Real-time tracking of virus evolution
Trevor Bedford (@trvrb)
9 Feb 2016
VIDD Seminar
Fred Hutch
Slides at bedford.io/talks/
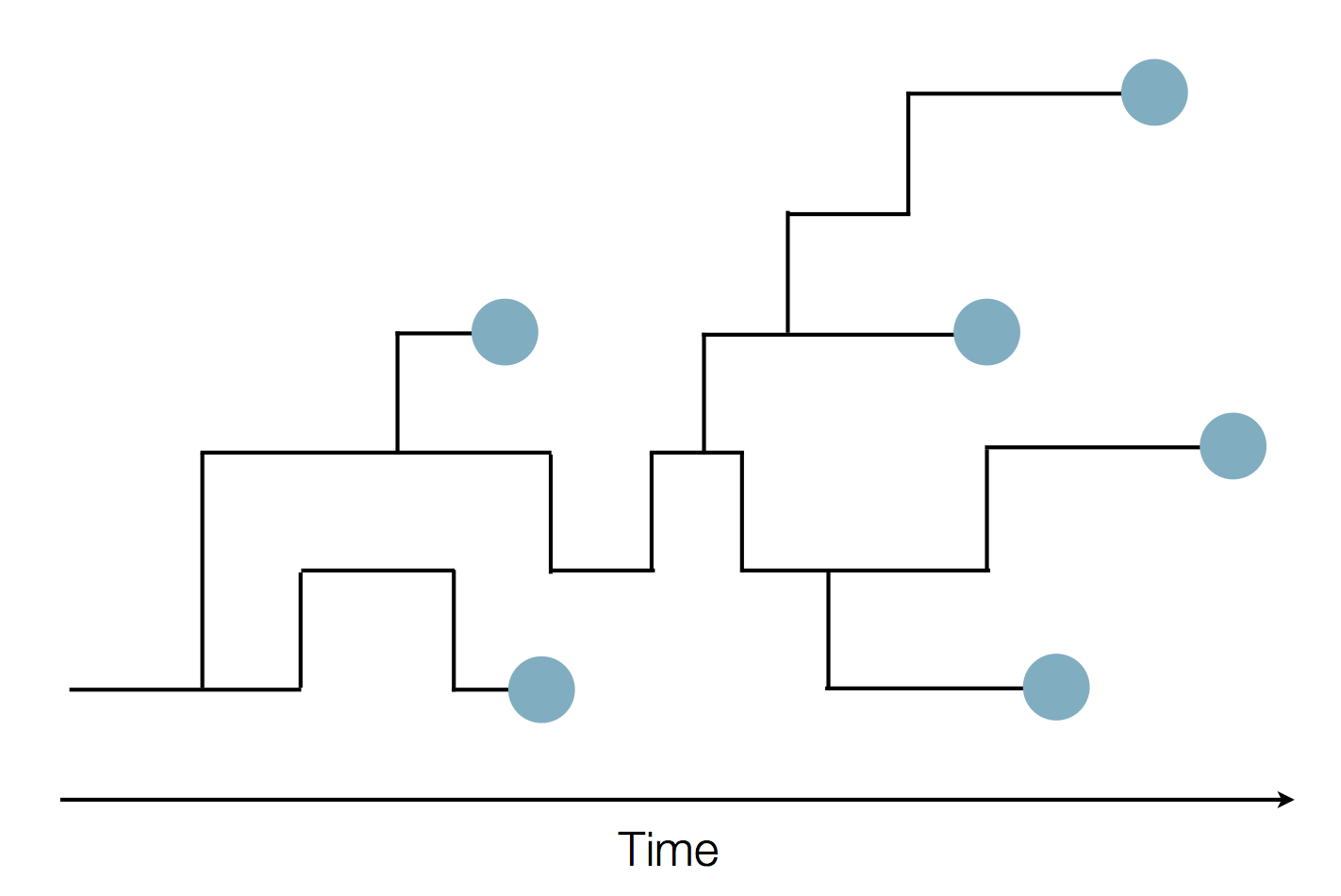
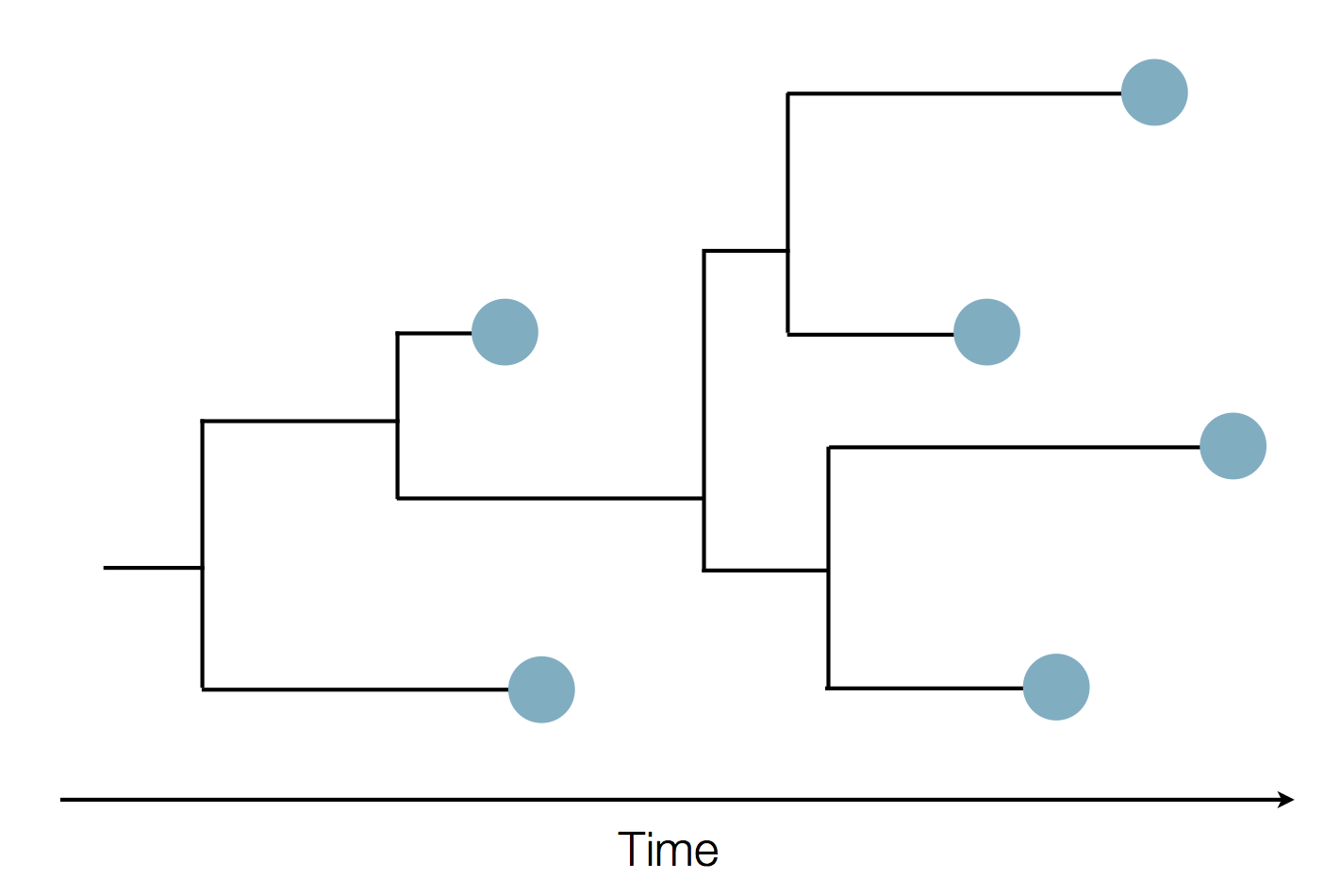
Phylogenies
Phylogenies describe history
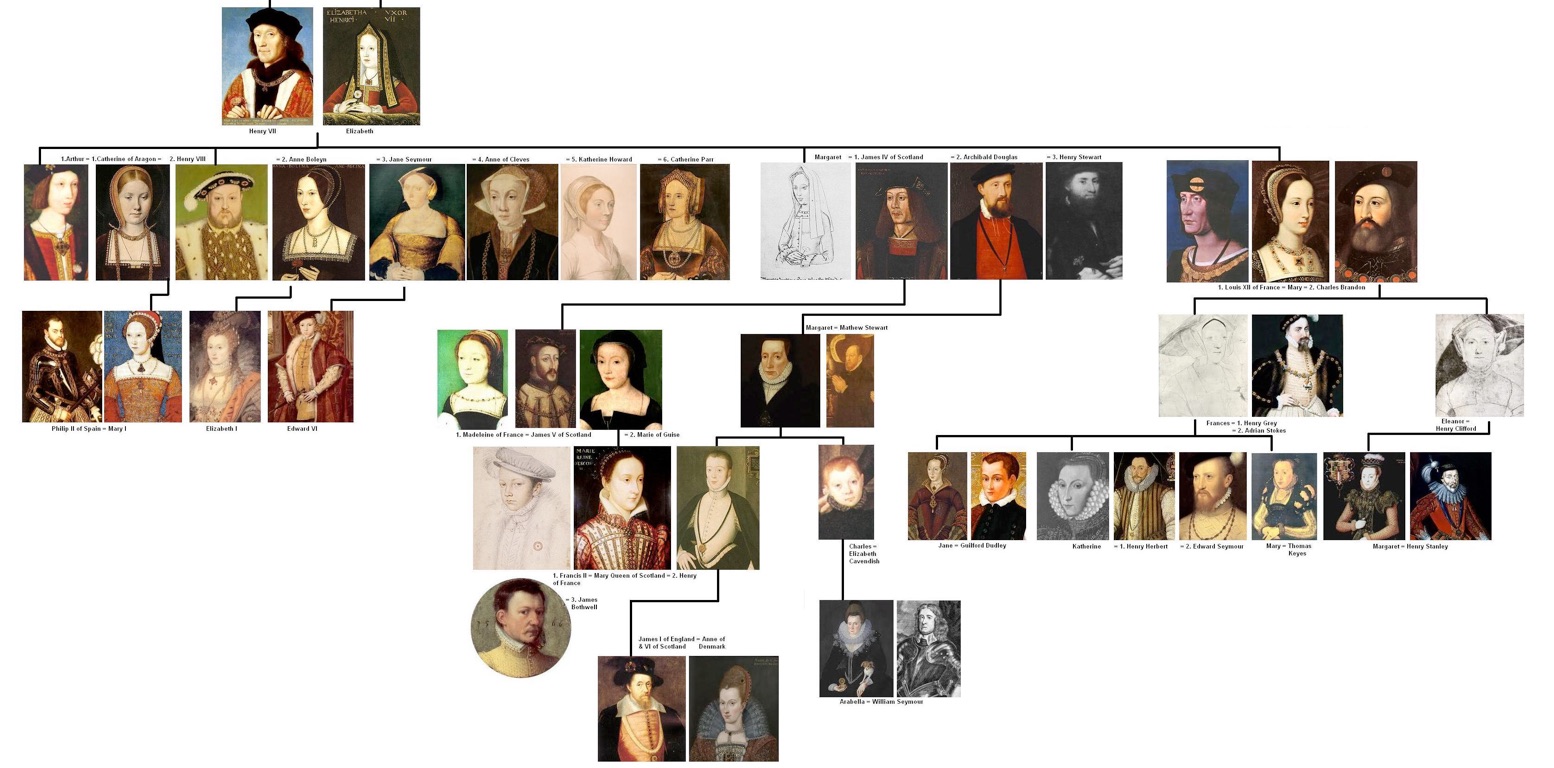
Phylogenies describe history
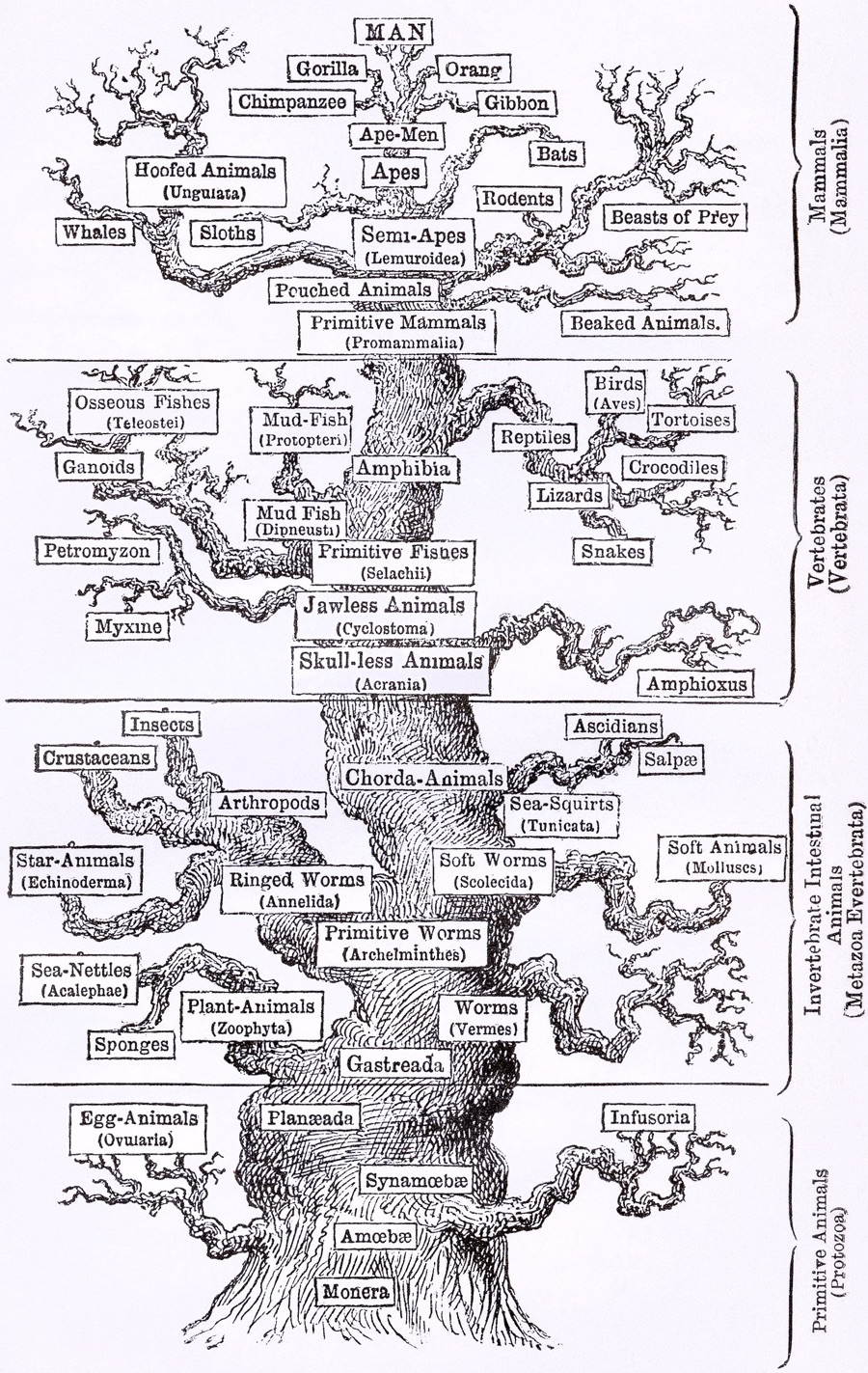
Phylogenies describe history
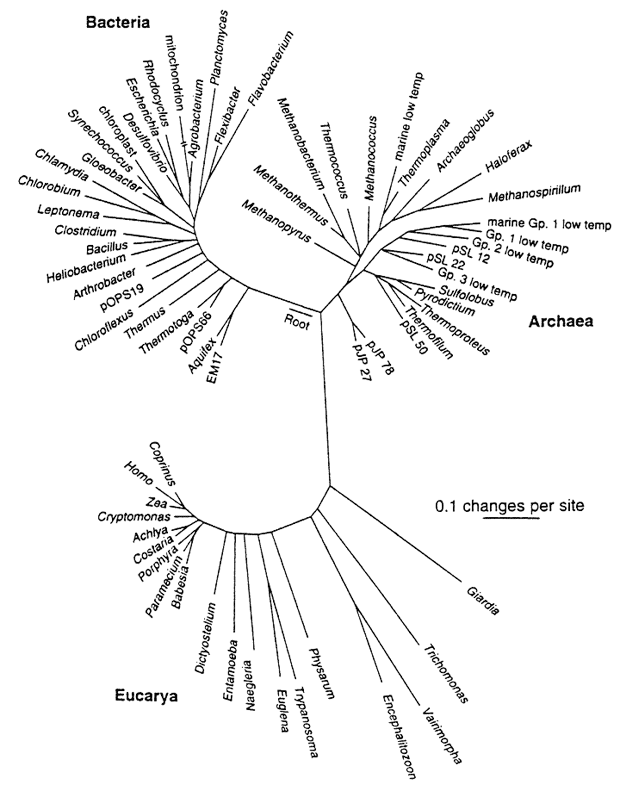
Phylogenies reveal process
Epidemics
