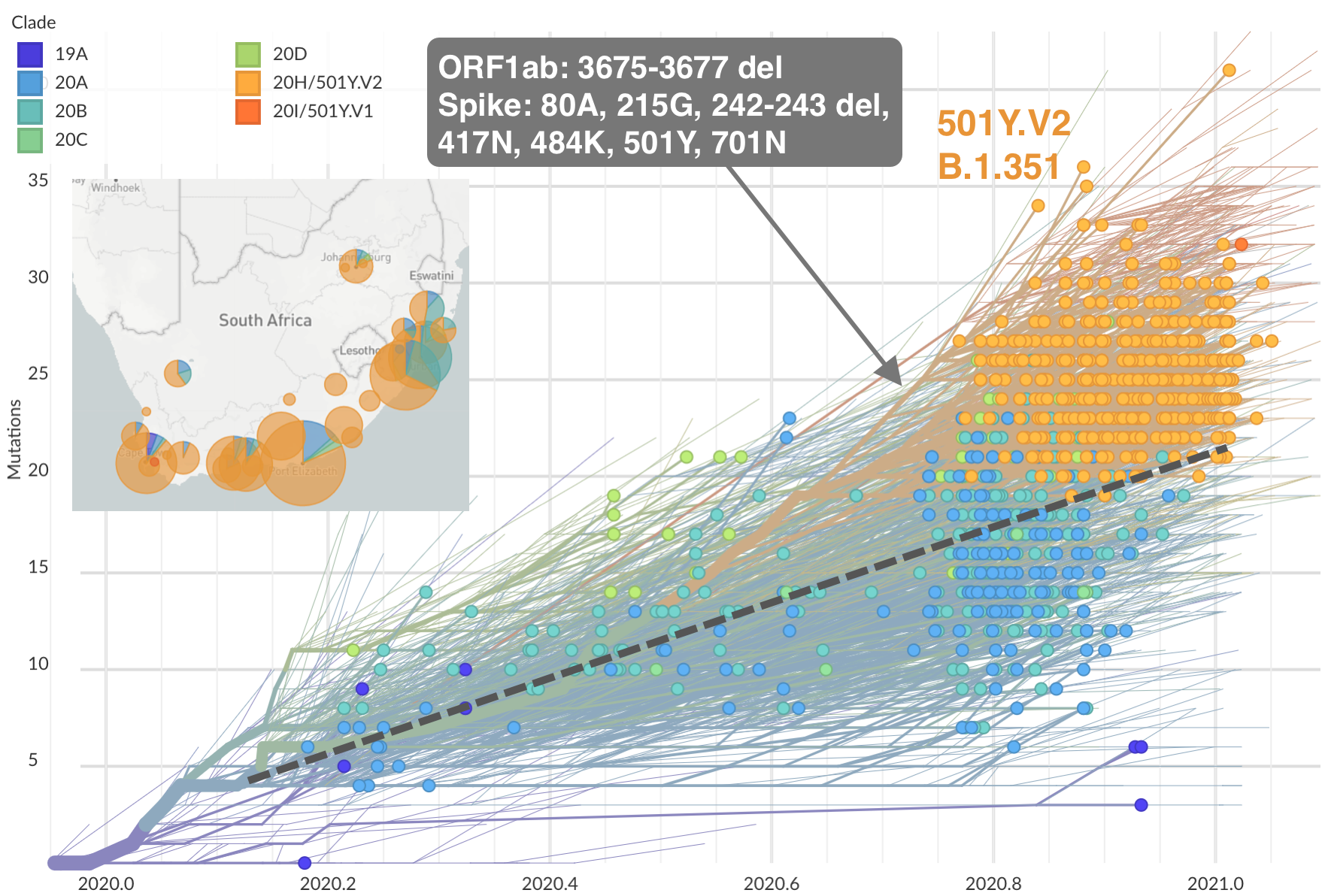
Emergence of Beta (B.1.351) in the South Africa
Emergence of Gamma (P.1) in the Brazil
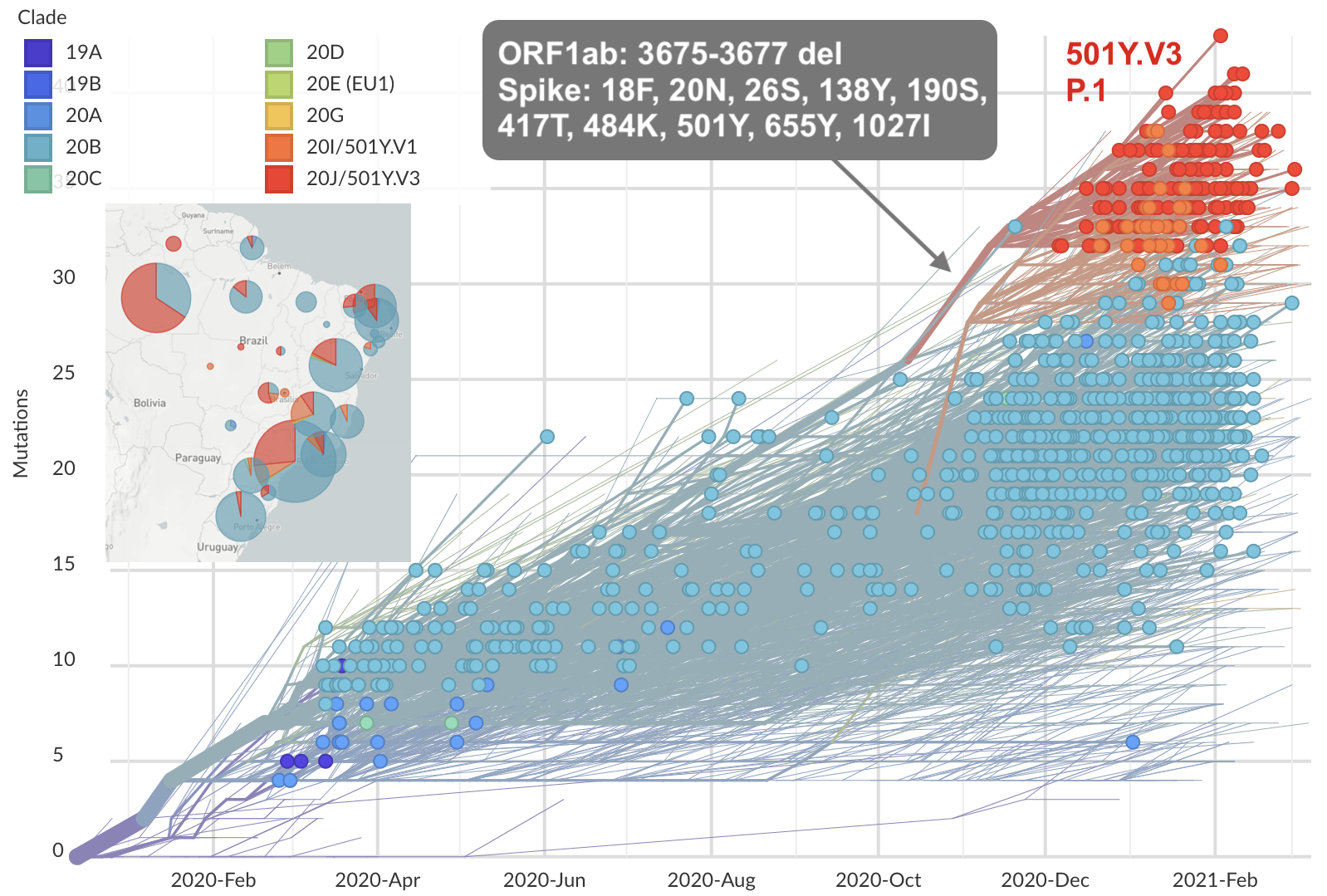
Working hypothesis of within-host evolution occurring during infection, driven by natural selection against host immunity
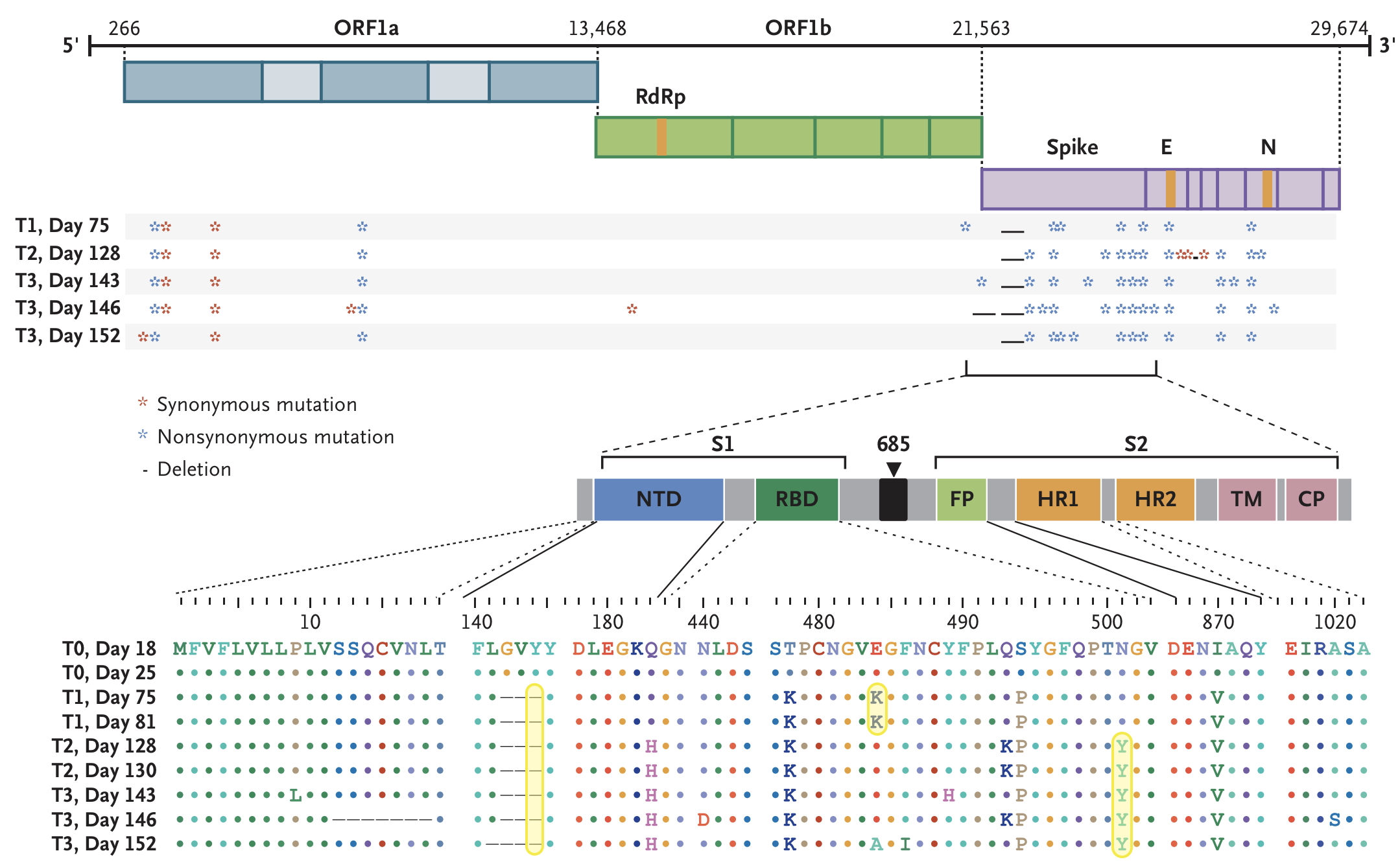
Rapid within-host evolution during persistent infection
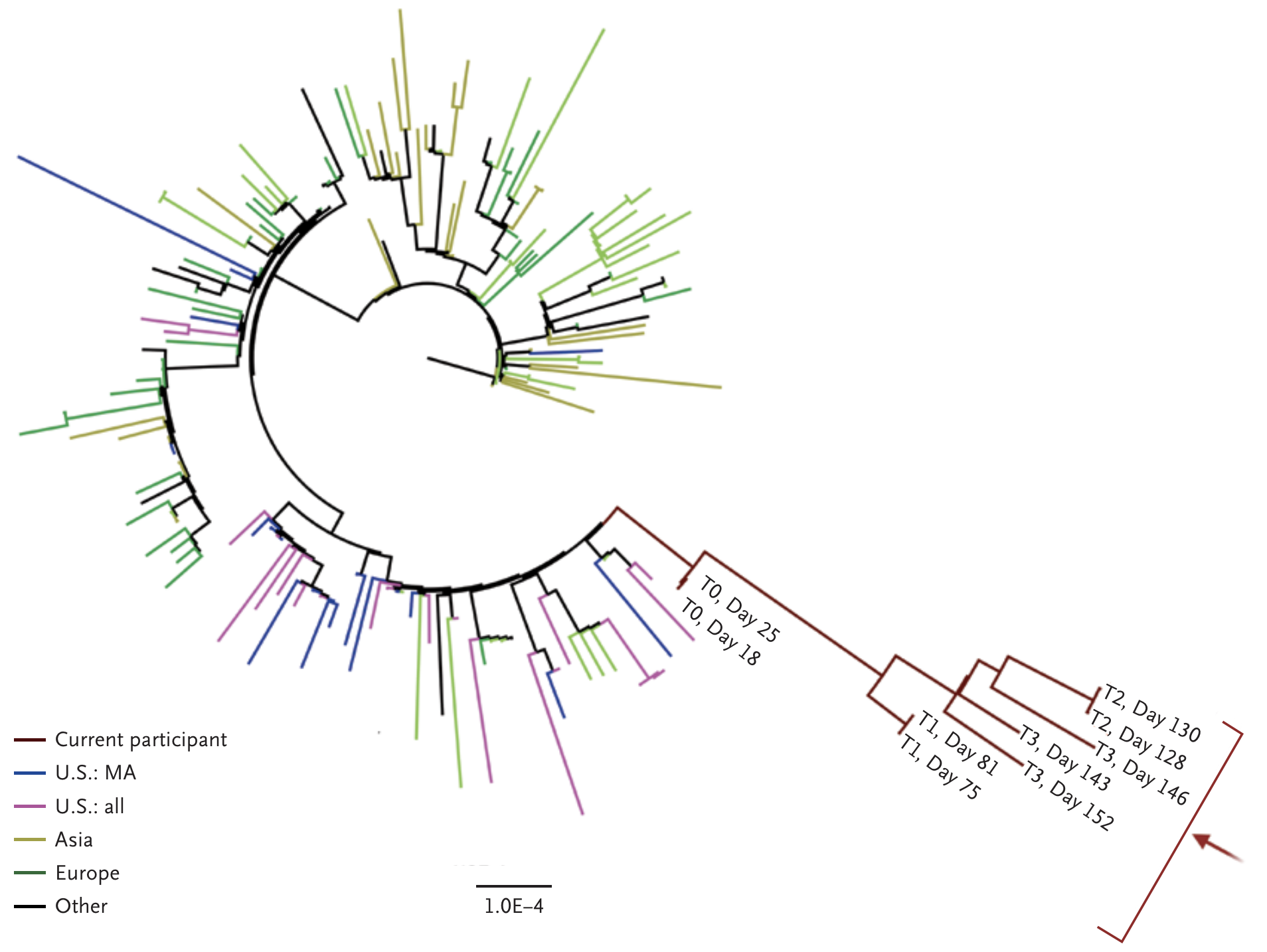
484K and 501Y observed during this evolution
Current circulation patterns
Spread of VOC / VOI lineages across the world
Delta is outcompeting other variants and is on track to sweep
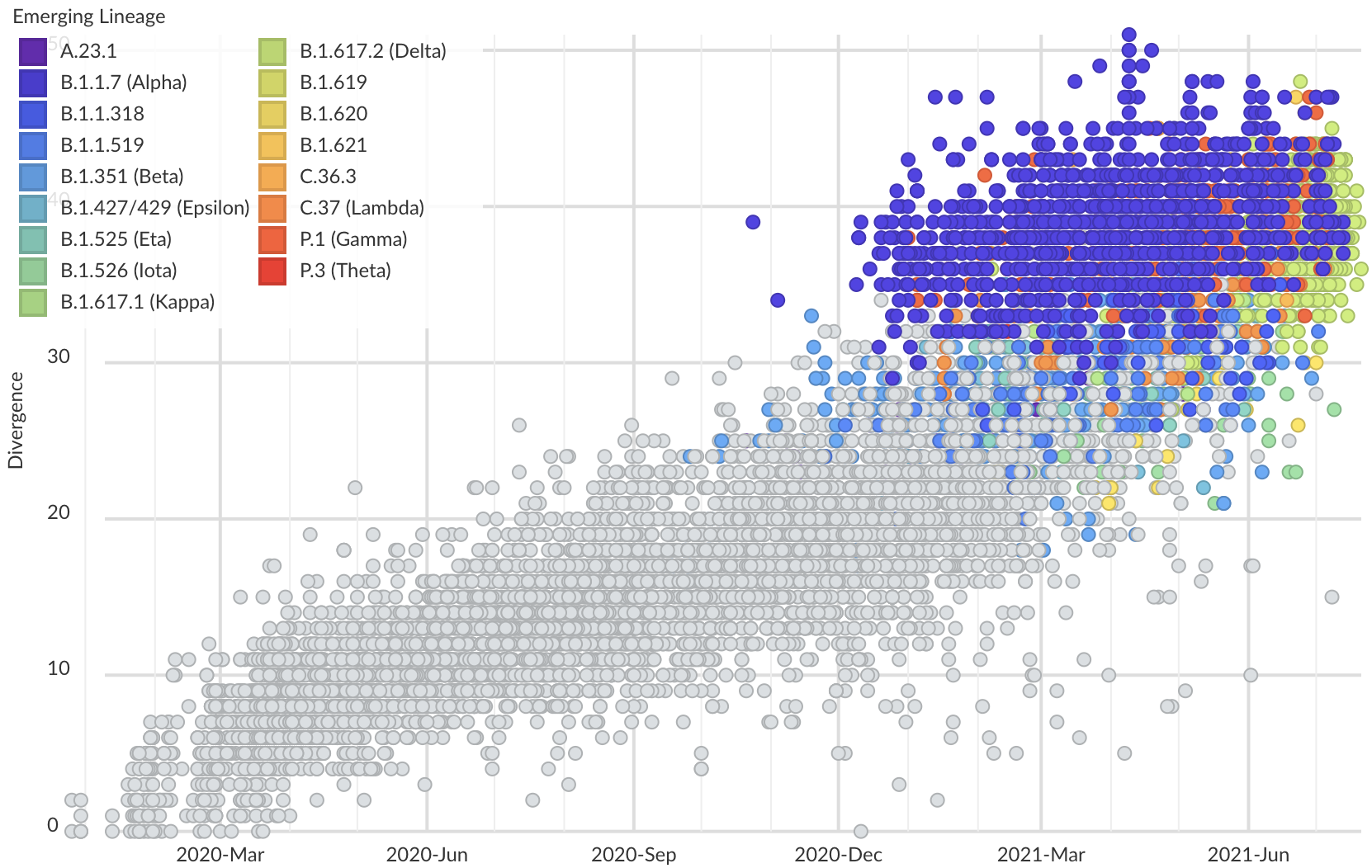
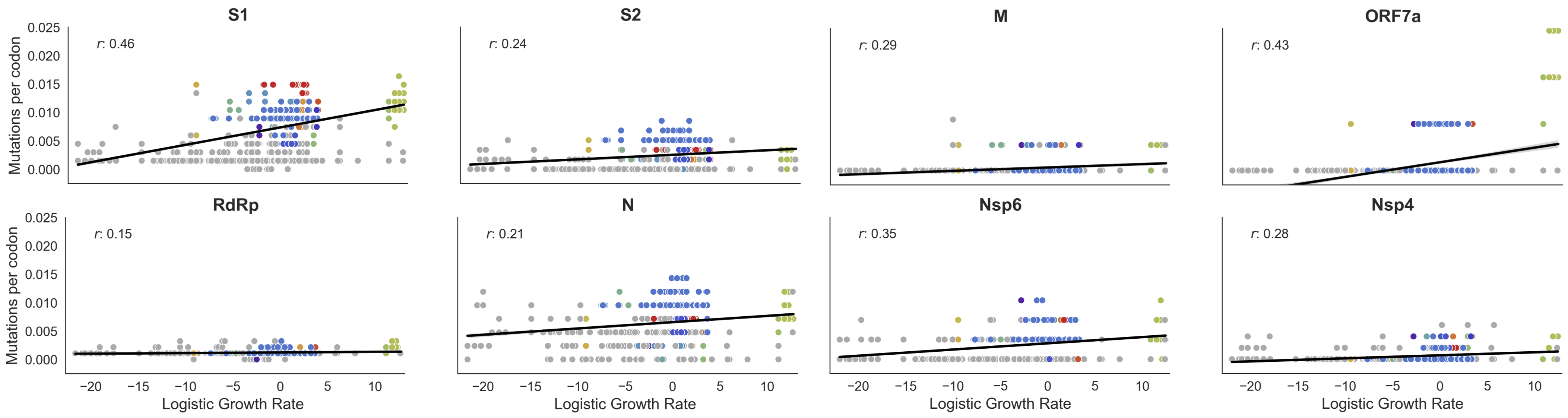
Variants show excess mutations across the genome