Epidemic process
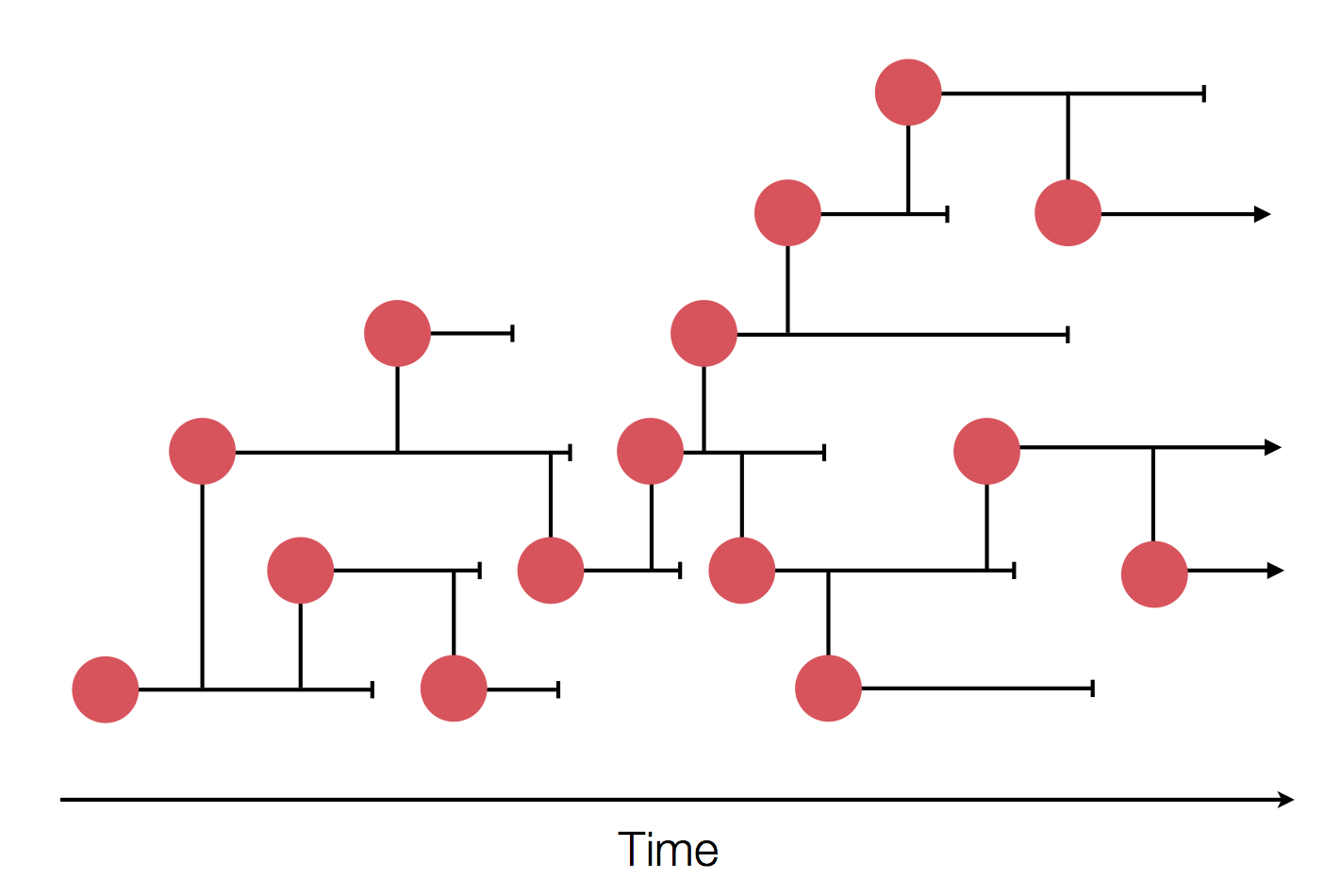
Sample some individuals
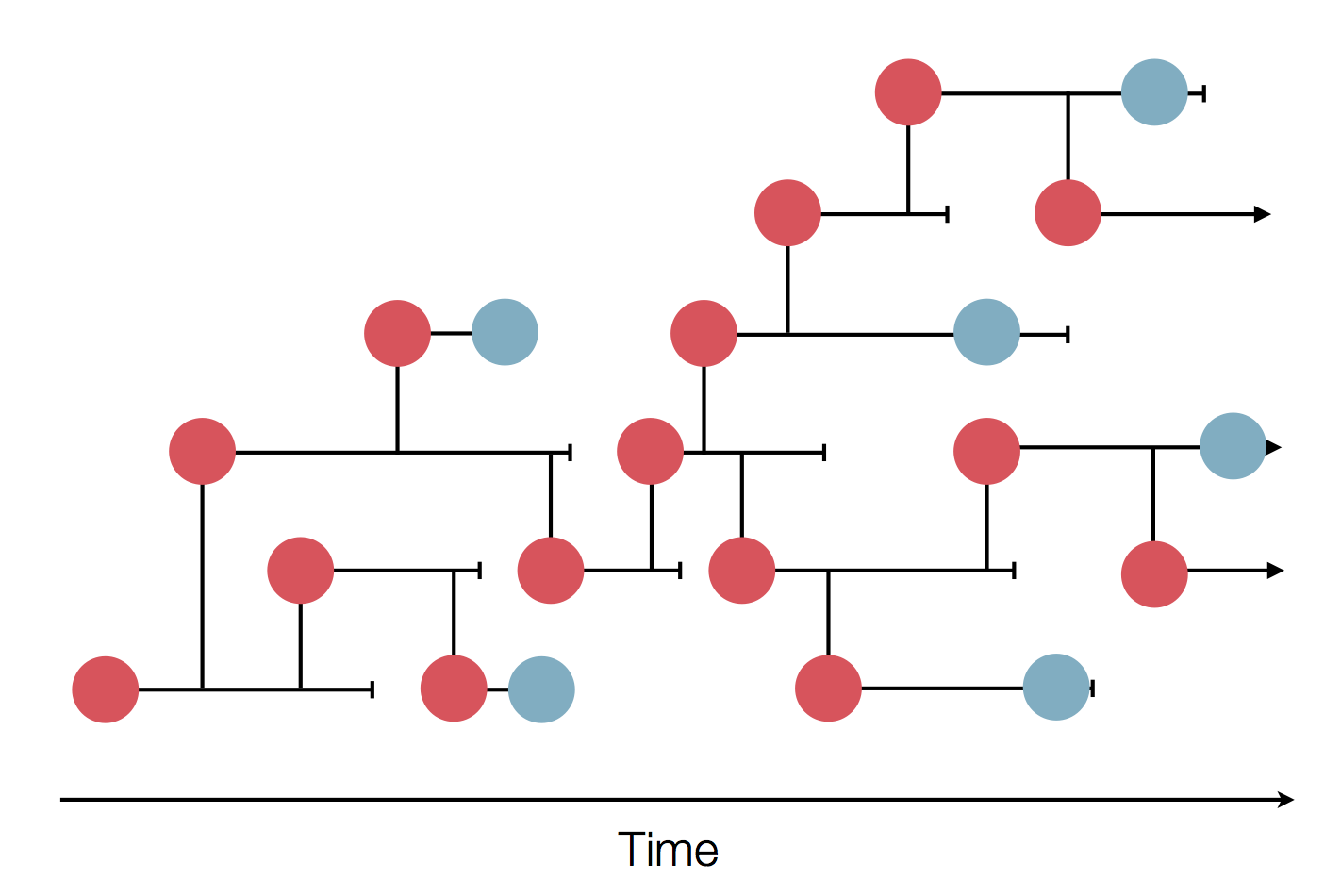
Sequence and determine phylogeny
Sequence and determine phylogeny
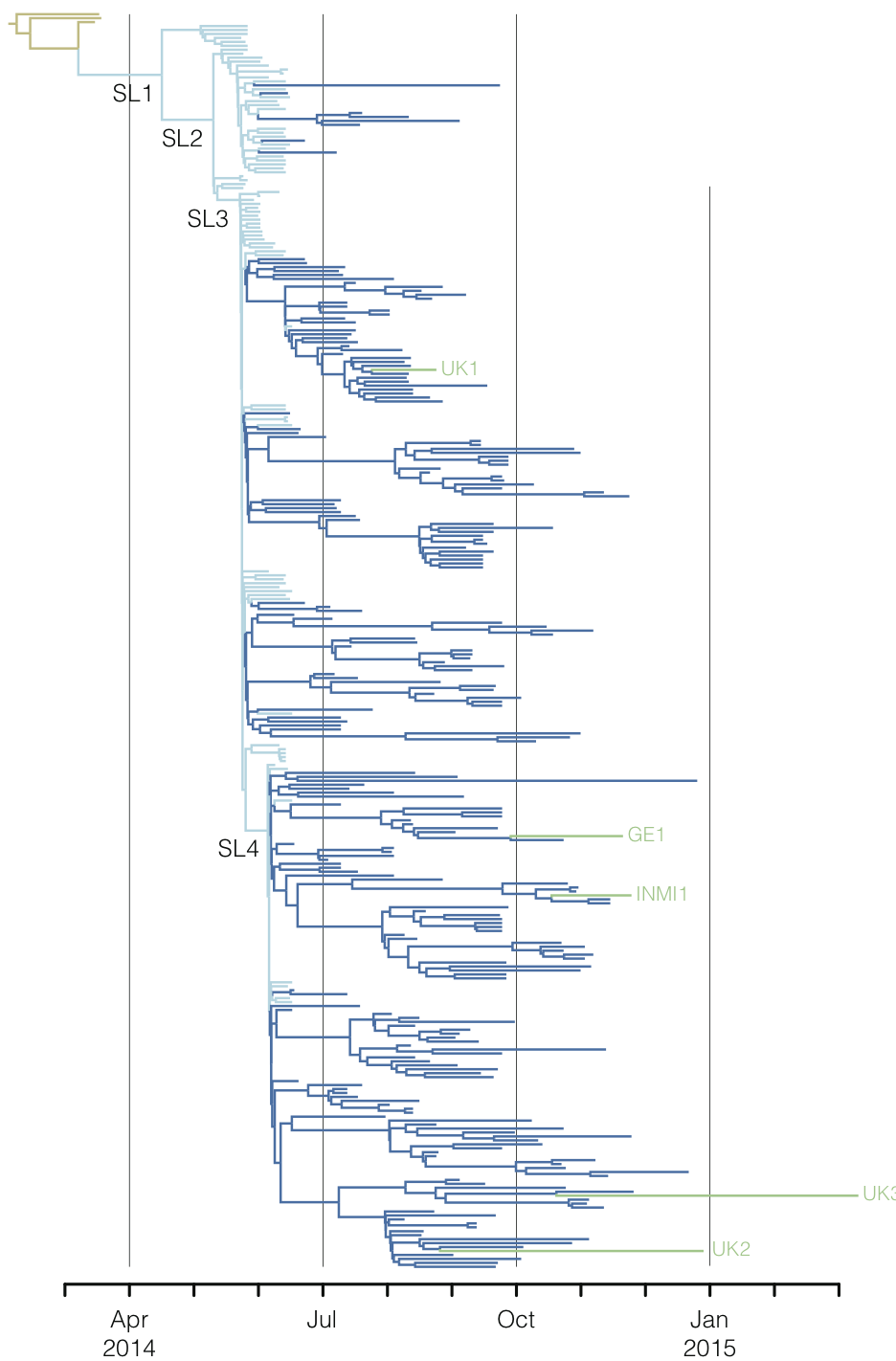
West African Ebola phylogeny
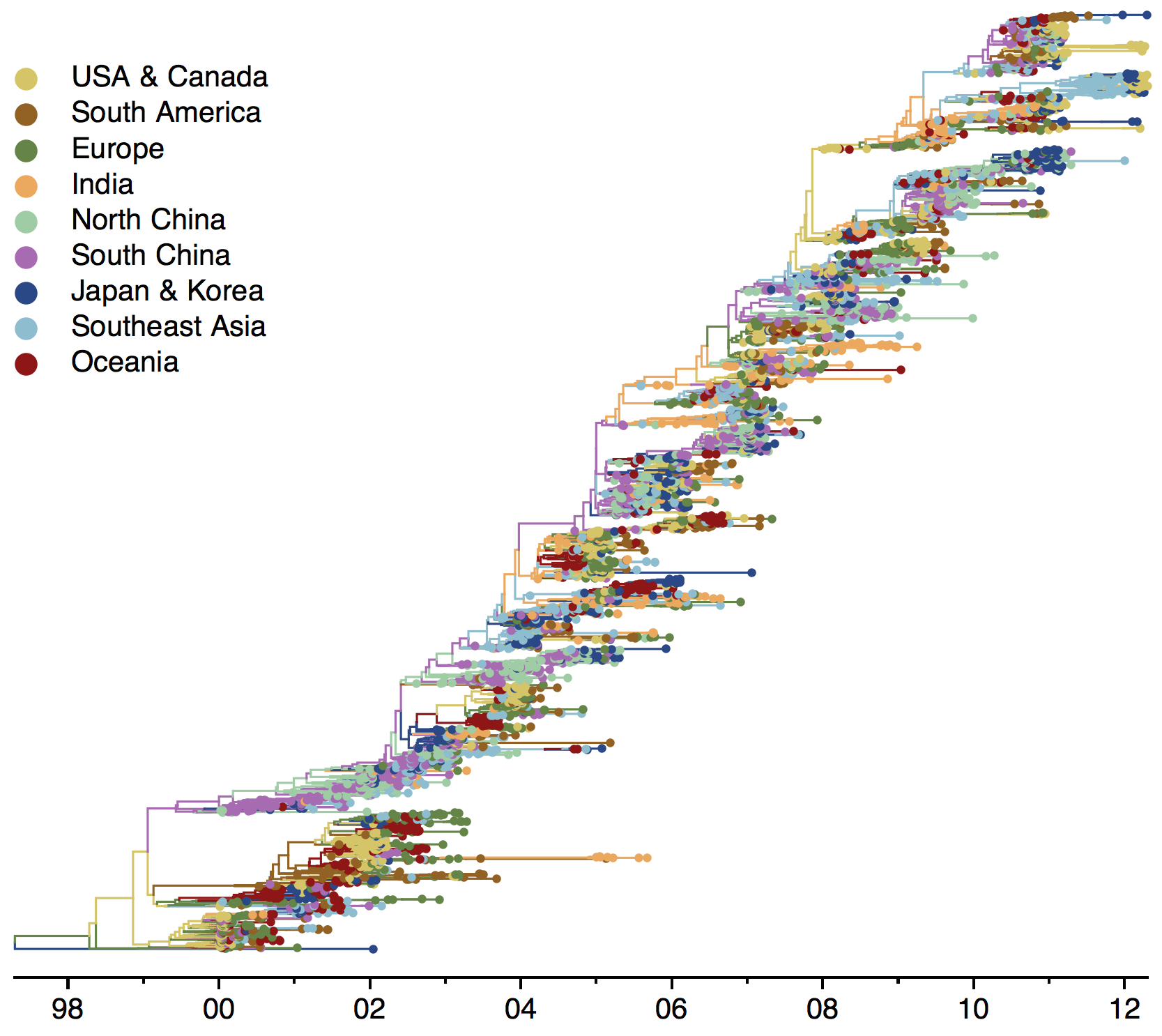
Global influenza phylogeny
Applications of evolutionary analysis for influenza vaccine strain selection and charting outbreak spread
Influenza
Previous research has focused on:
- Antigenic drift
- Geographic circulation
Influenza virus
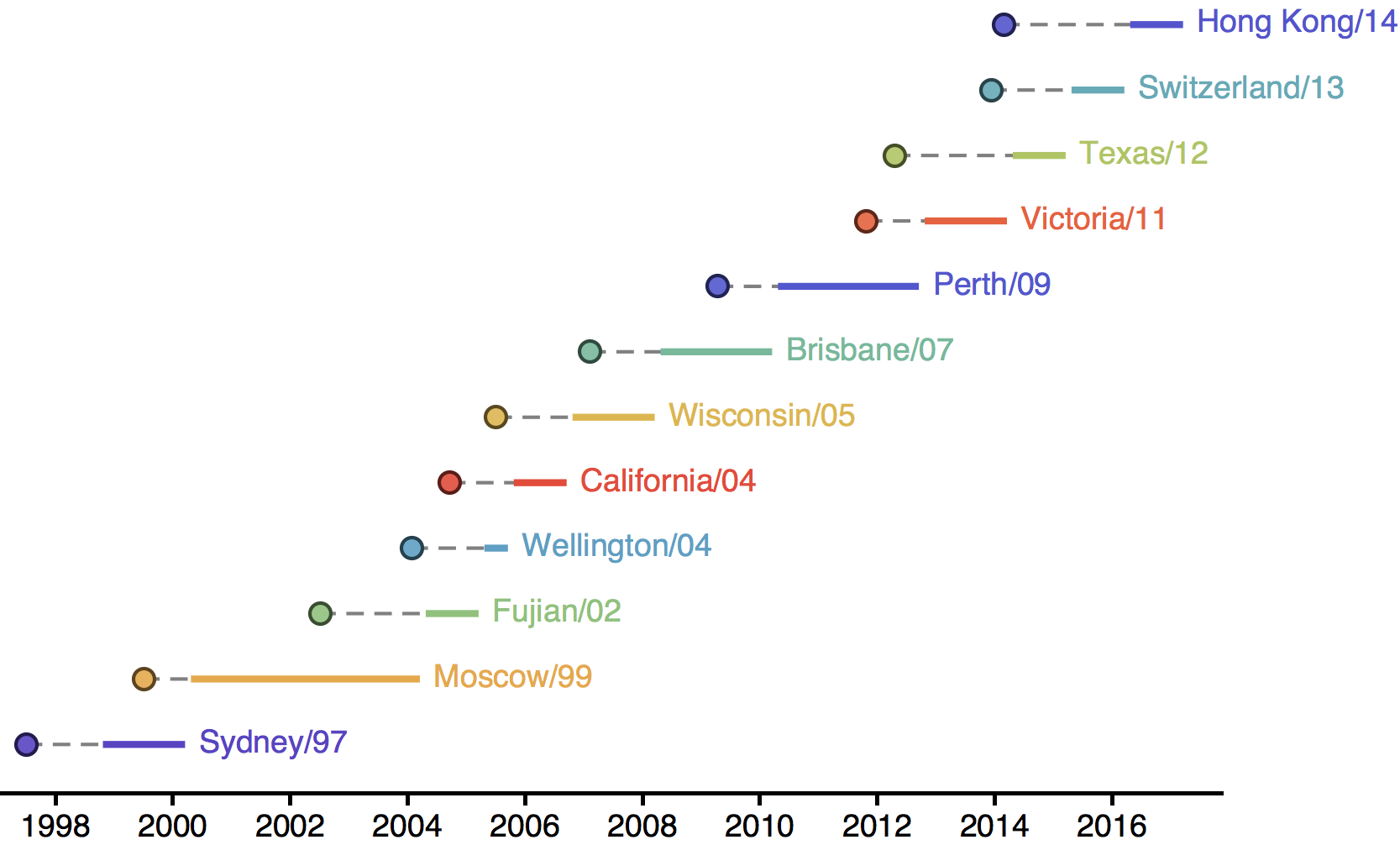
Influenza H3N2 vaccine updates
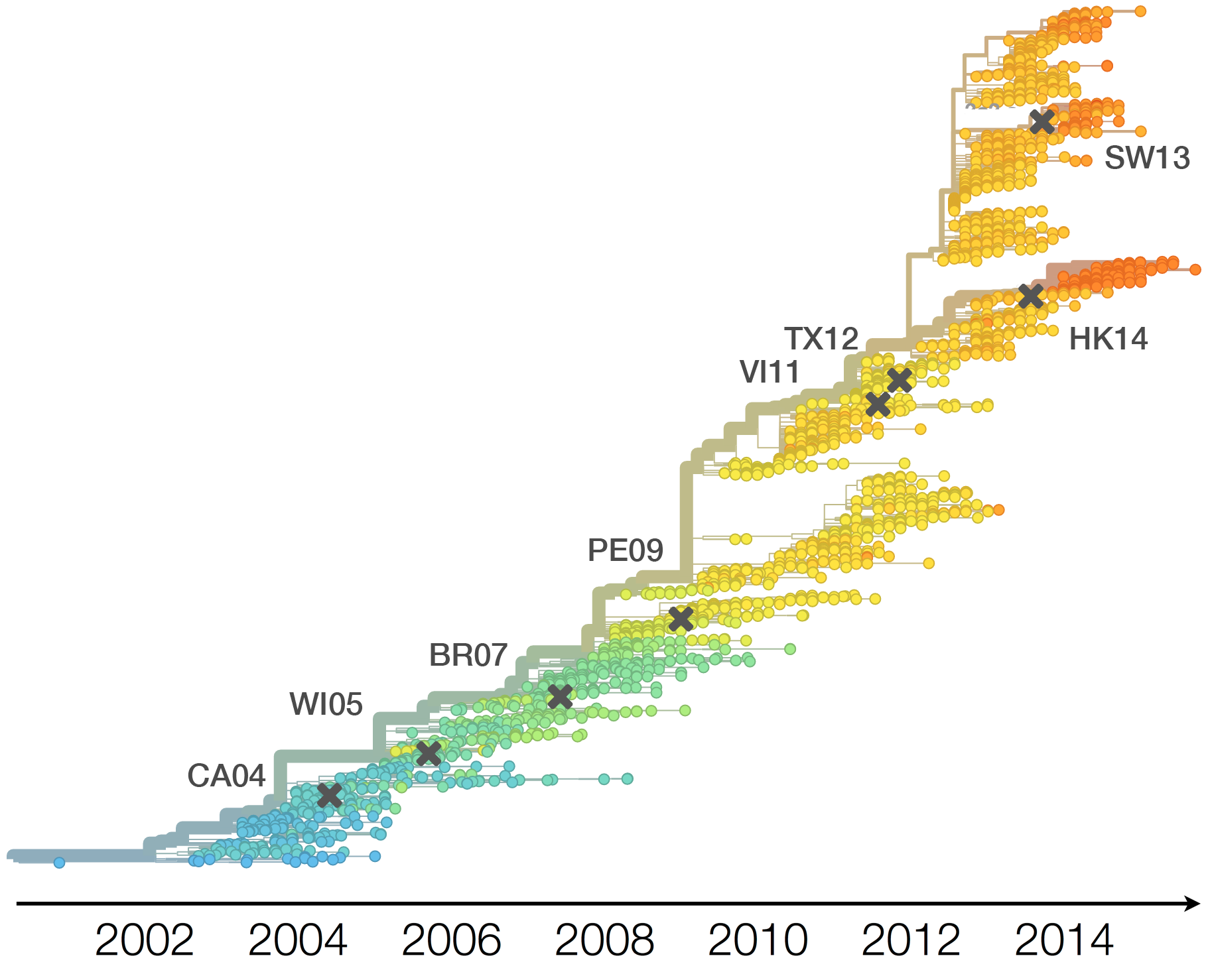
H3N2 phylogeny showing antigenic drift
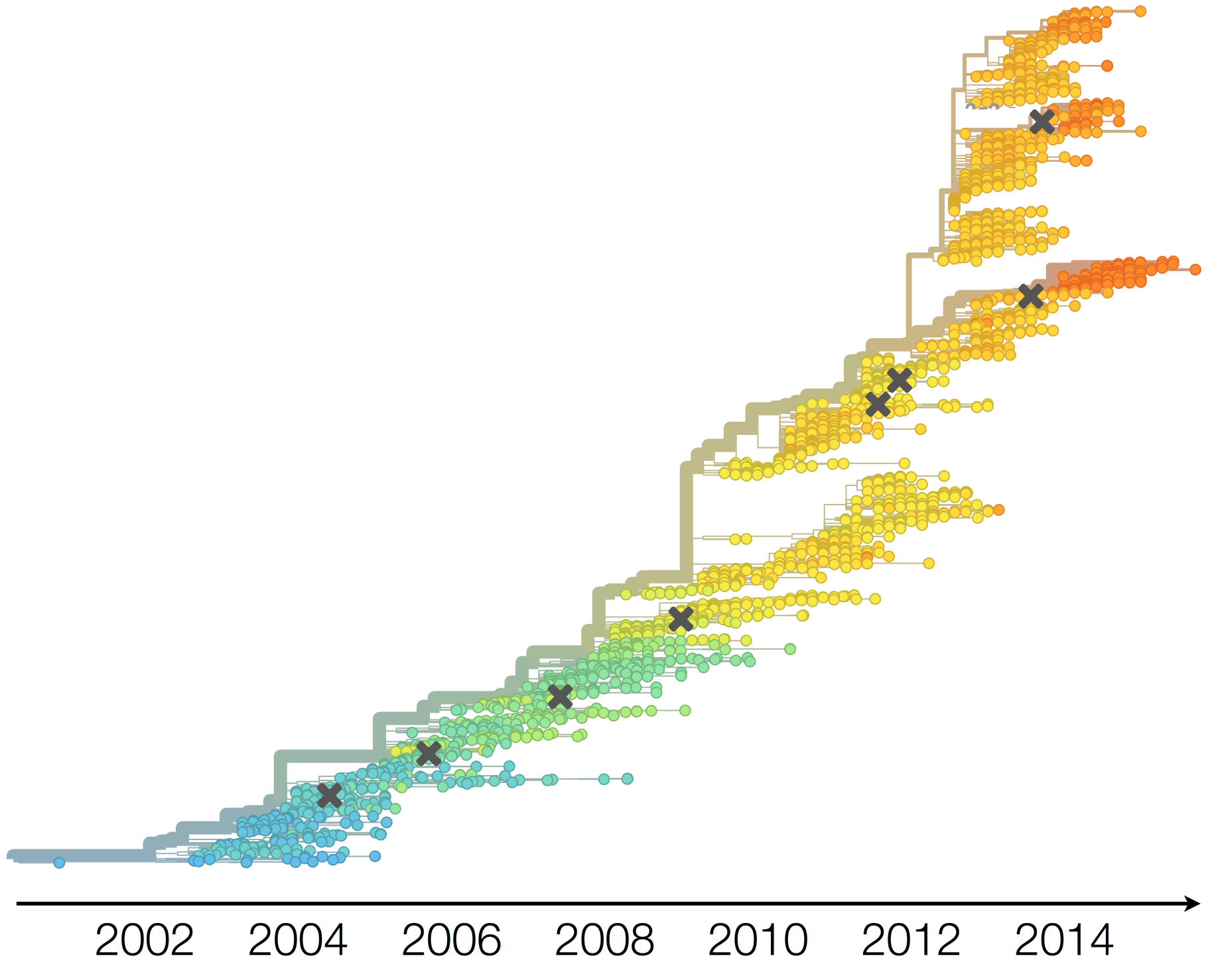
H3N2 phylogeny showing antigenic drift
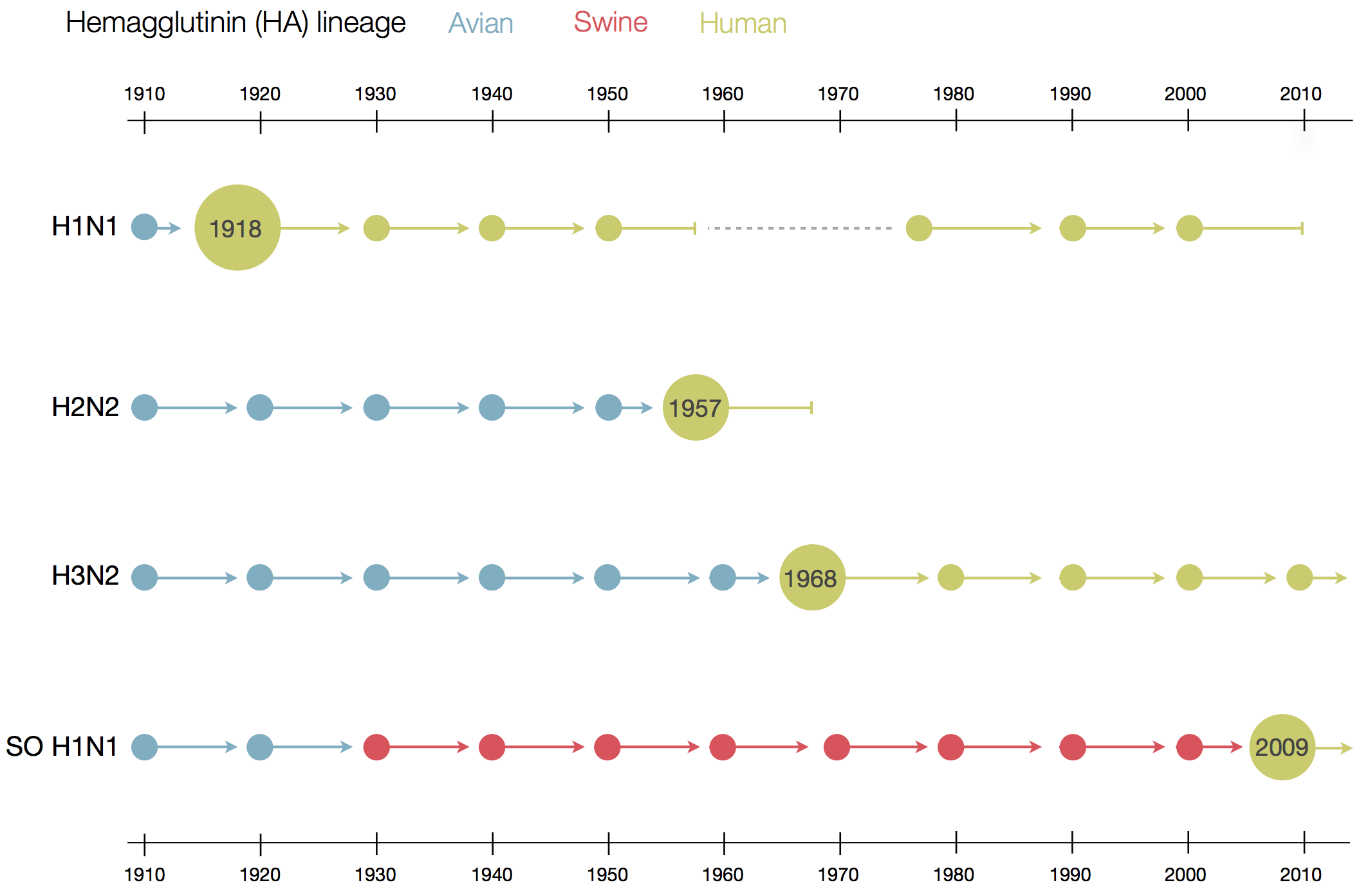
Flu pandemics caused by host switch events
Influenza B does not have pandemic potential
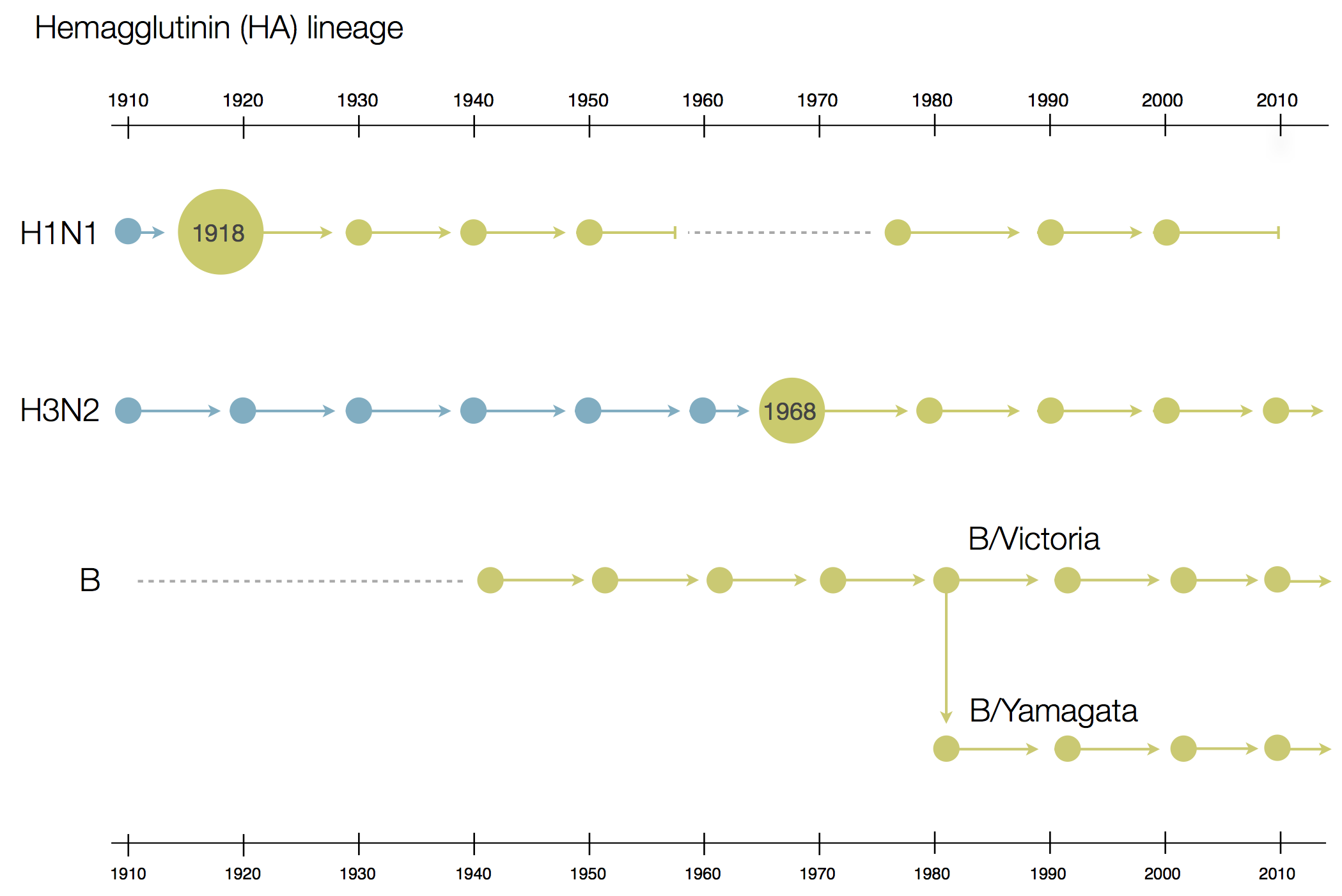