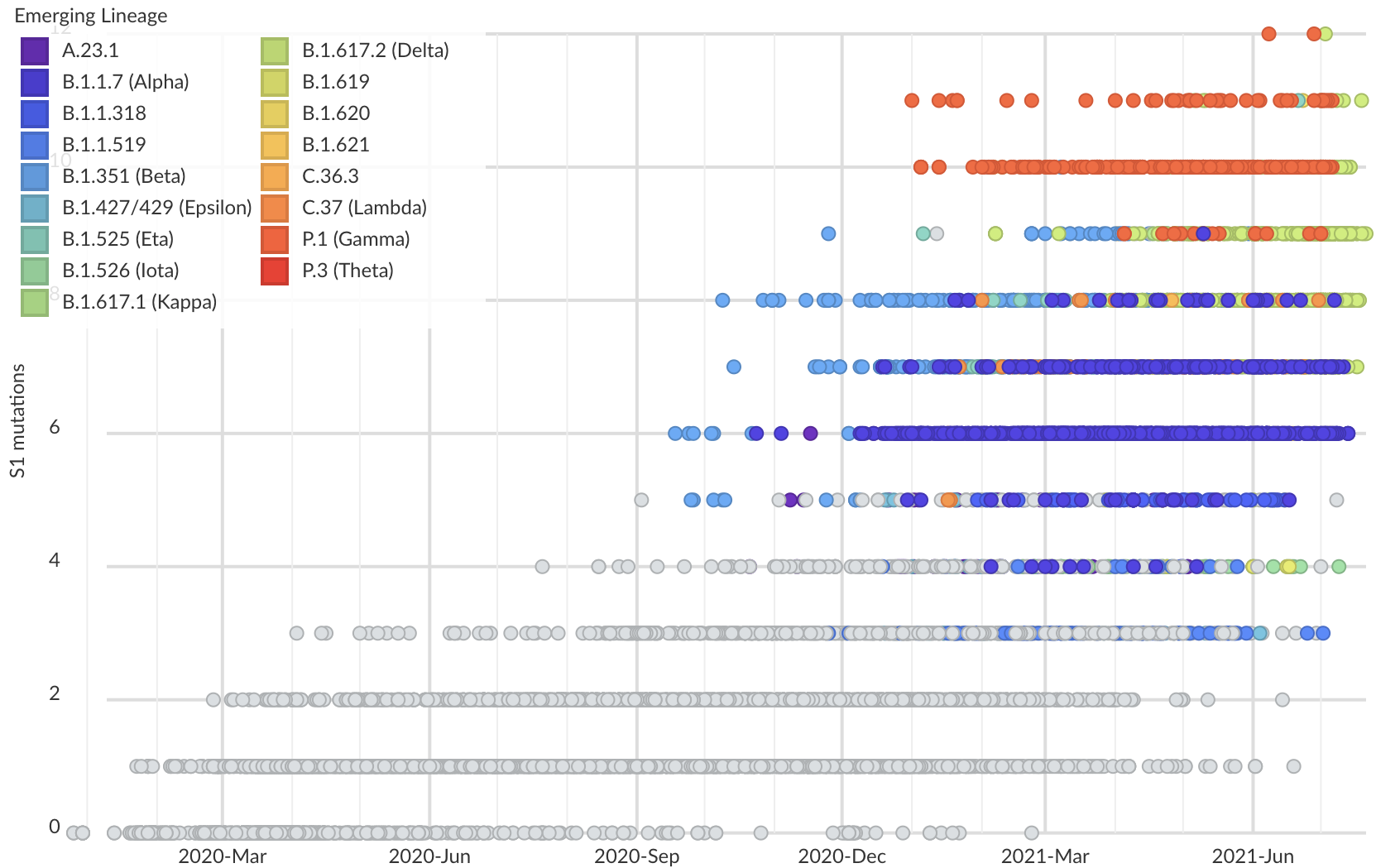
But show most substantial excess in the S1 domain of spike
Assessing adaptive evolution
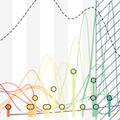
Rapid and parallel adaptive mutations in spike S1 drive clade success in SARS-CoV-2
 Katie Kistler,
Katie Kistler,
 John Huddleston
John Huddleston
Phylogeny of 10k genomes equitably sampled in space and time
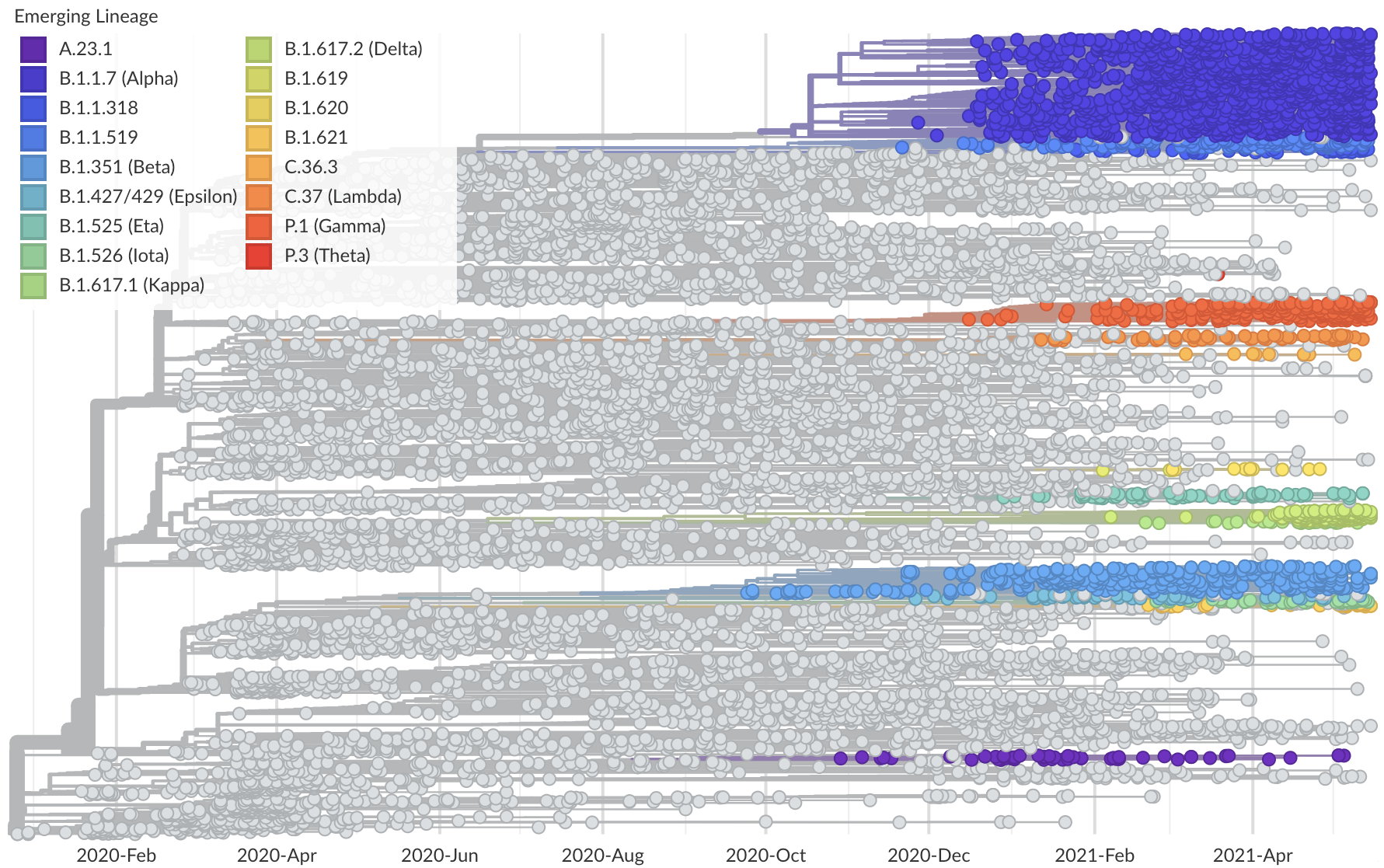
Measure clade growth as a proxy for viral fitness
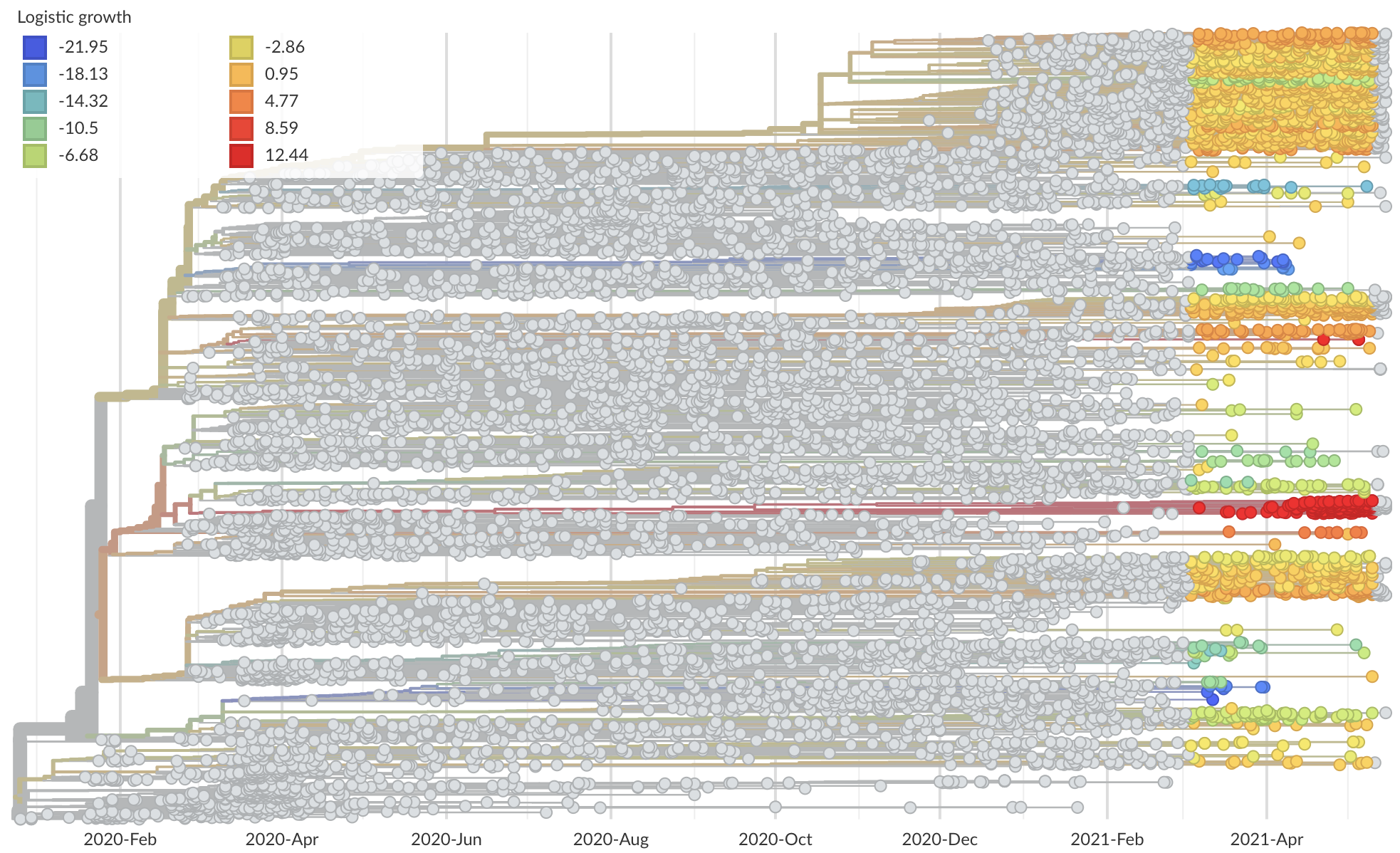
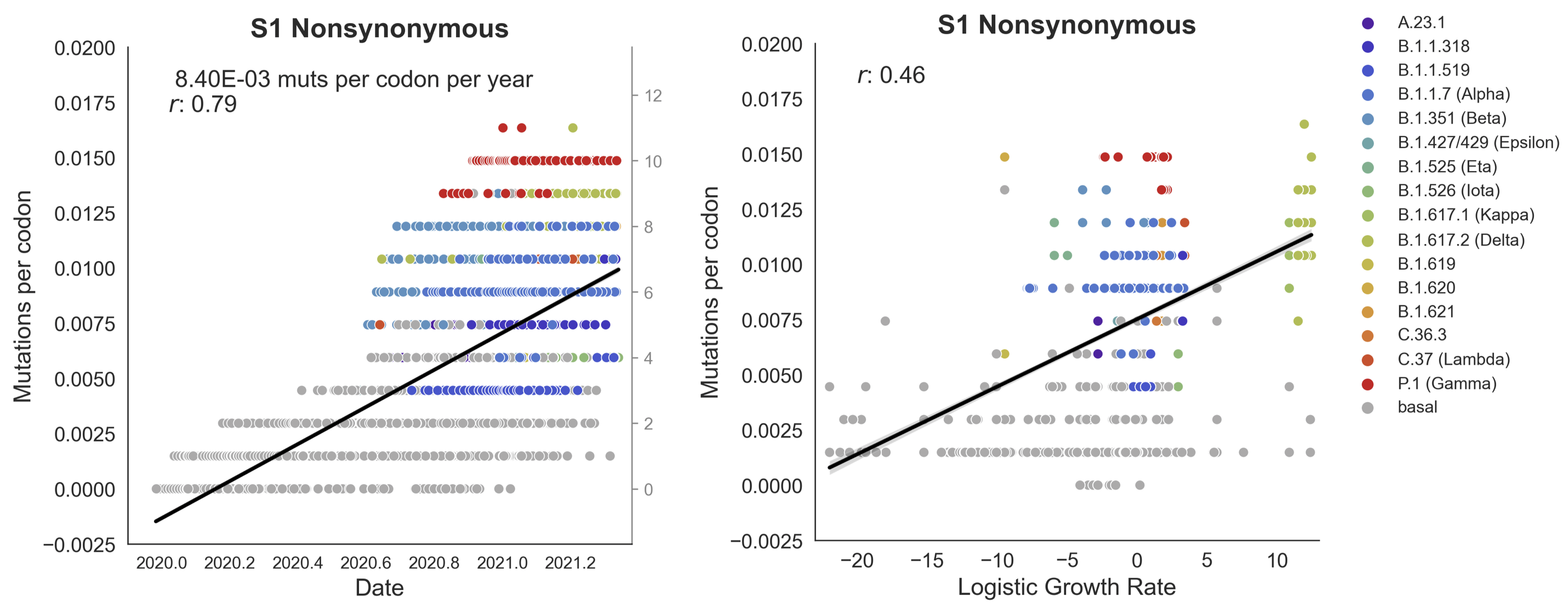
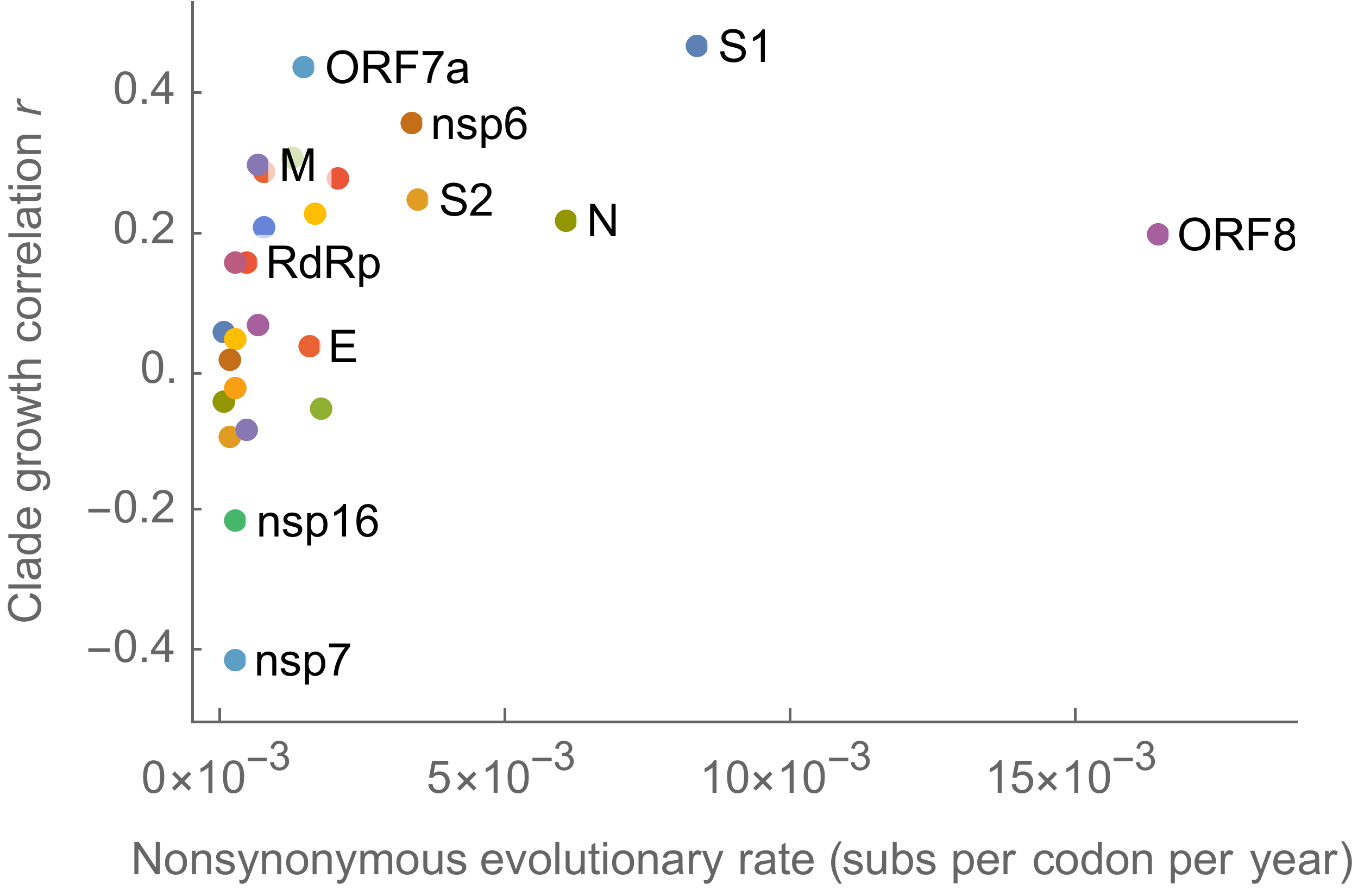
Clades with more S1 nonsynonymous mutations grow faster
This correlation is absent in control regions
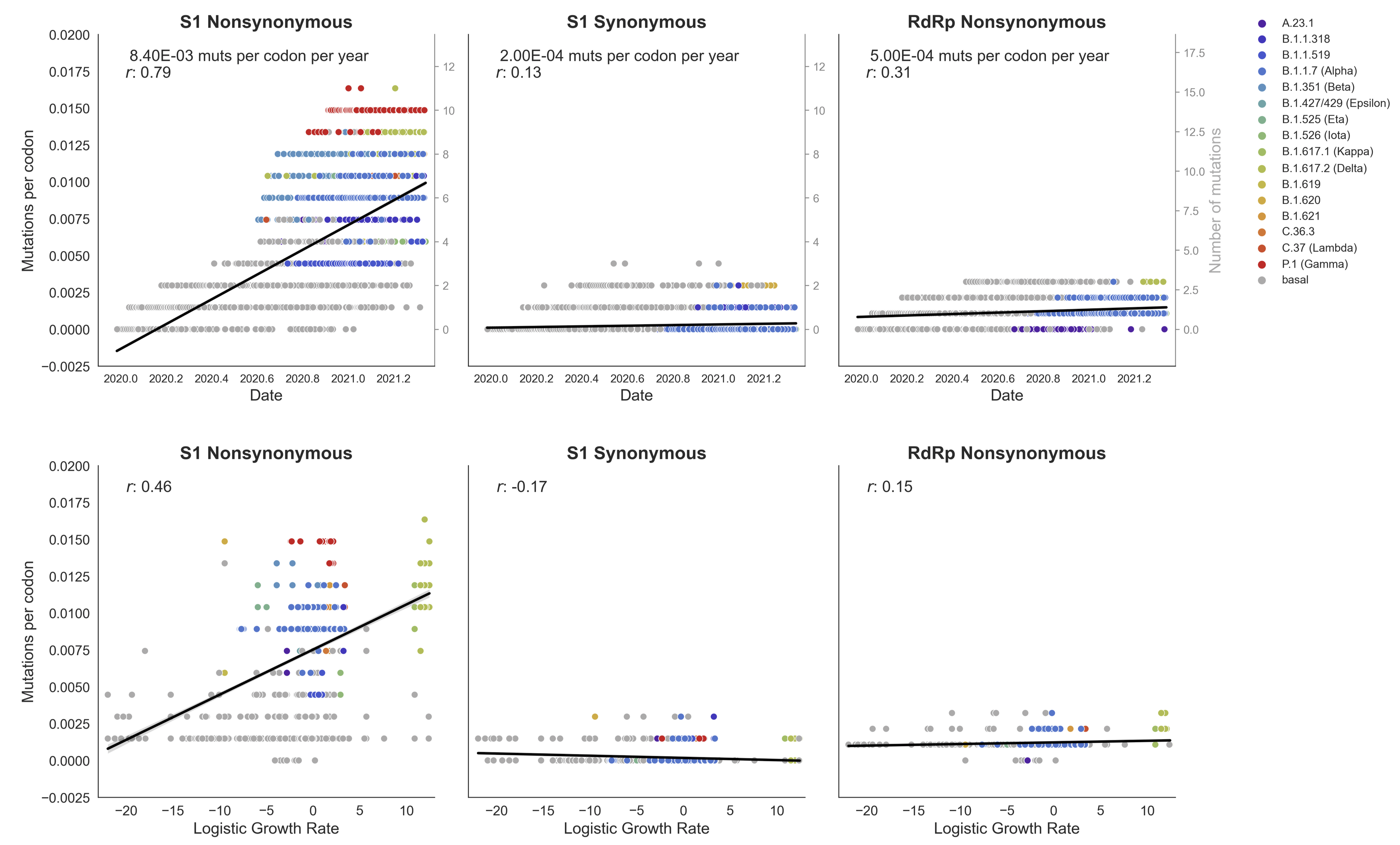
This correlation is stronger in S1 than other genes
S1 is quickly evolving and highly correlated with clade growth
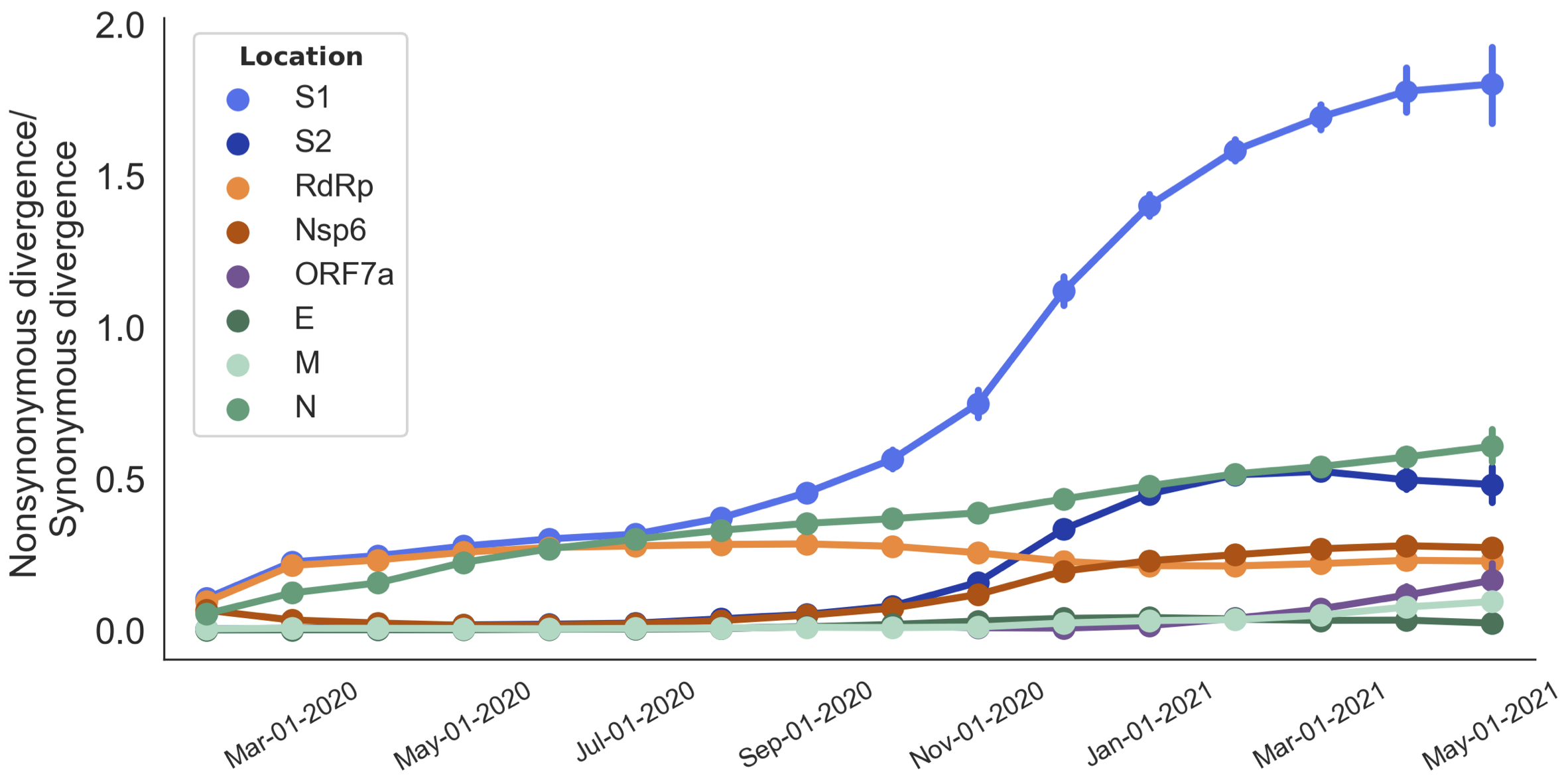
dN/dS through time further highlights adaptive evolution
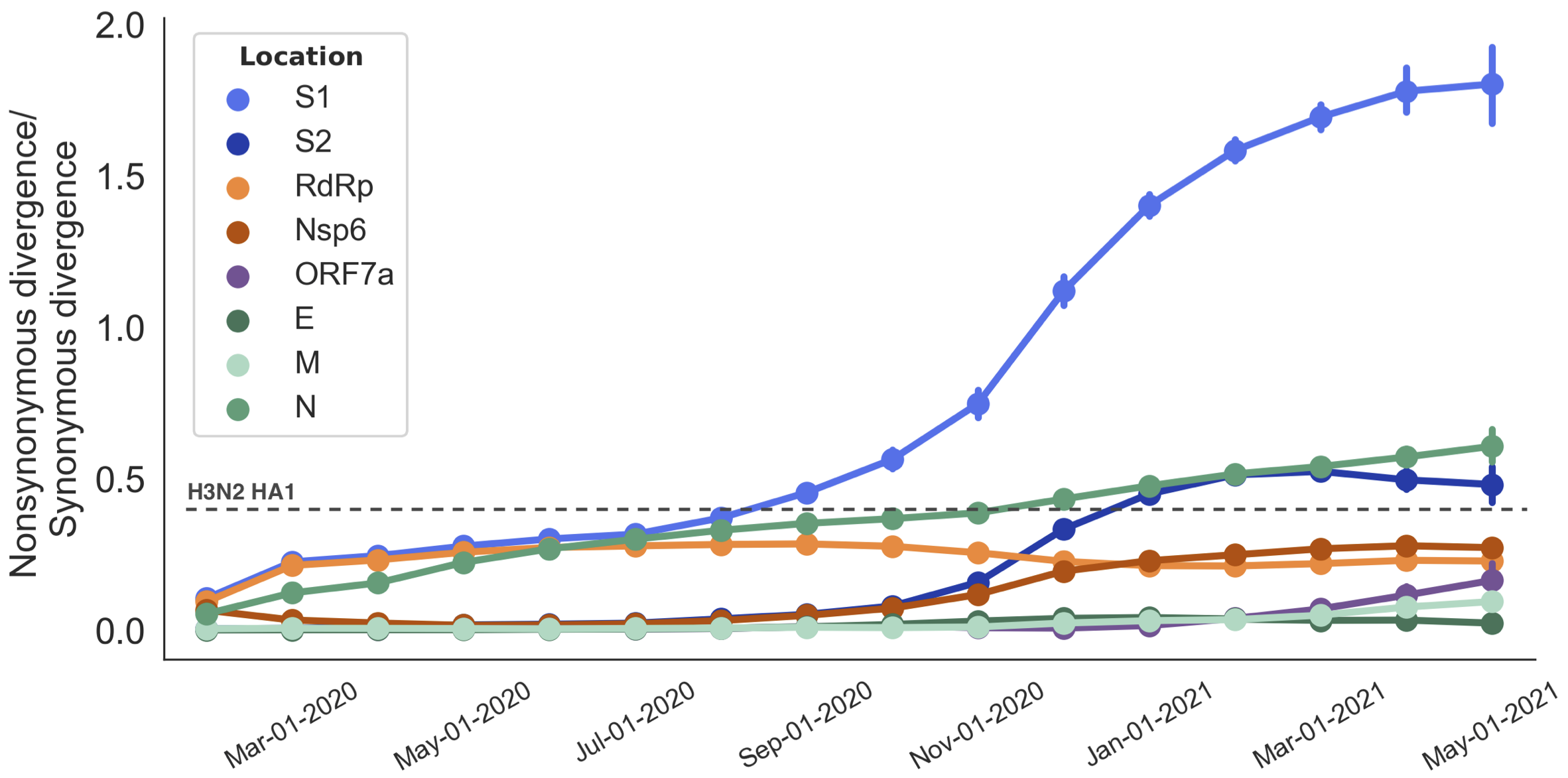
Rapid pace of adaptive evolution relative to H3N2 influenza
S1 mutations cluster phylogenetically, suggesting bursts of evolution
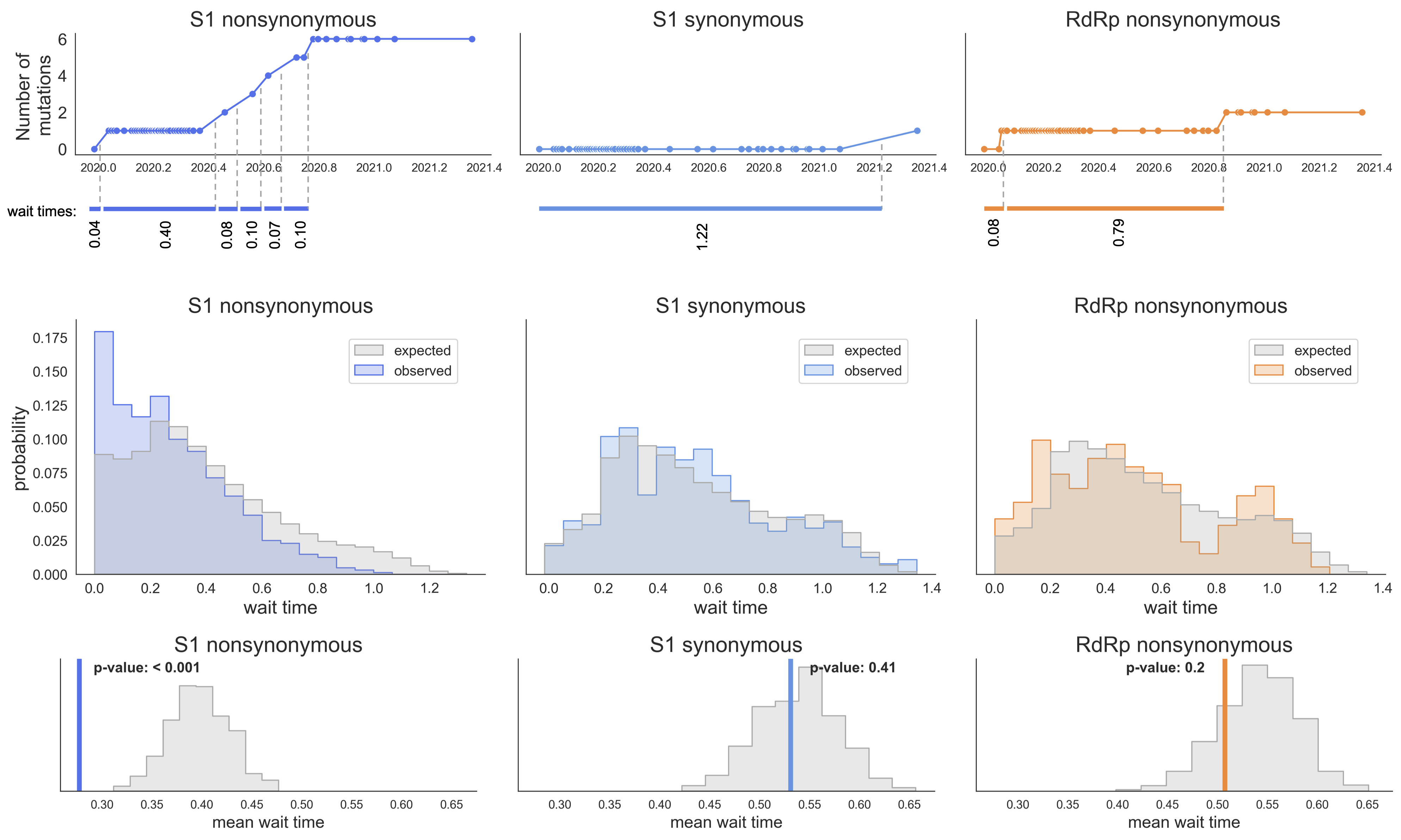
Many S1 mutations show strong convergent evolution
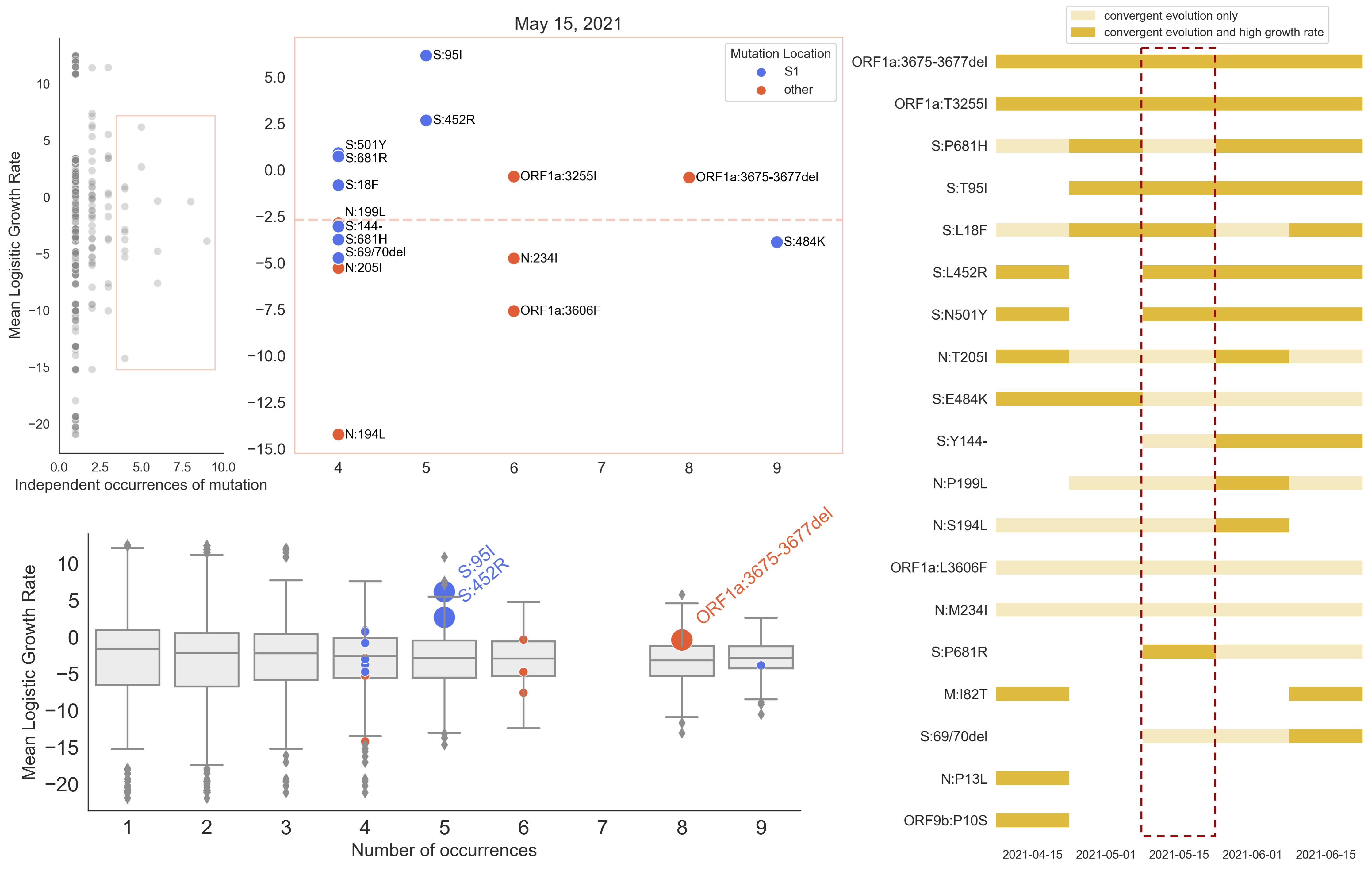
Mutations in S1 arising via within-host pressures result increase viral fitness and are enriched in the viral population by natural selection
Although selection has not been primarily for antigenic drift, observed level of adaptability suggests its potential