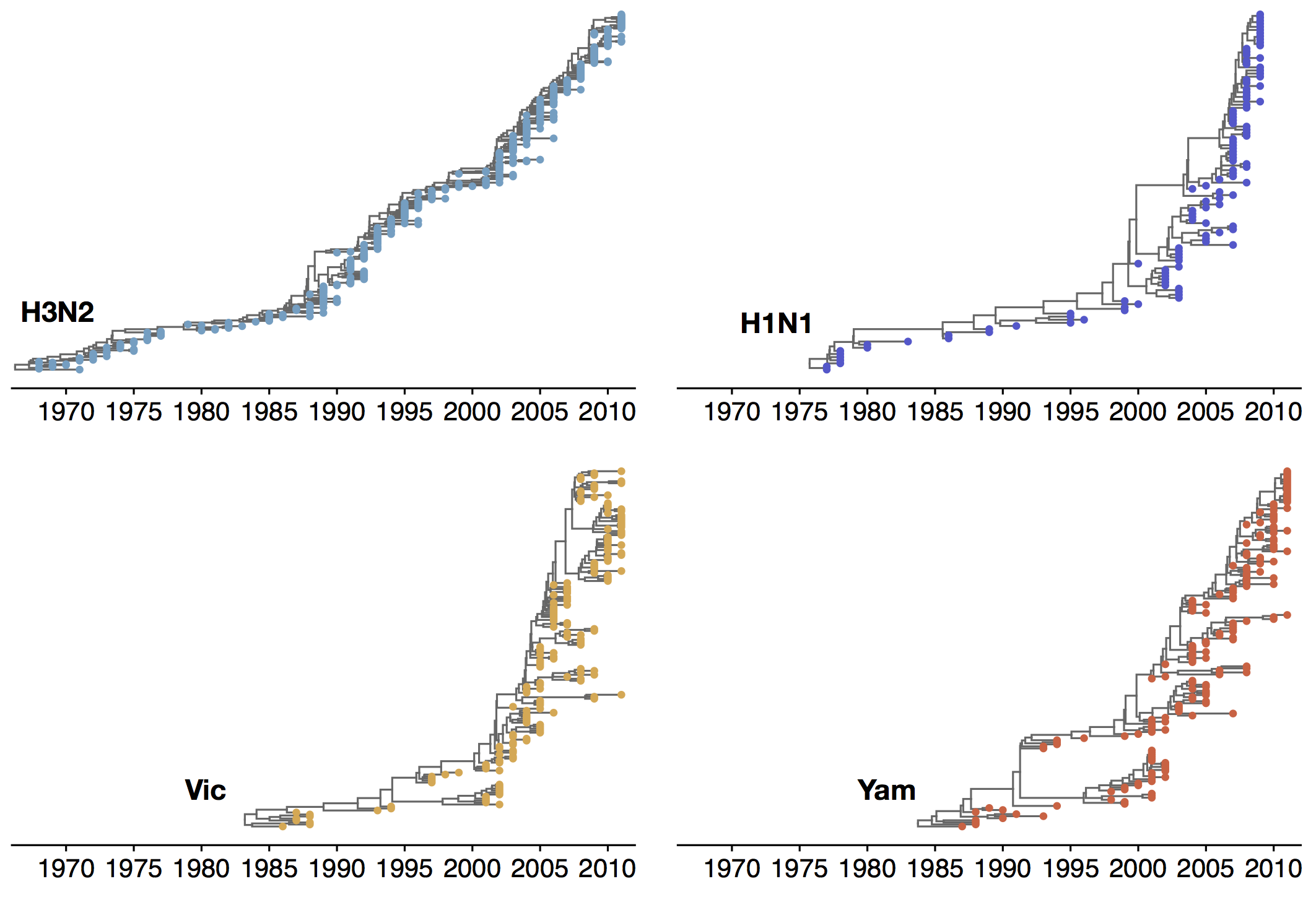
Phylogenetic trees of different influenza lineages
Antigenic drift
with Andrew Rambaut, Marc Suchard and others
Bedford et al 2014. Integrating influenza antigenic dynamics
with molecular evolution. eLife.
Influenza hemagglutination inhibition (HI) assay
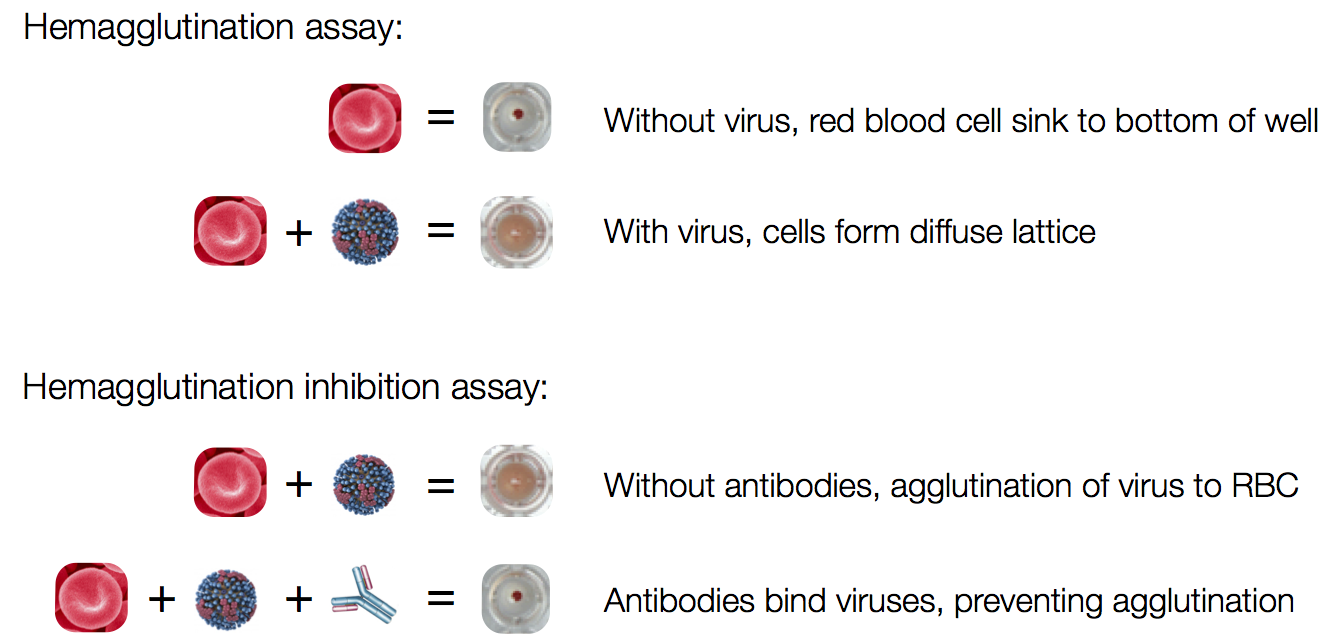
HI measures cross-reactivity across viruses
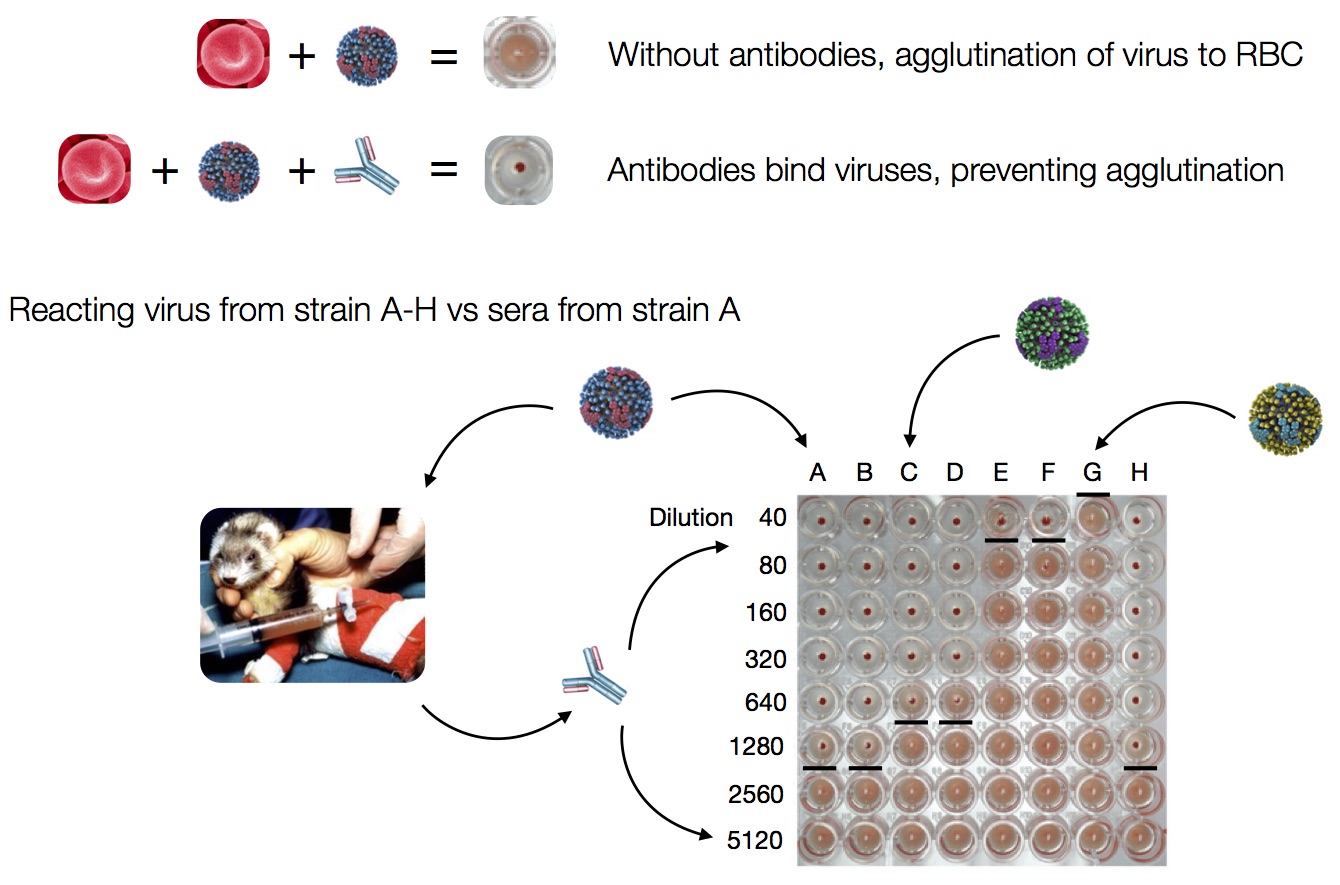
Data in the form of table of maximum inhibitory titers
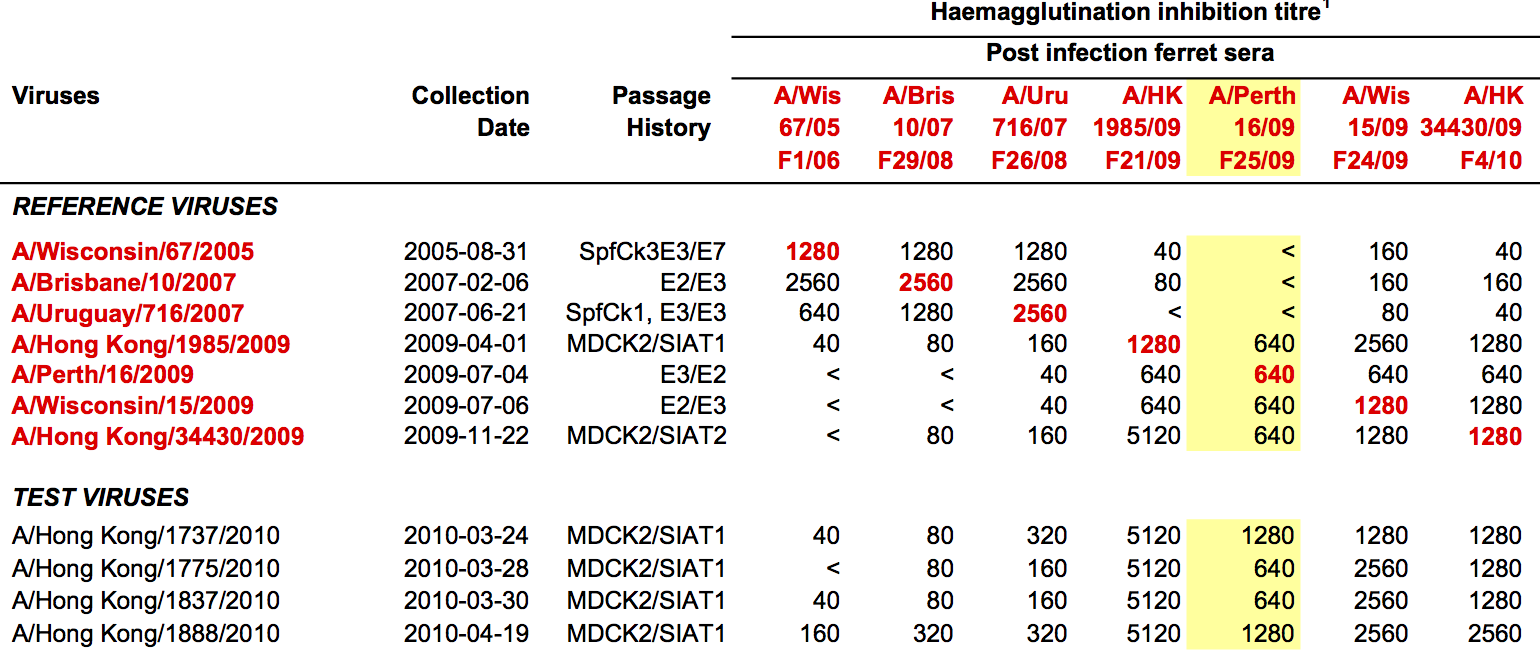
Compiled HI data difficult to work with
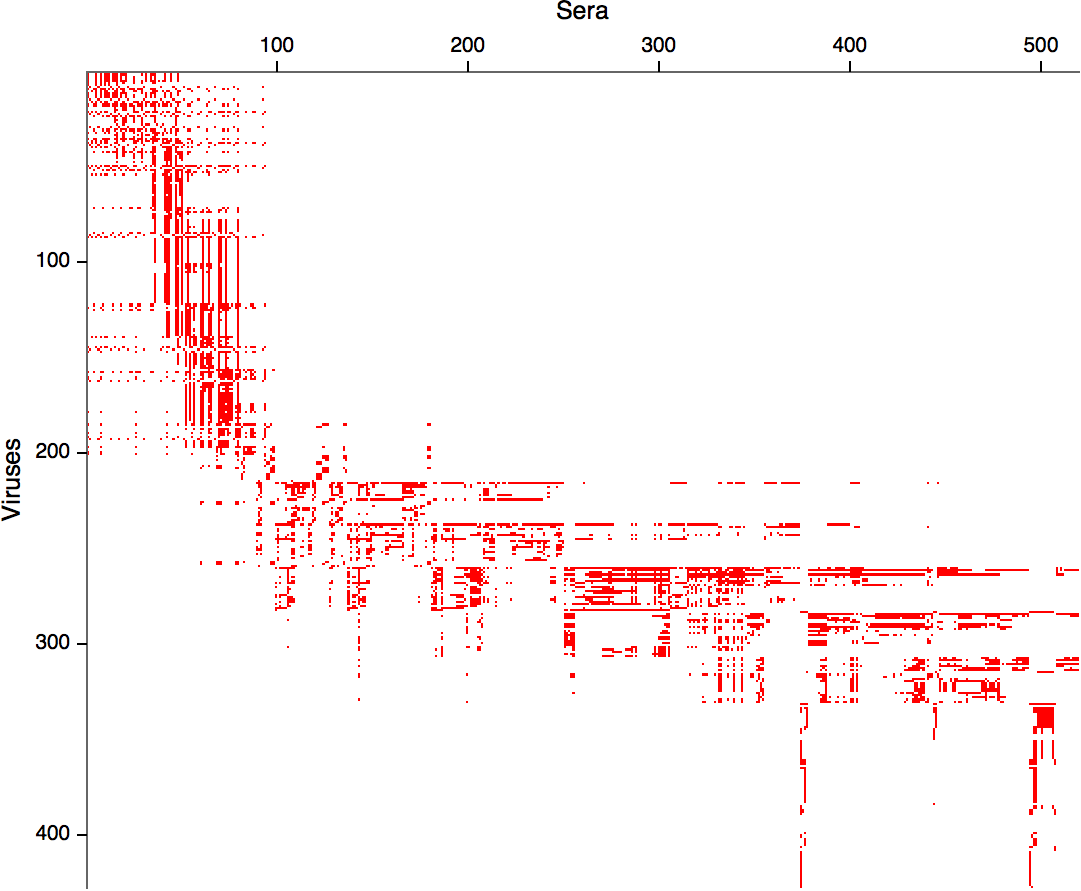
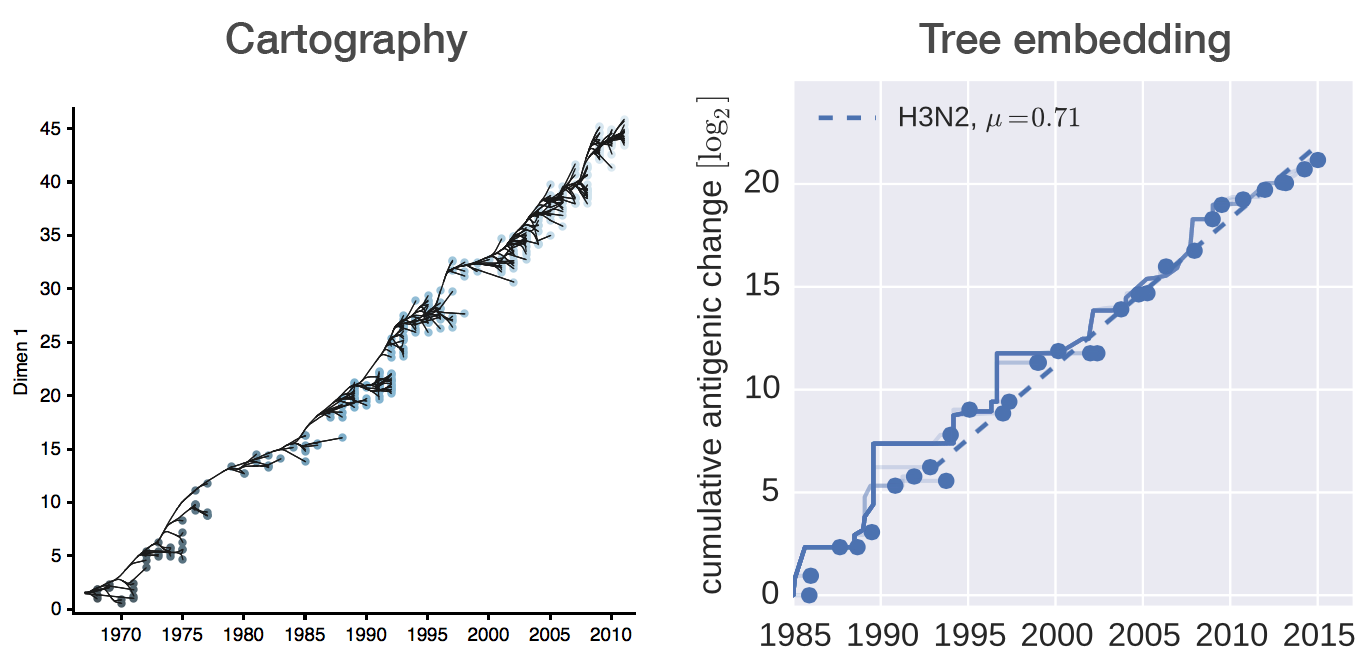
Antigenic cartography positions viruses and sera to recapitulate titer values
Antigenic cartography positions viruses and sera to recapitulate titer values
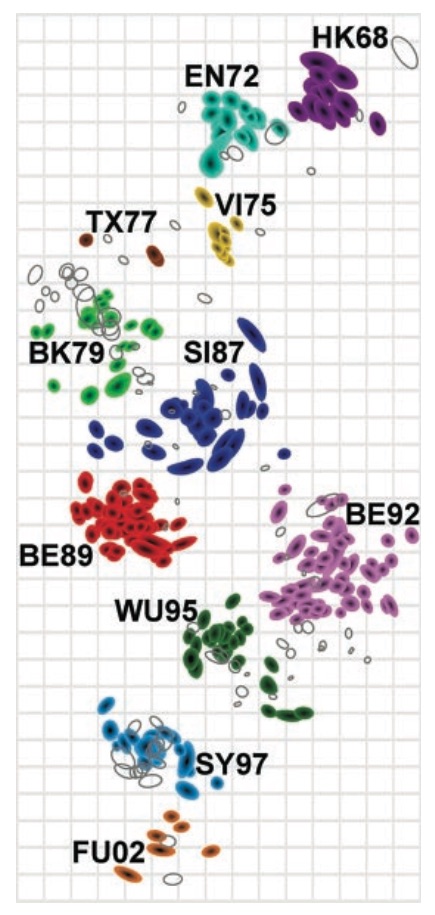
Combine phylogeny and HI data to estimate a joint antigenic map
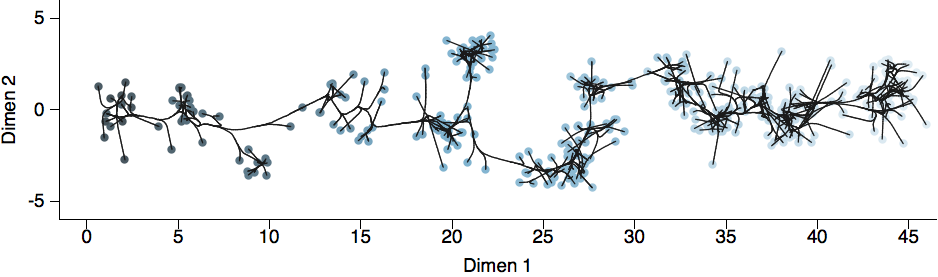