Variant transmission dynamics
Multiple approaches to modeling fitness differences between circulating variants
Multinomial logistic regression models work well on frequencies
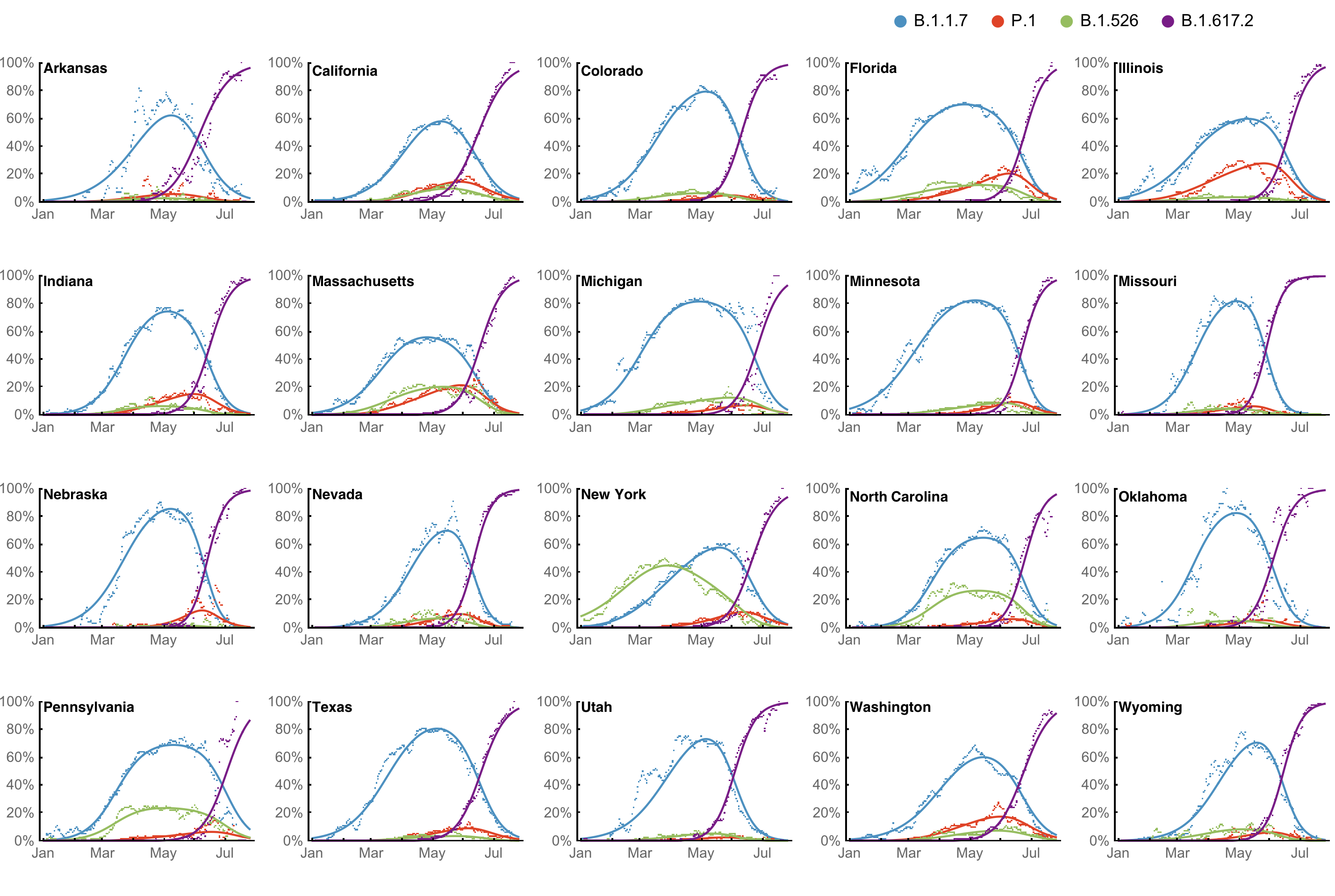
However, frequency of a variant may rise while cases fall
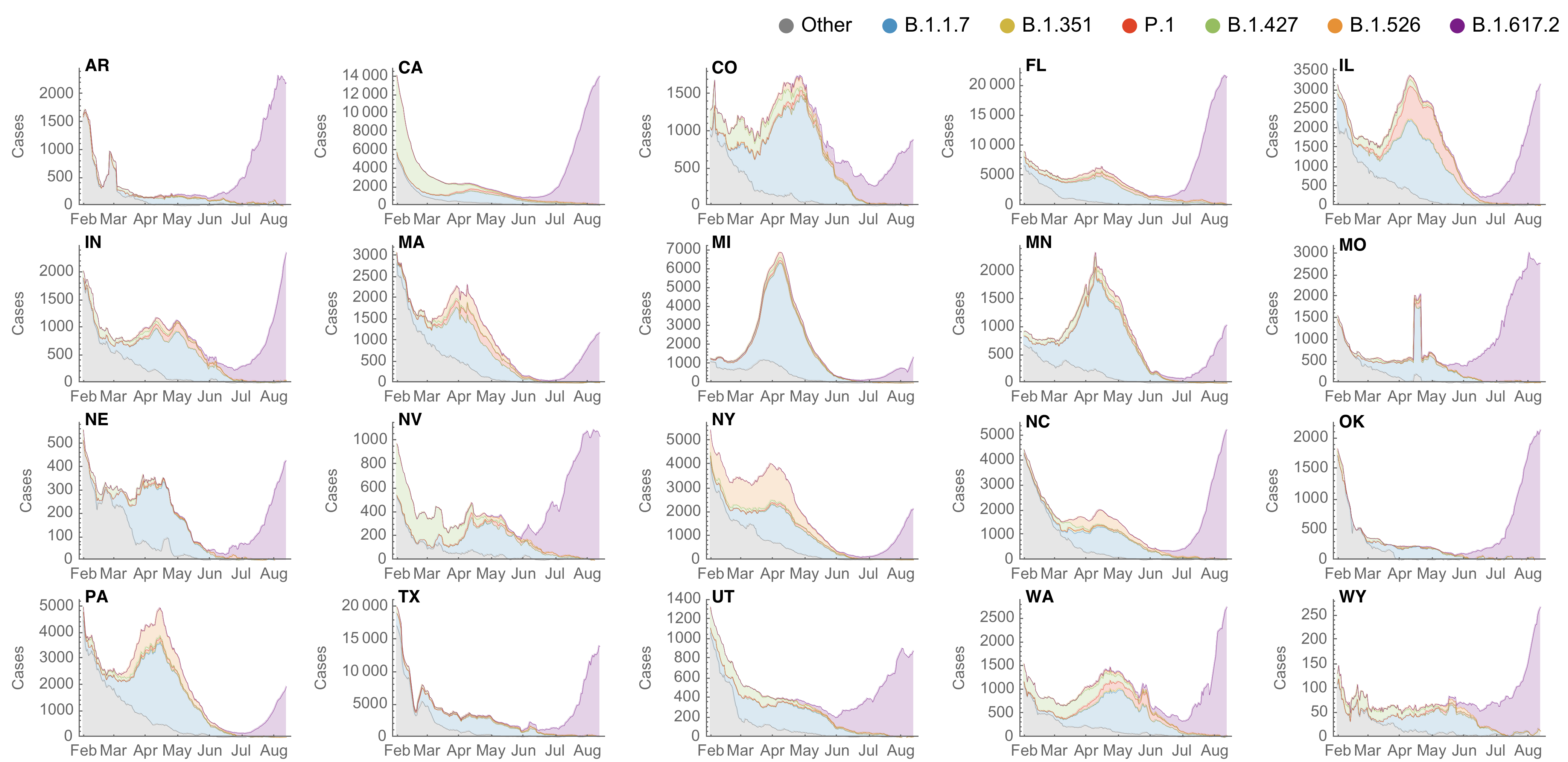
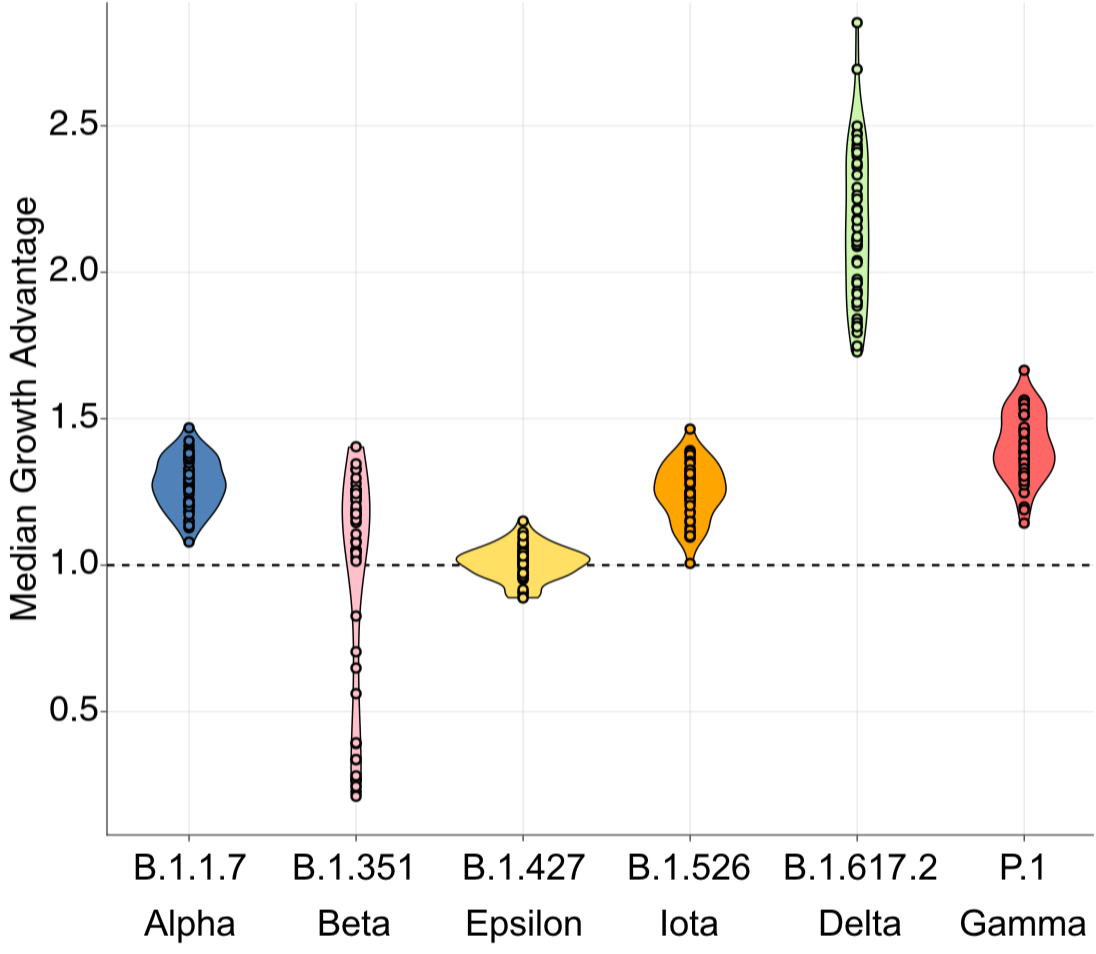
SARS-CoV-2 variant dynamics across US states show consistent differences in transmission rates
 Marlin Figgins
Marlin Figgins
Estimation of variant-specific Rt through time using state-level data
State-level case counts are partitioned based on frequencies of sequenced cases
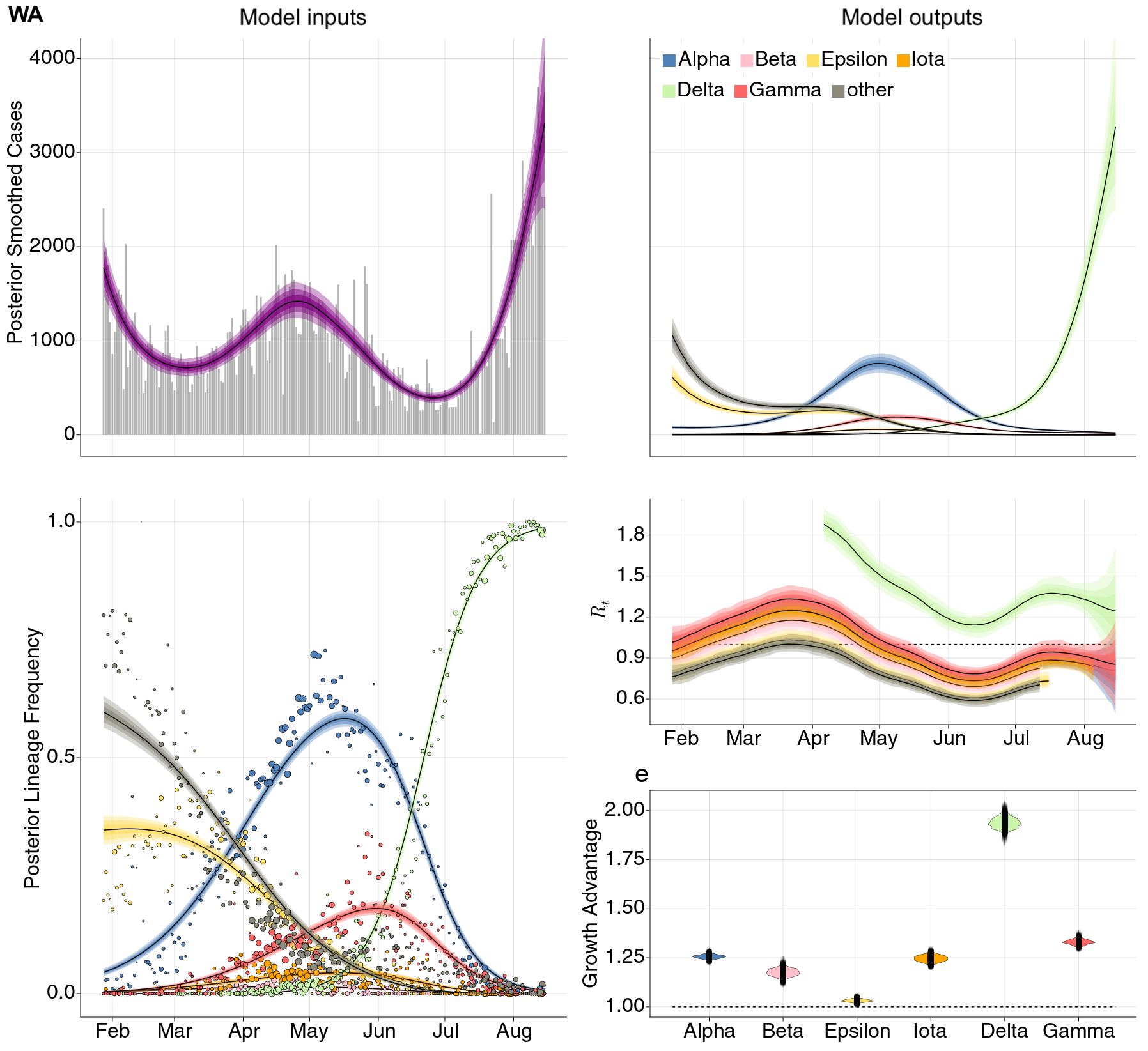
Differences in intrinsic Rt across variants, but all trending downwards