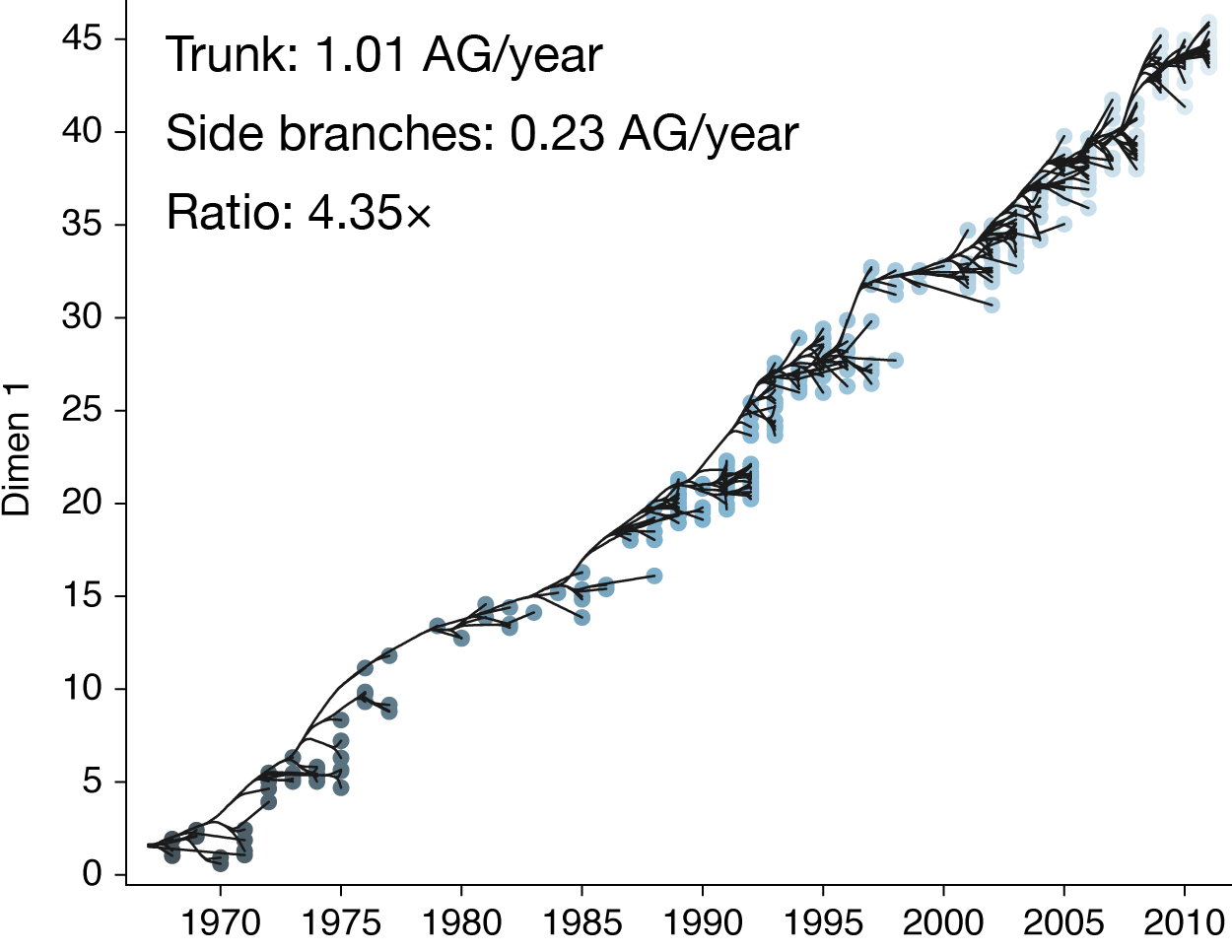
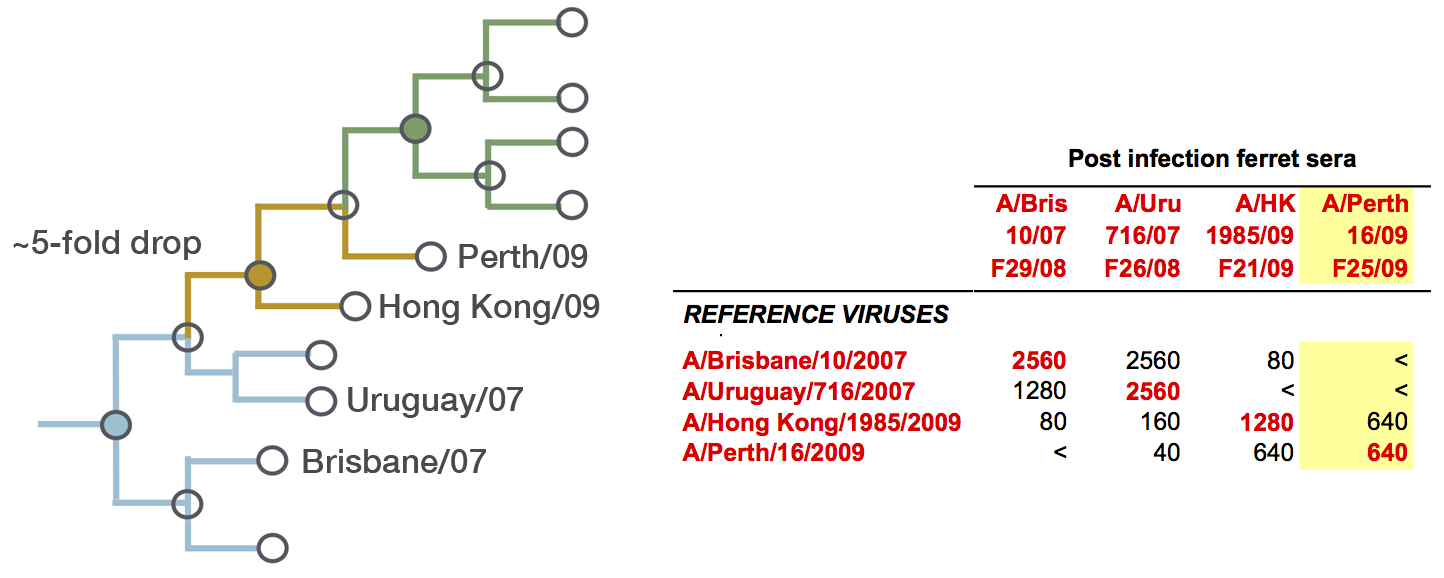
Drift results from selective advantage of antigenically novel lineages
Phylogenetic trees of different influenza lineages
Antigenic phenotype across lineages
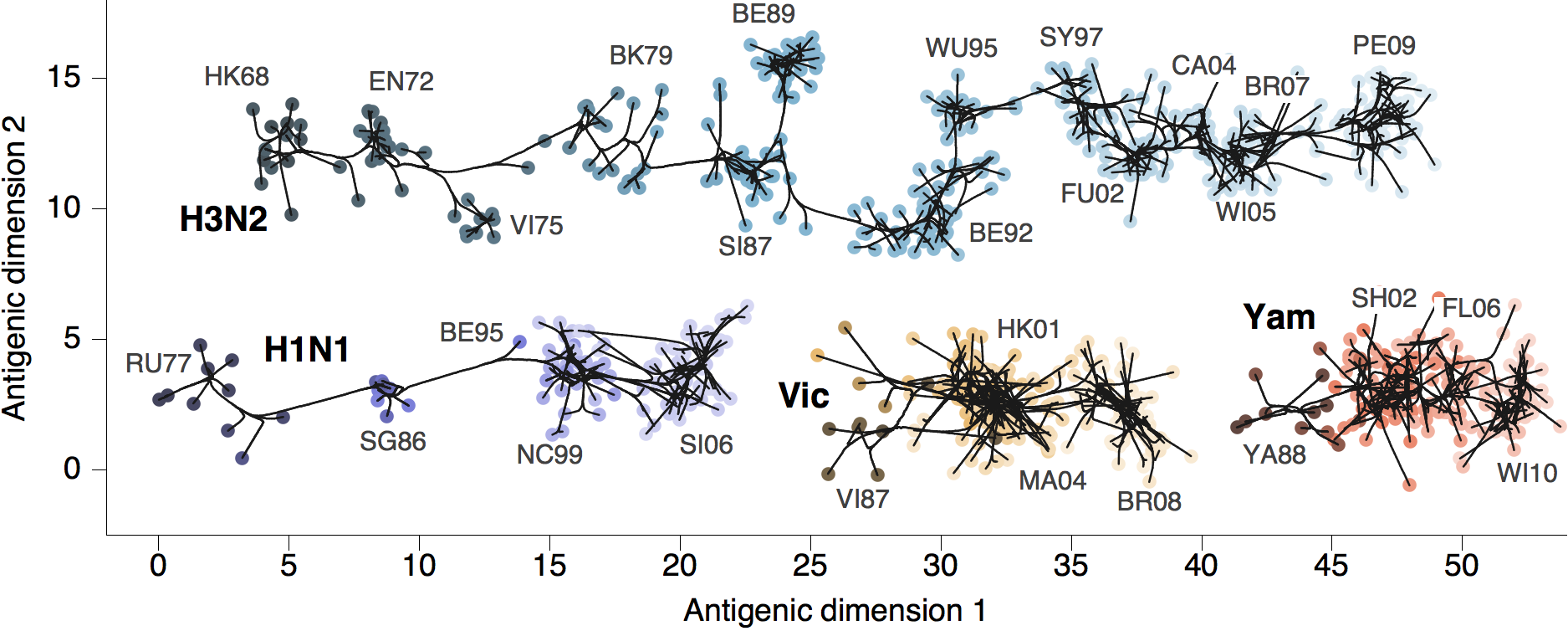
Antigenic drift across lineages
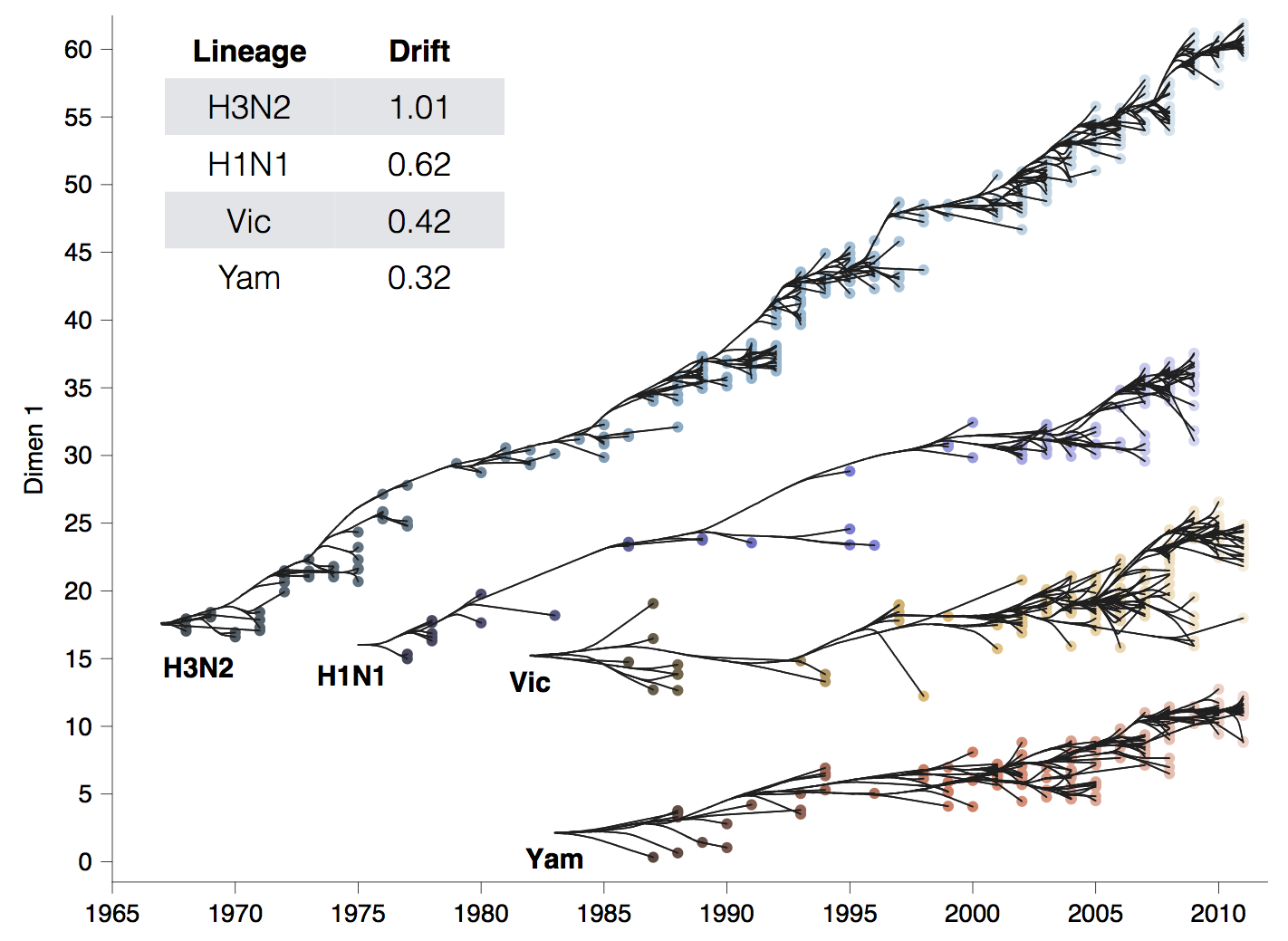
First study to embed a model of the process of antigenic evolution on a phylogeny. More than just description.