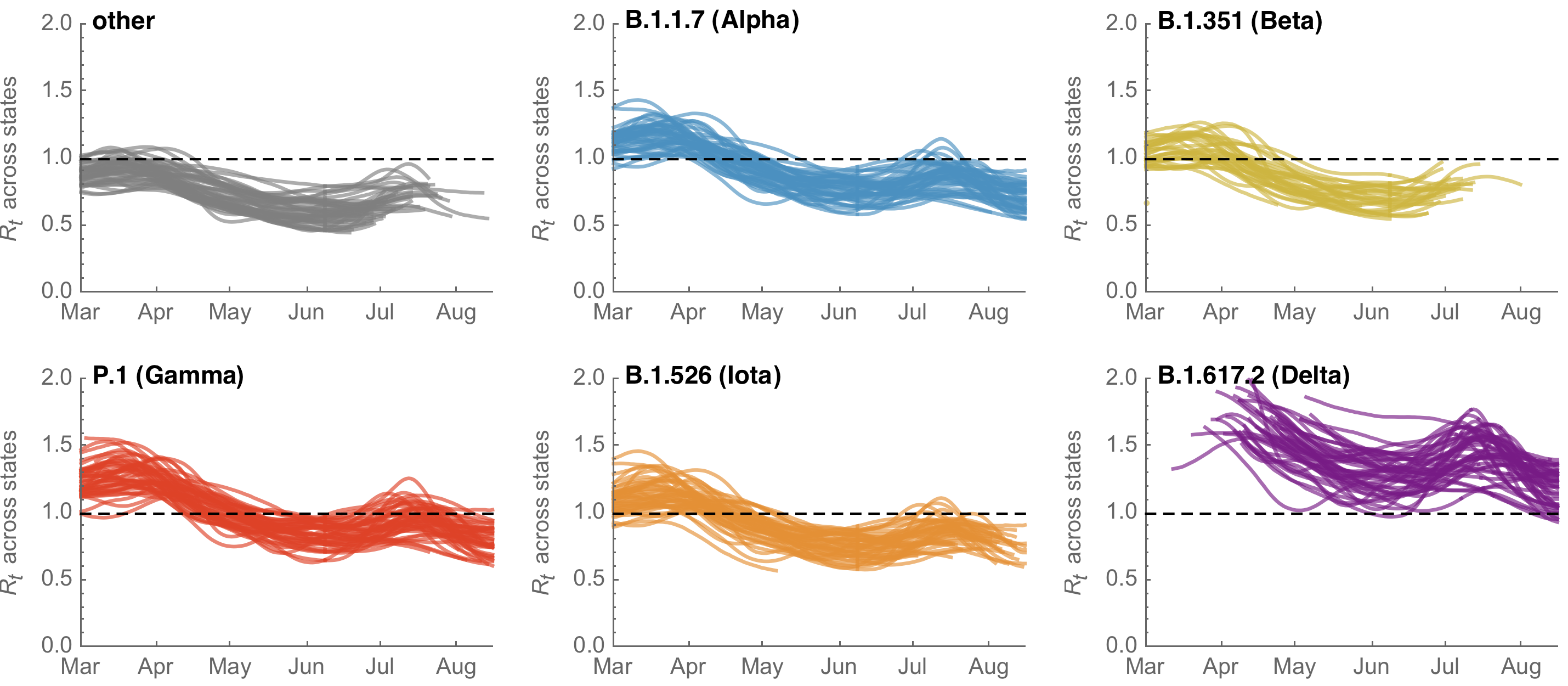
By August 15 only Delta had Rt consistently greater than 1
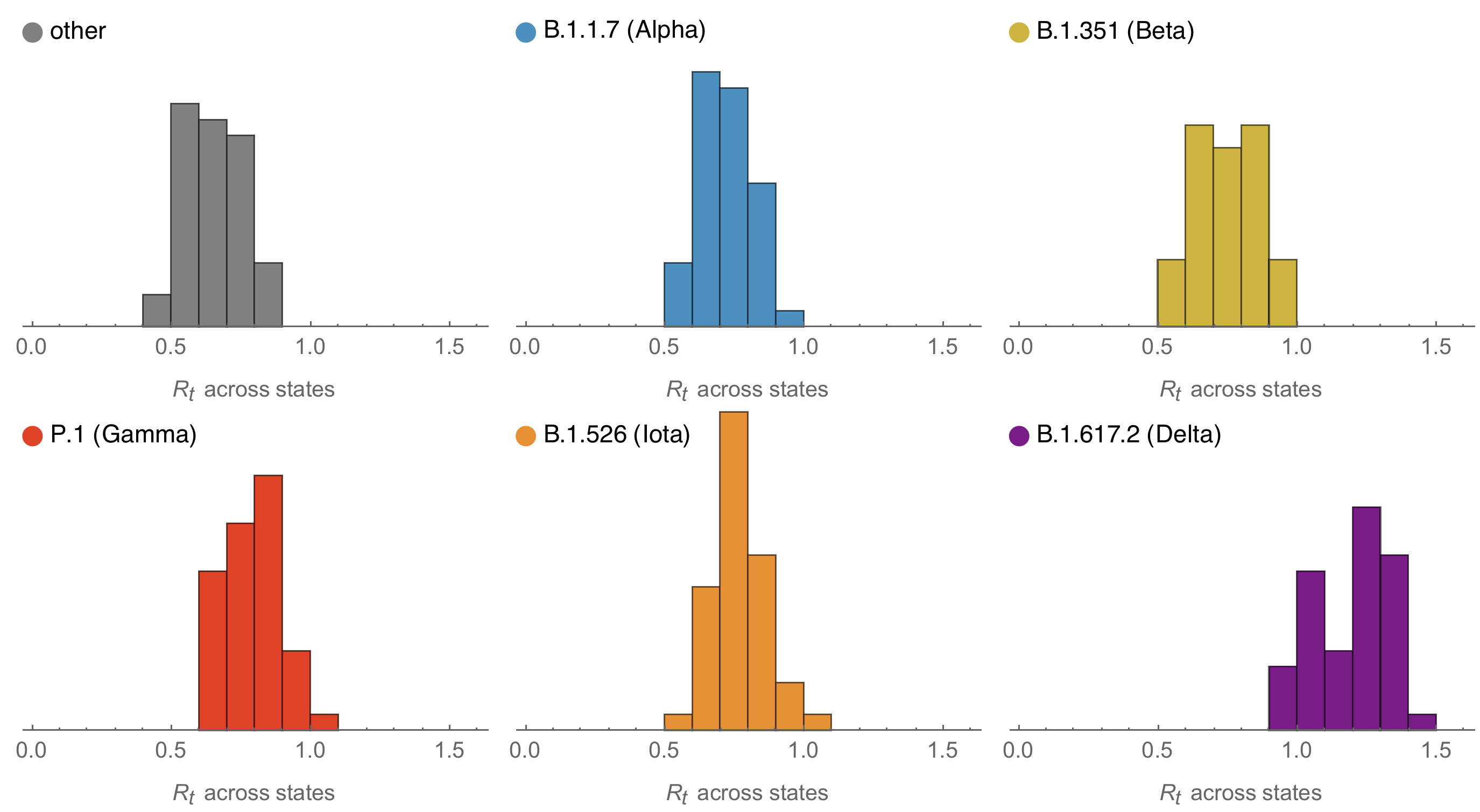
Consistent differences in variant-specific transmission rate across states
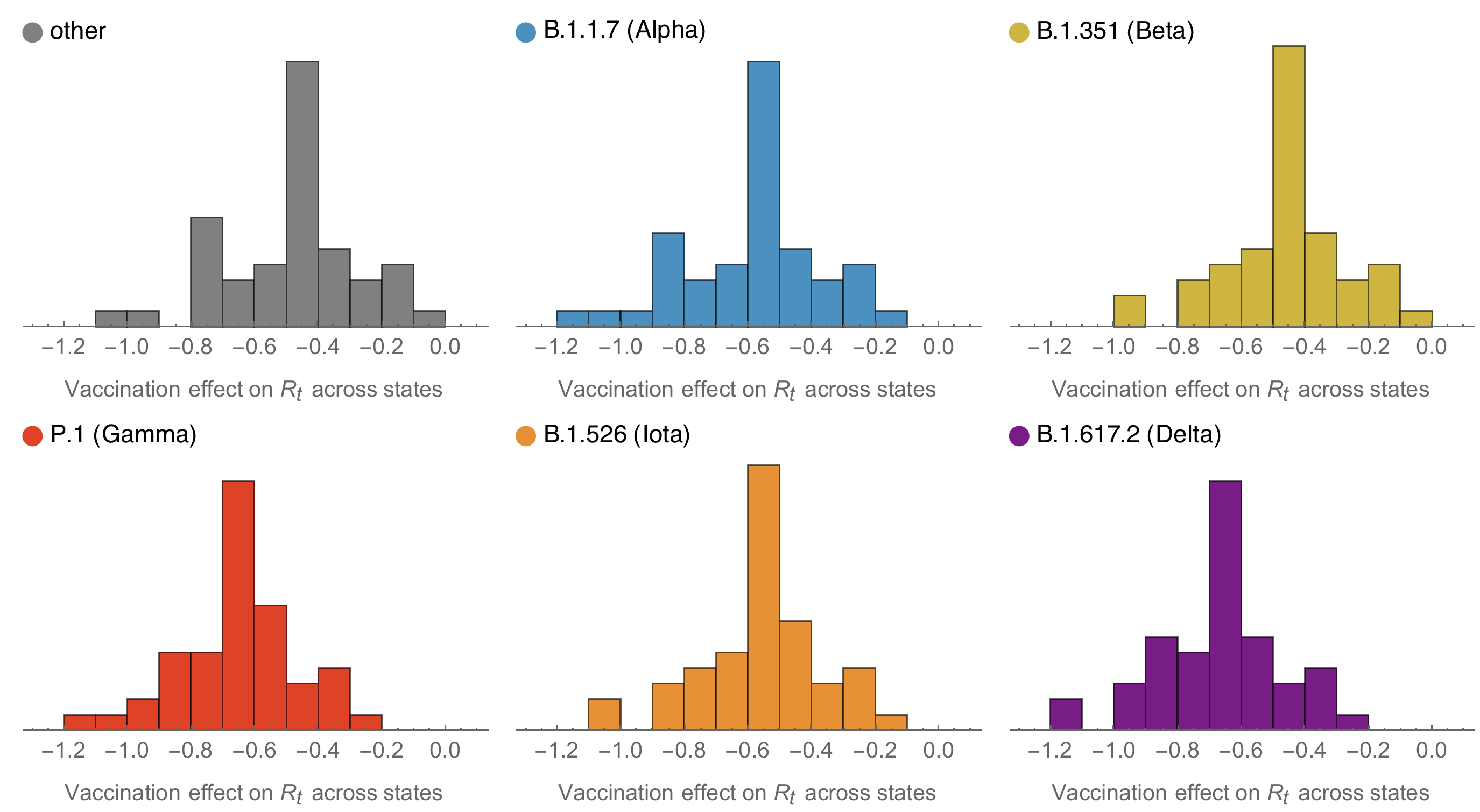
Consistent reductions in variant-specific Rt from vaccination
Future work would ideally tie together granular empirical estimates of viral fitness from frequency data together with mutational and phenotypic predictors to learn what is driving variant success
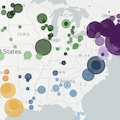
Genomic surveillance
Sequences generated and shared at an unprecedented pace with 630k genomes shared in Aug 2021 alone
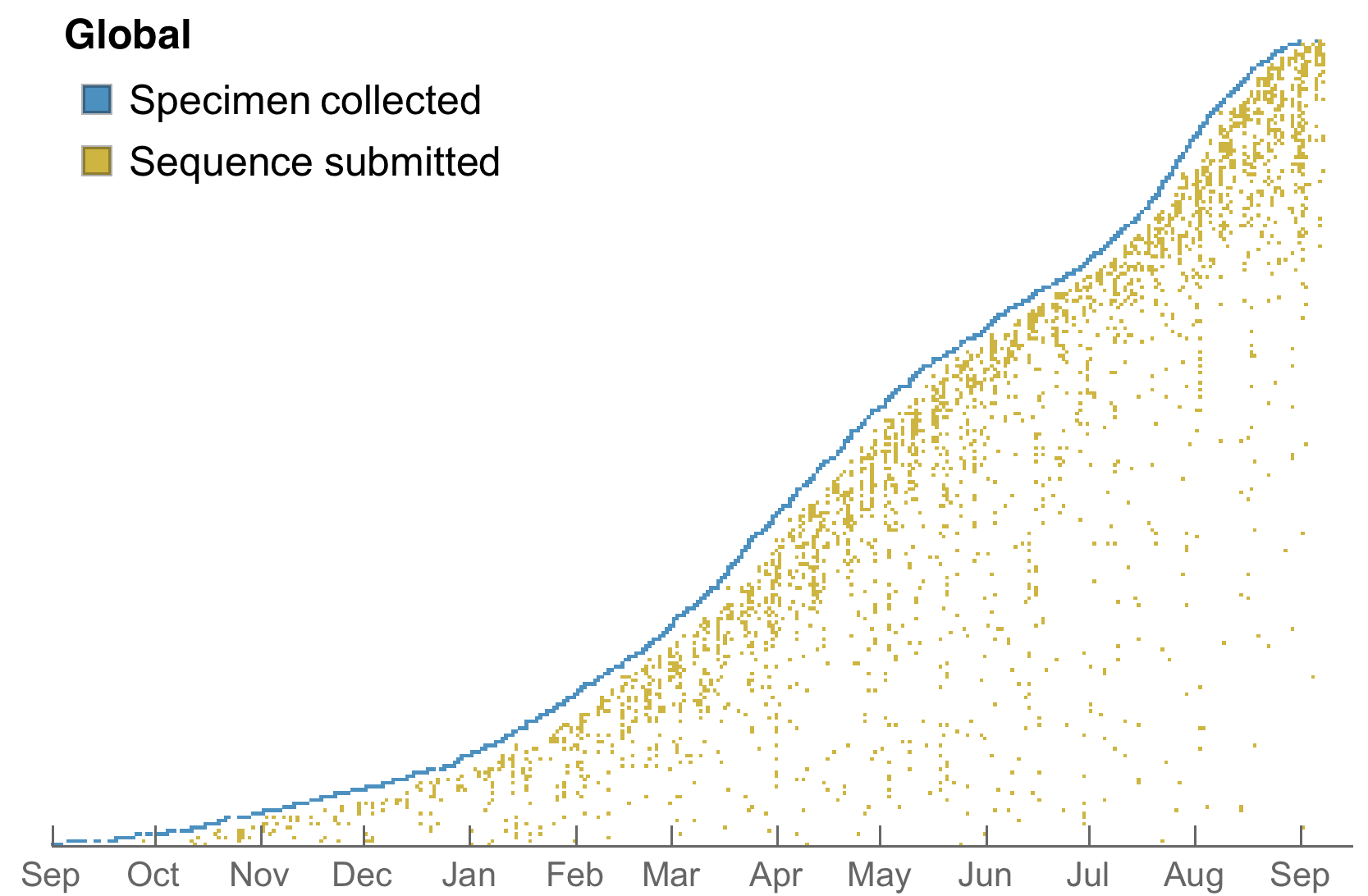