Geographic circulation
with Colin Russell, Philippe Lemey and many others
Seasonality in influenza
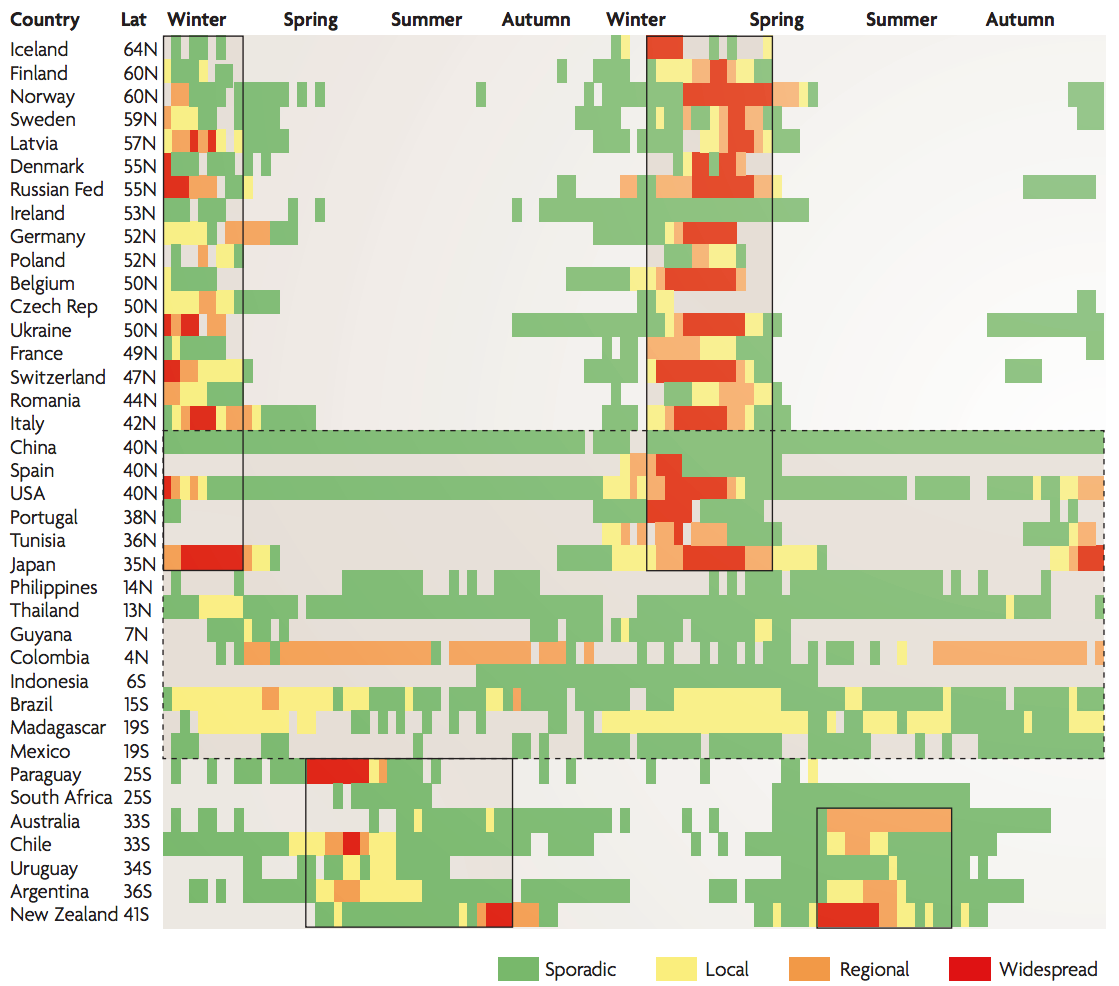
Sample H3N2 from around the world
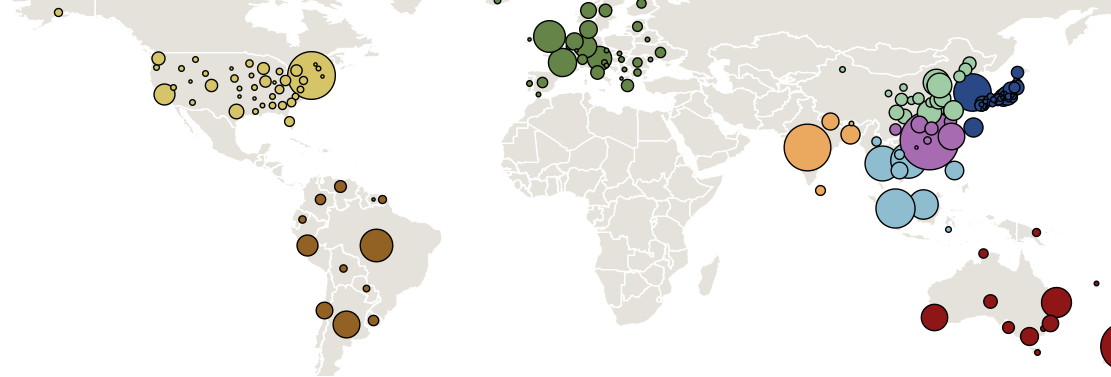
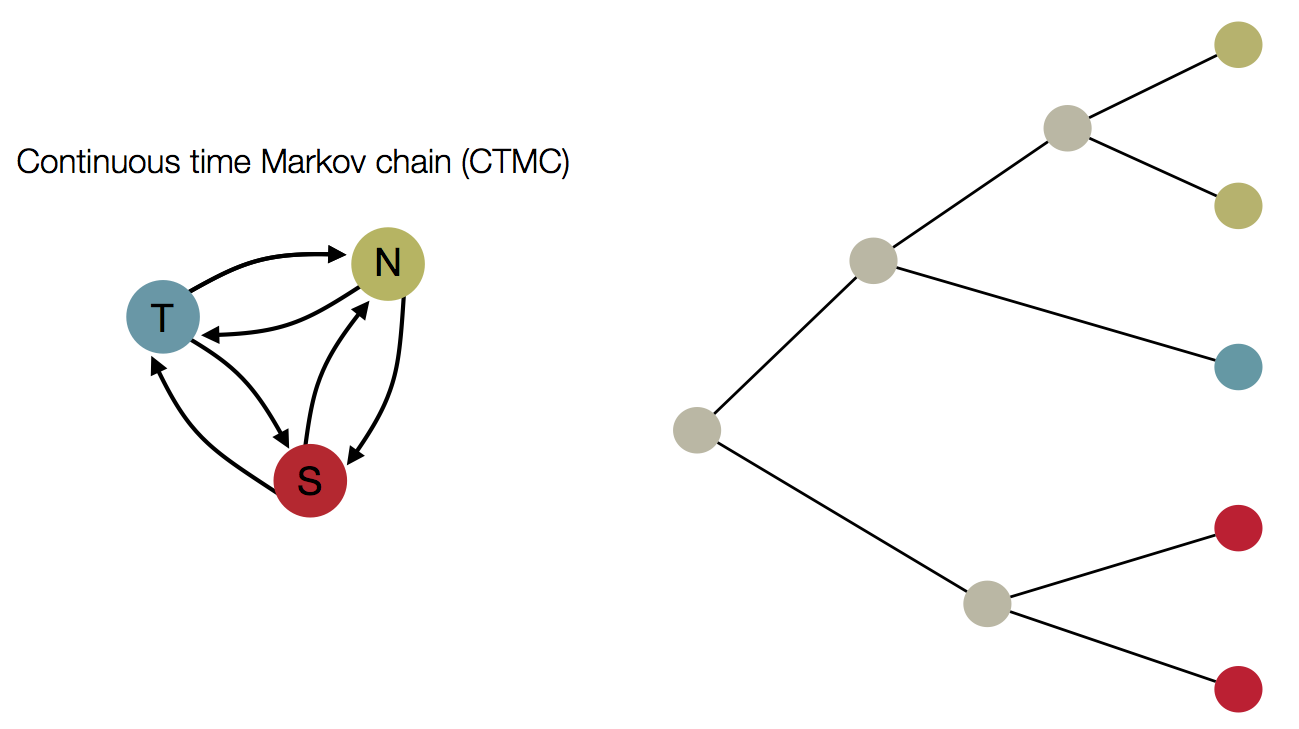
Treating geographic state as an evolving character
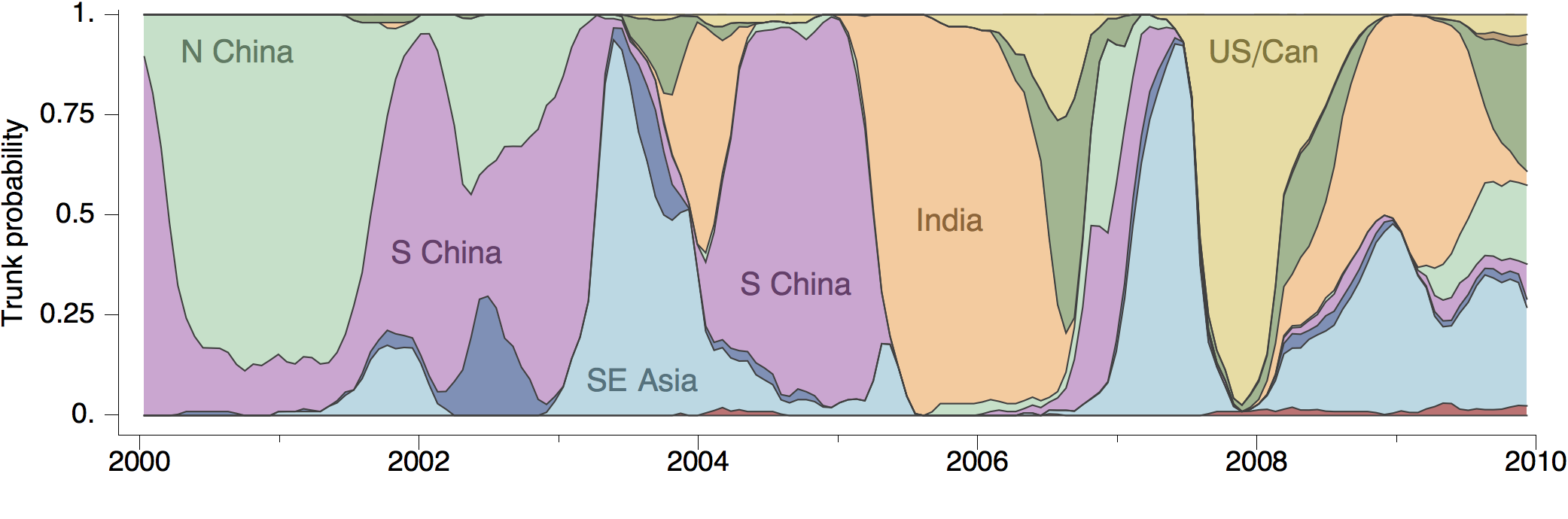
Phylogeny of H3 with geographic history
Infer geographic transition matrix
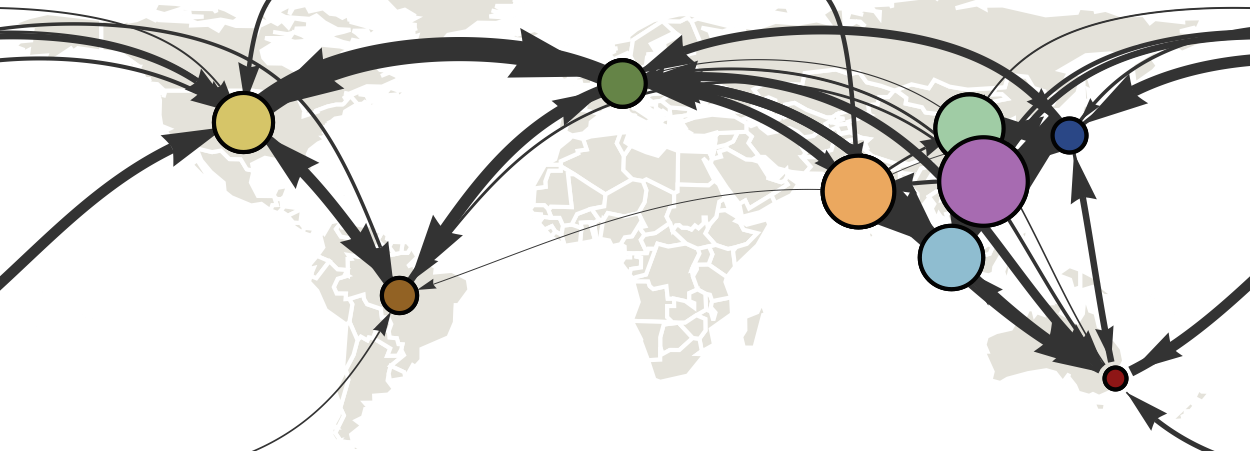