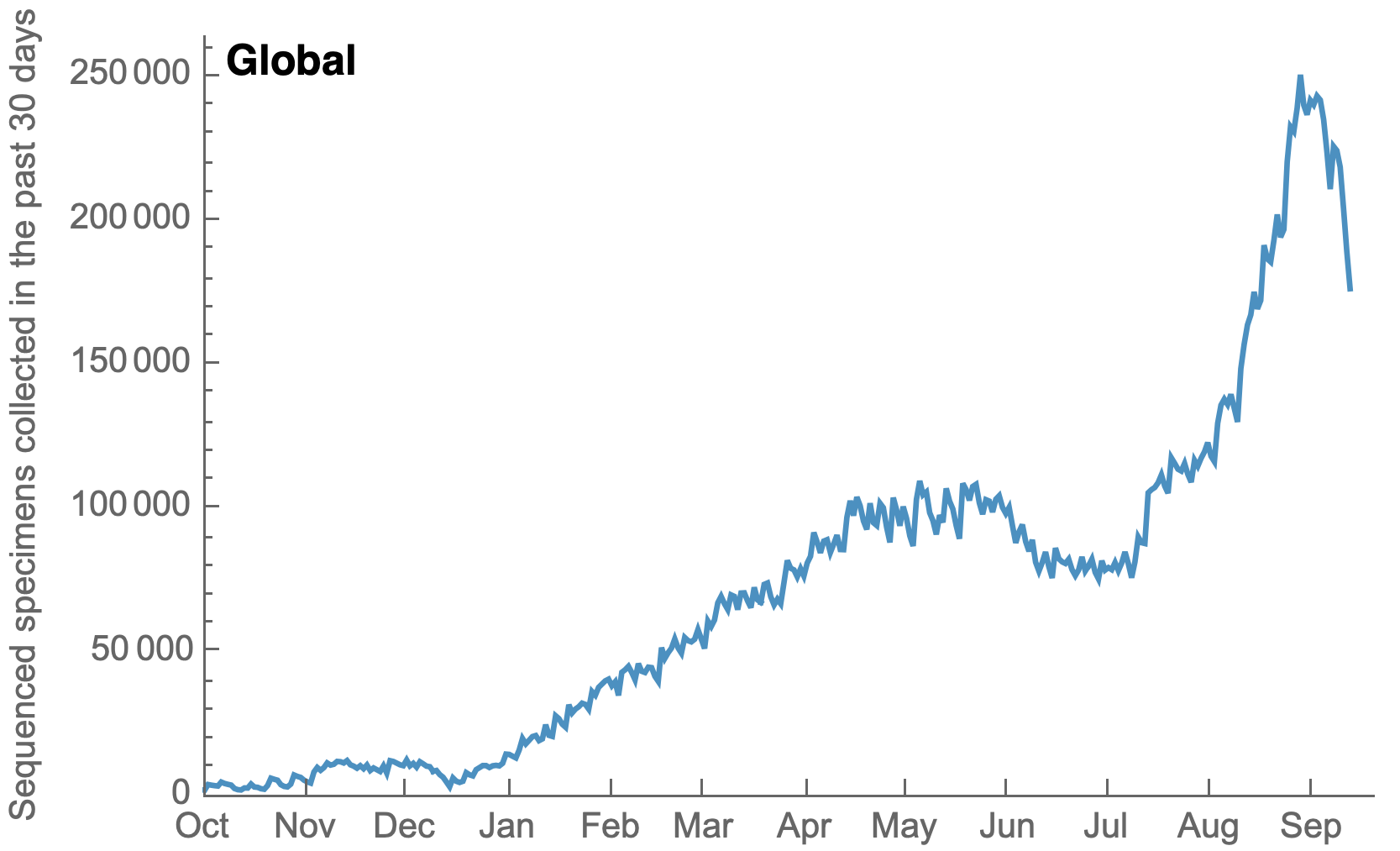
My favorite metric is number of sequences available from samples collected in the past 30 days
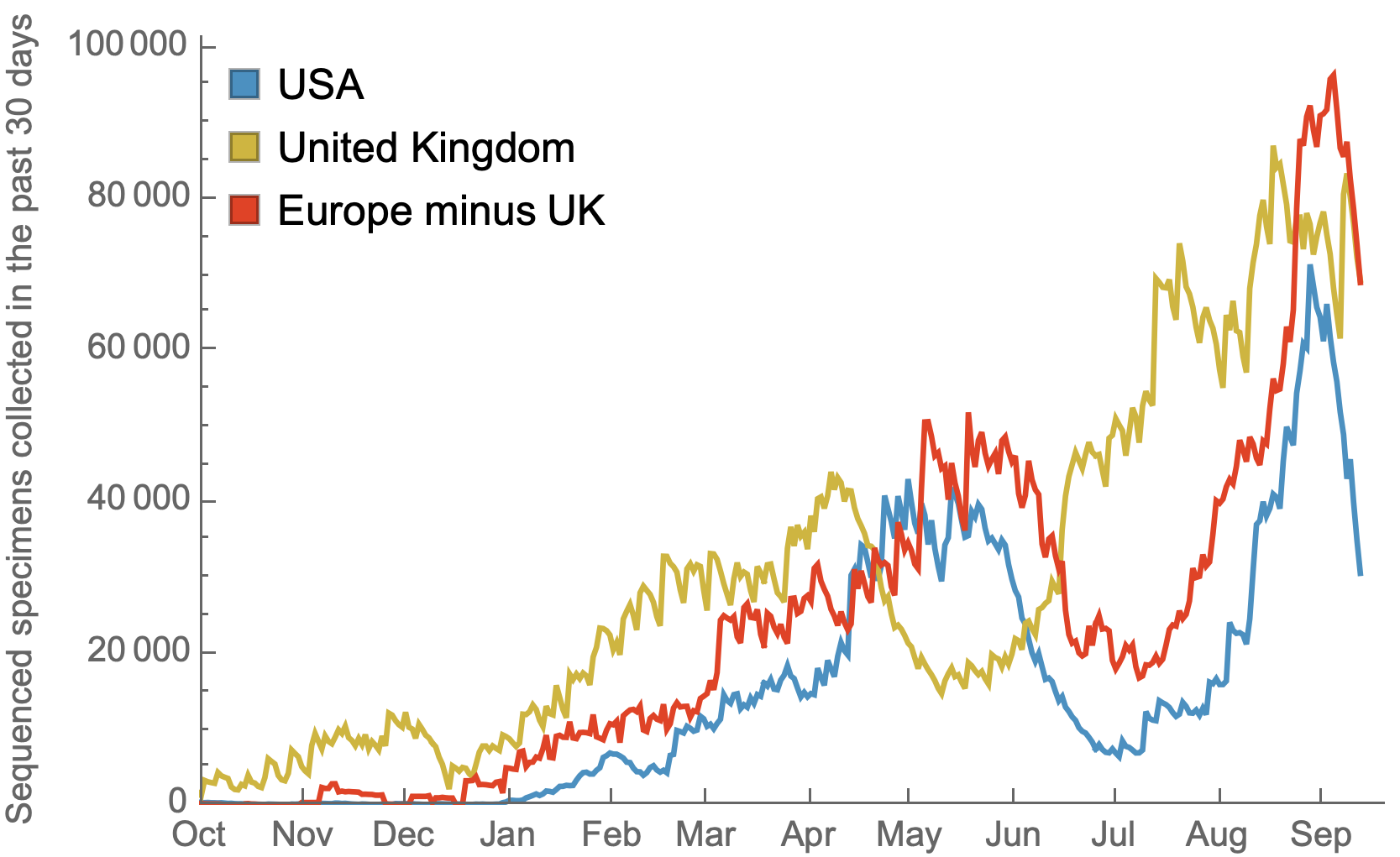
US, UK and Europe all have stunning genomic surveillance programs
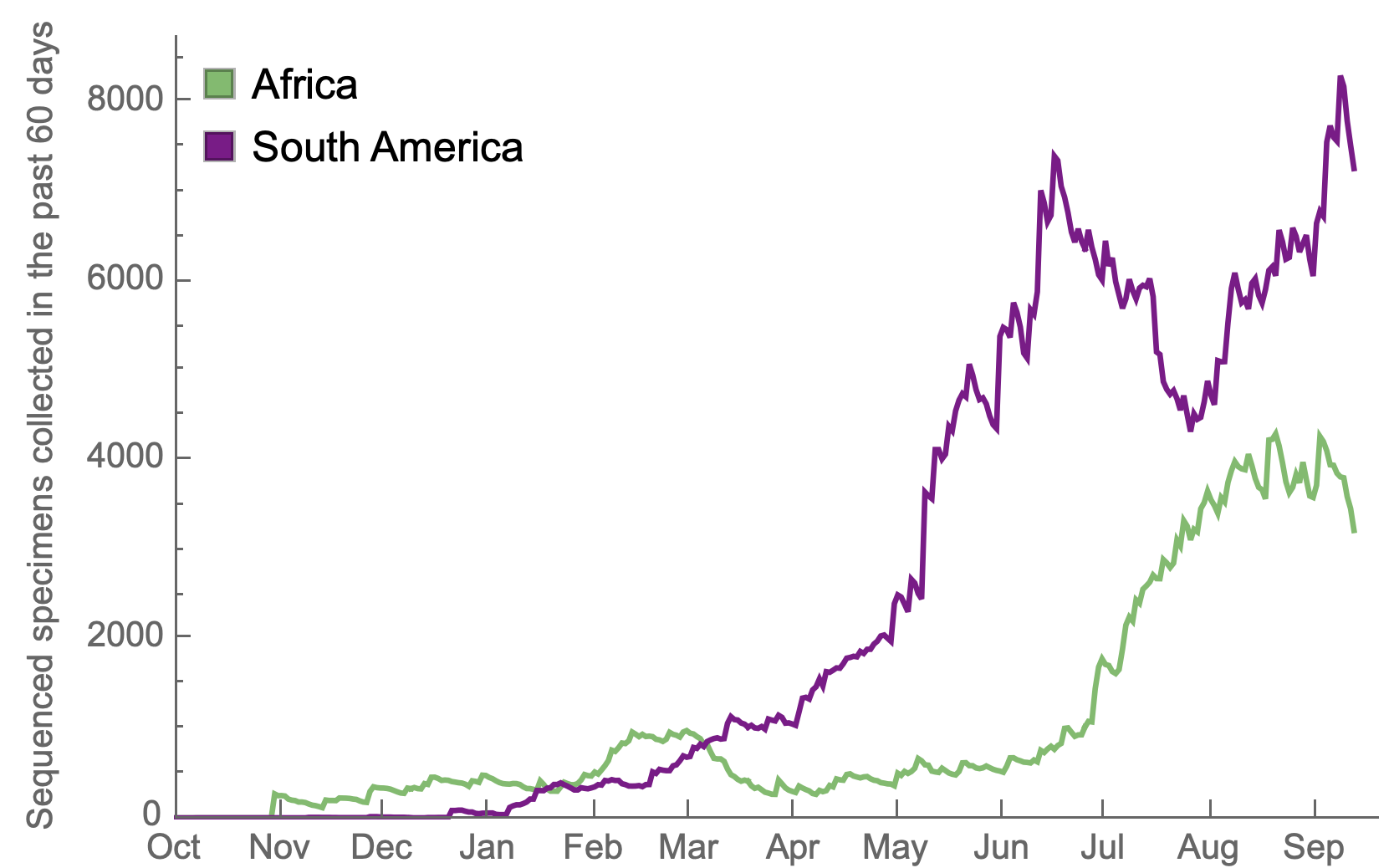
Africa and South America have picked up dramatically but there are still geographic gaps
Global genomic surveillance aimed at rapid turnaround times important to catch new variants early
In depth genomic surveillance tied to data systems allows investigation of variant severity, secondary attack rate, etc...
Future perspectives
Broad expectations based on comparison with flu
- R0 of Delta is perhaps ~5 compared to R0 of flu of ~2. At same rates of evolution and waning expect more COVID circulation.
- Rates of adaptive sequence evolution in SARS-CoV-2 have been about ~5X that of H3N2 flu. Expect this to slow down, but suggests that virus will be generally quite adaptable.
- IFR of COVID is roughly comparable to flu once you have prior immunity granting durable memory responses.
1, 2 and 3 suggest a virus that circulates like flu does at perhaps higher levels (given R0), but individual infections aren't much more severe in terms of mortality than H3N2 influenza. Expect perhaps 50-100K deaths in the US each year.
SARS-CoV-2 scientific (and prosaic) priorities
- Forecasting evolution and ensuring vaccine updates match circulating viruses
- Universal vaccine to engender broader immune response and/or to target more conserved epitopes
- Plentiful testing, contact tracing and a public health apparatus
- Treatments, ventilation, etc...
Acknowledgements
SARS-CoV-2 genomic epi: Data producers from all over the world, GISAID and the Nextstrain team
Bedford Lab:
![]() John Huddleston,
John Huddleston,
![]() James Hadfield,
James Hadfield,
![]() Katie Kistler,
Katie Kistler,
![]() Louise Moncla,
Louise Moncla,
![]() Maya Lewinsohn,
Maya Lewinsohn,
![]() Thomas Sibley,
Thomas Sibley,
![]() Jover Lee,
Jover Lee,
![]() Cassia Wagner,
Cassia Wagner,
![]() Miguel Paredes,
Miguel Paredes,
![]() Nicola Müller,
Nicola Müller,
![]() Marlin Figgins,
Marlin Figgins,
![]() Eli Harkins,
Eli Harkins,
![]() Denisse Sequeira
Denisse Sequeira