Air travel predicts migration rates
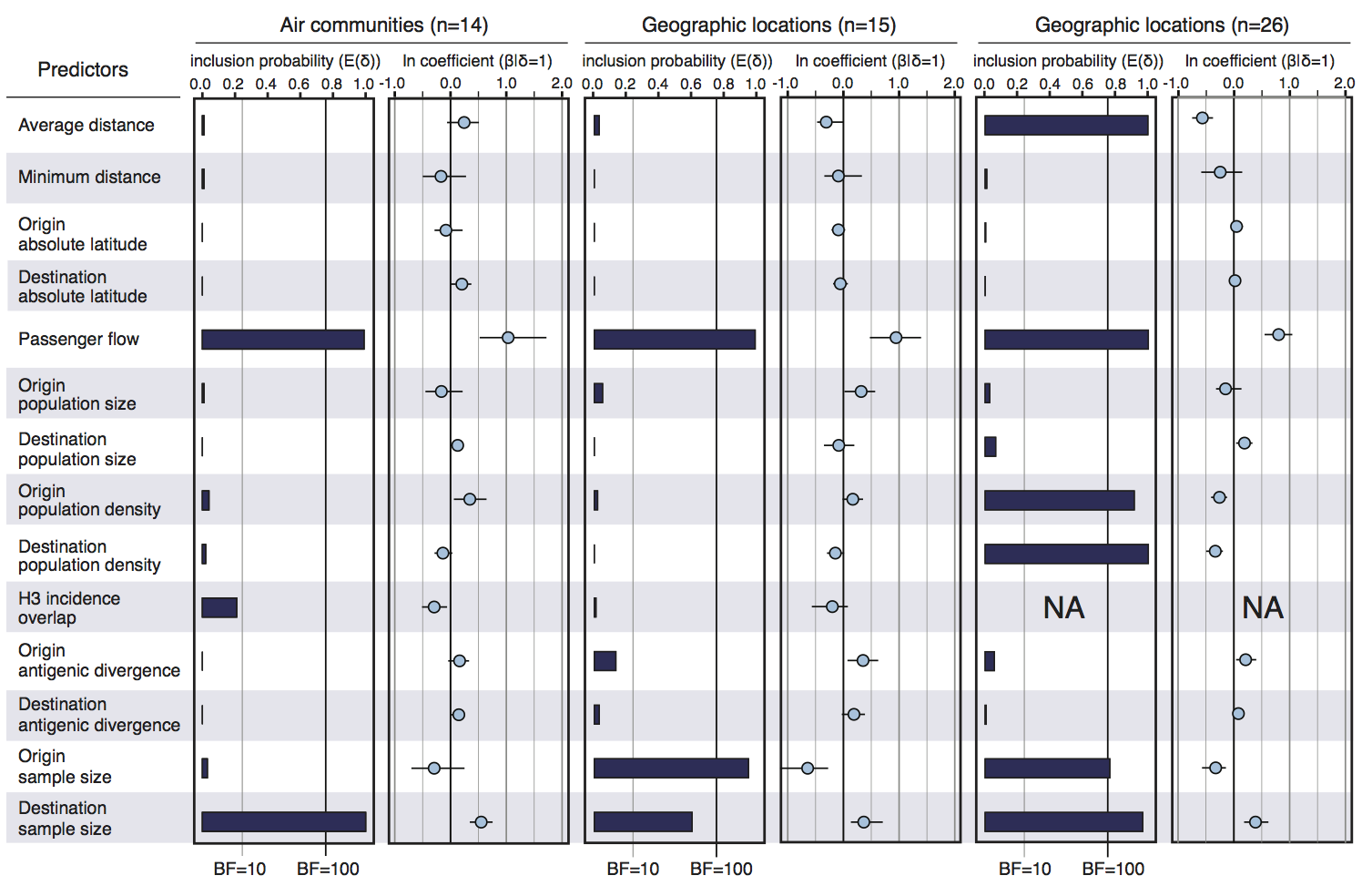
Geographic location of phylogeny trunk
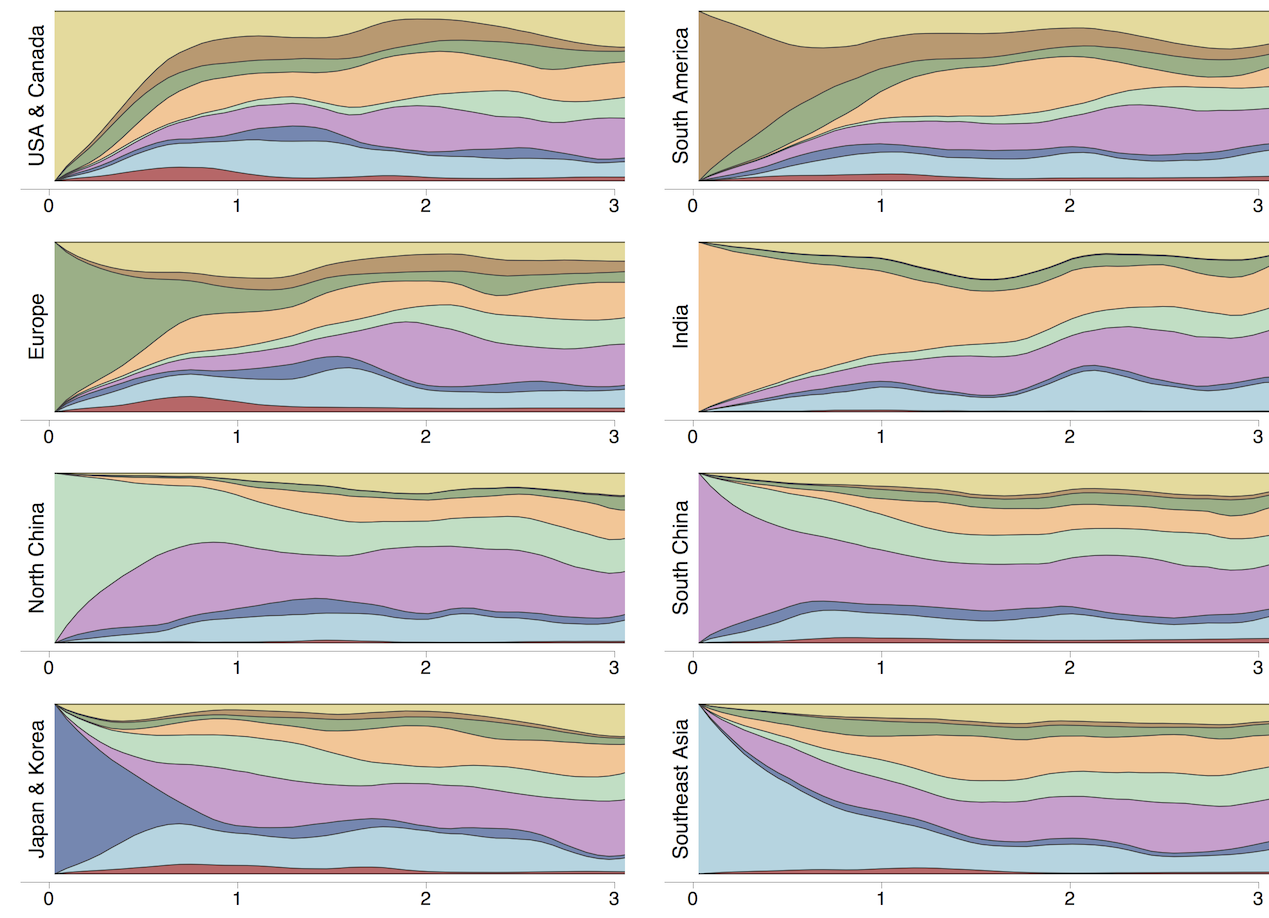
Region-specific ancestry
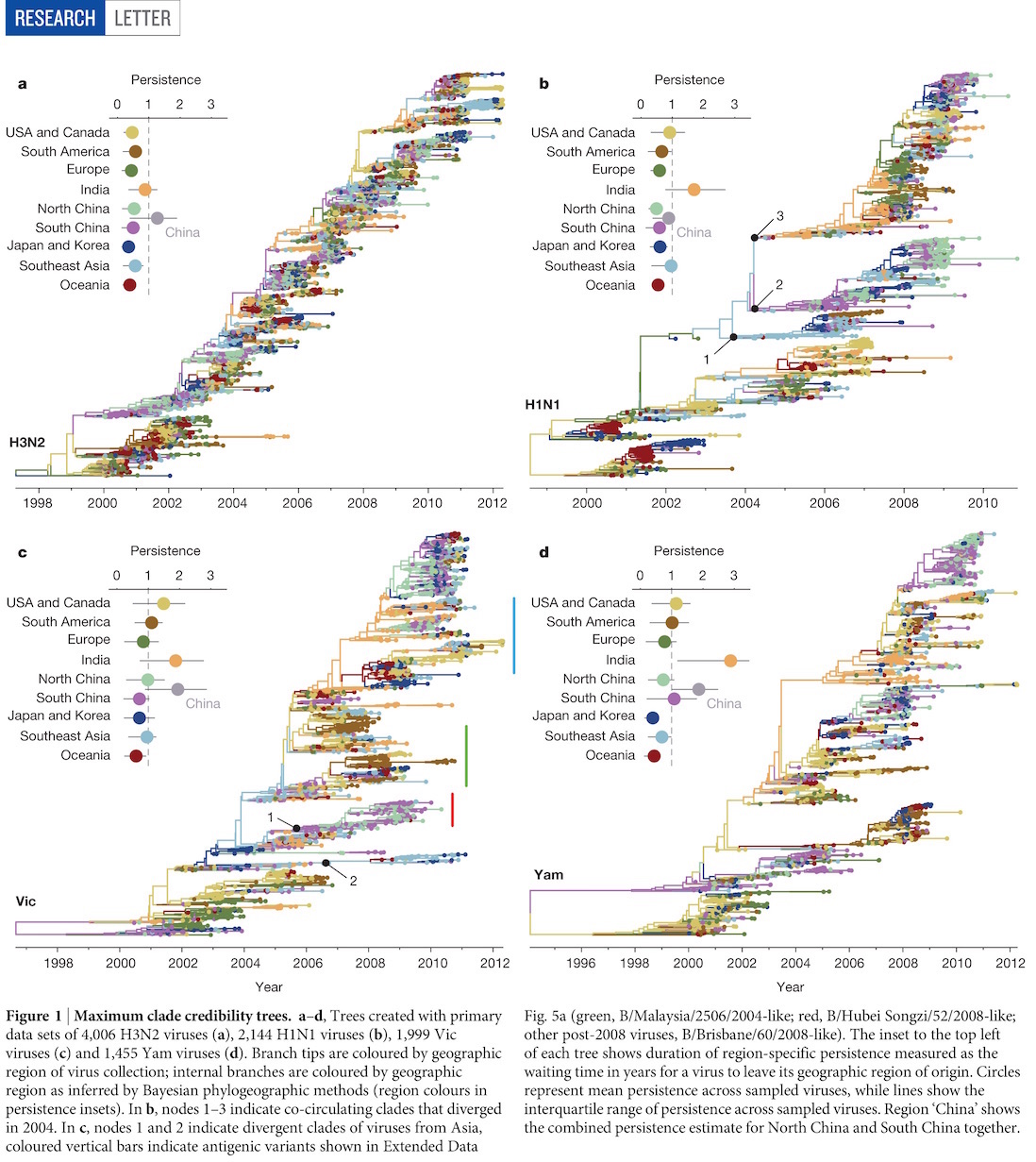
Phylogenies across subtypes / lineages
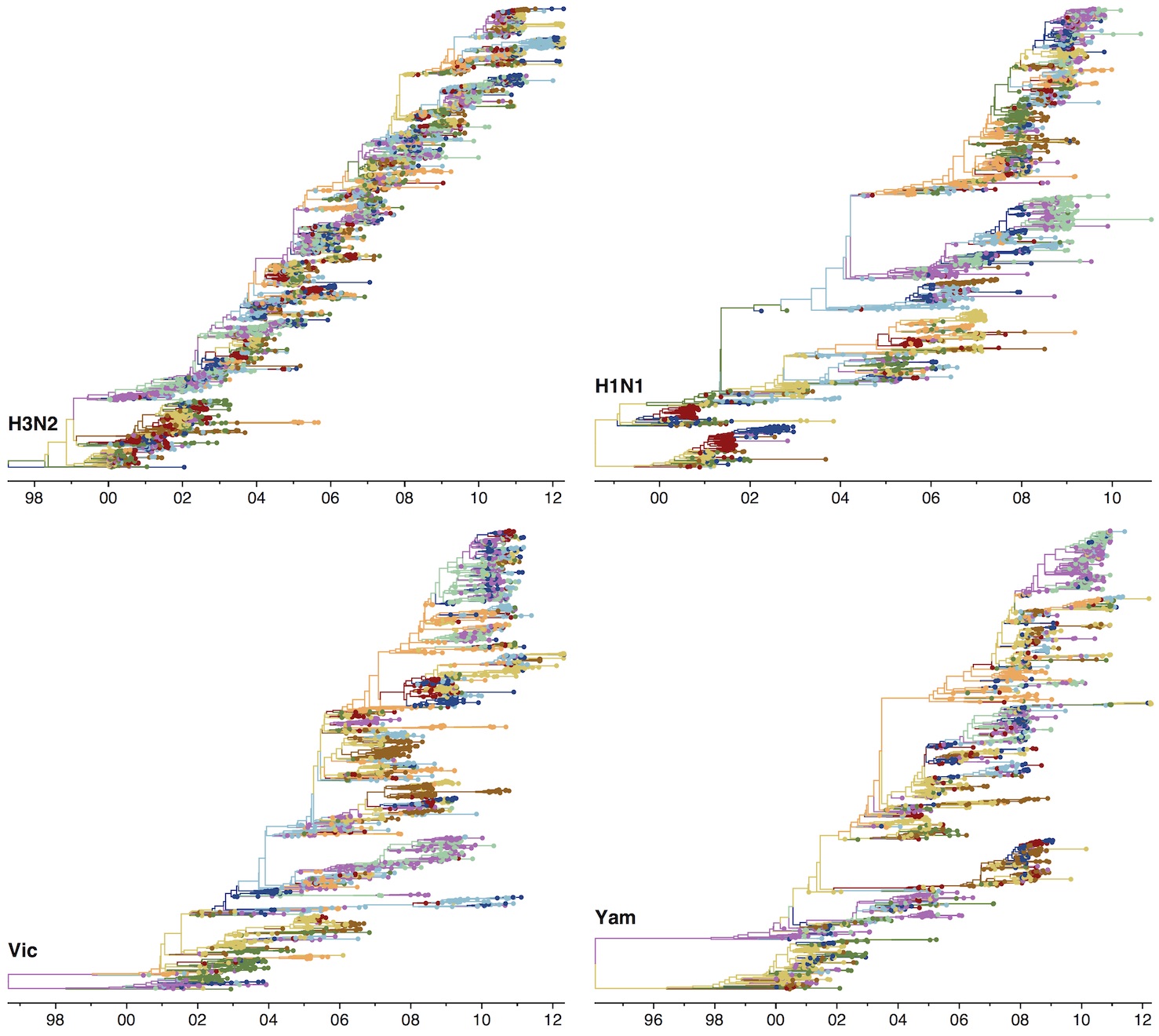
H3N2 phylogeny
H1N1 phylogeny
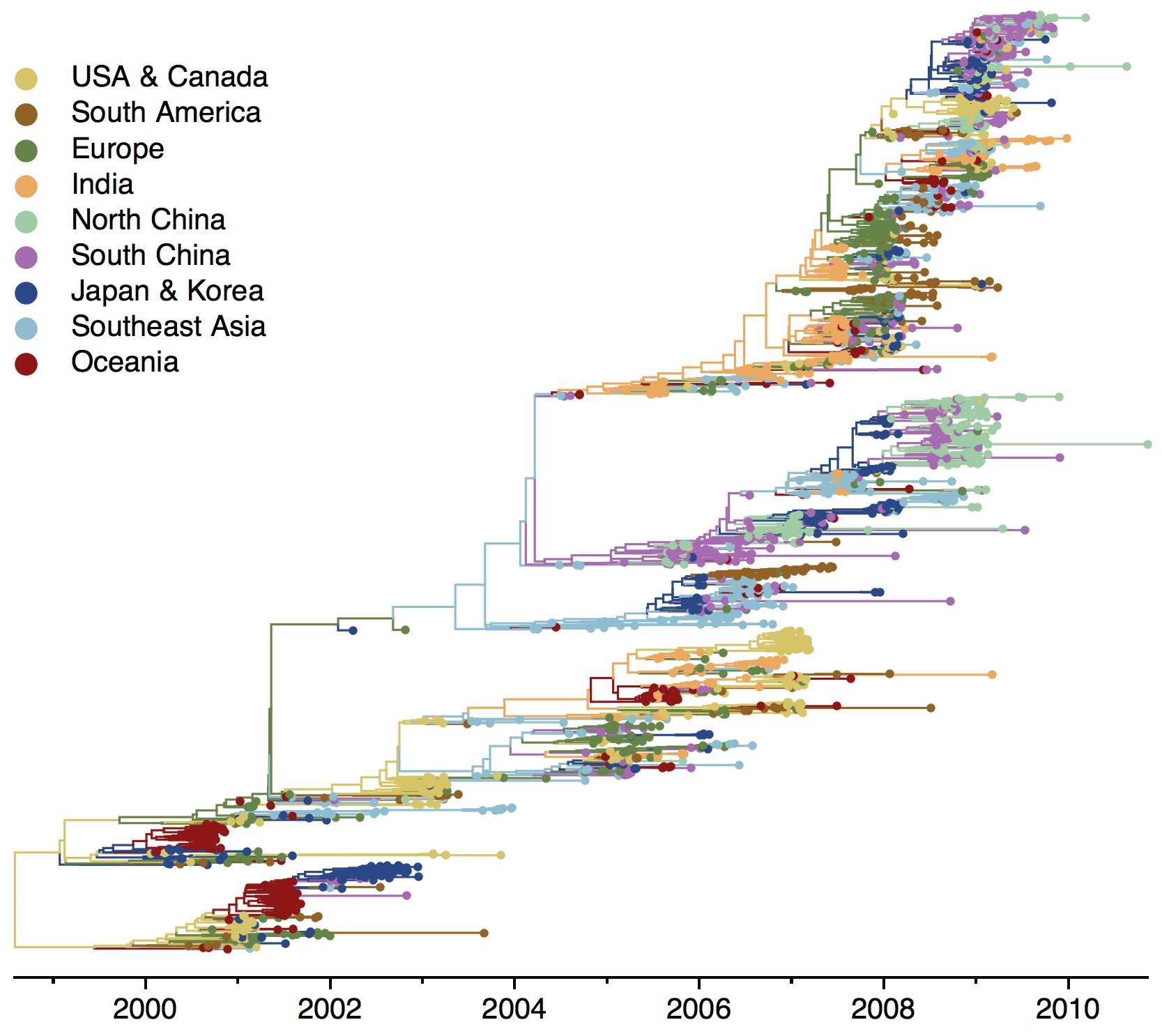
B/Vic phylogeny
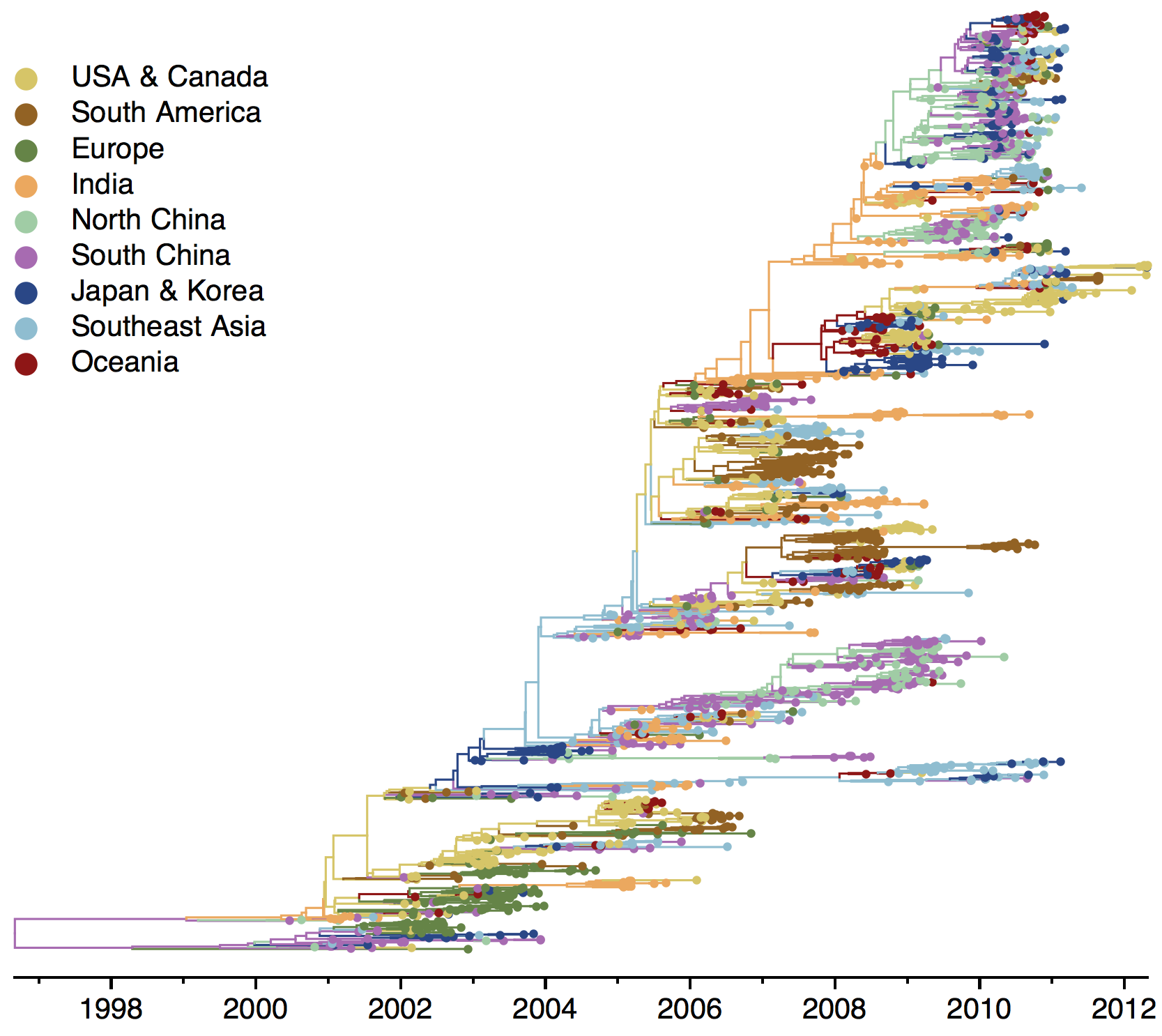
B/Yam phylogeny
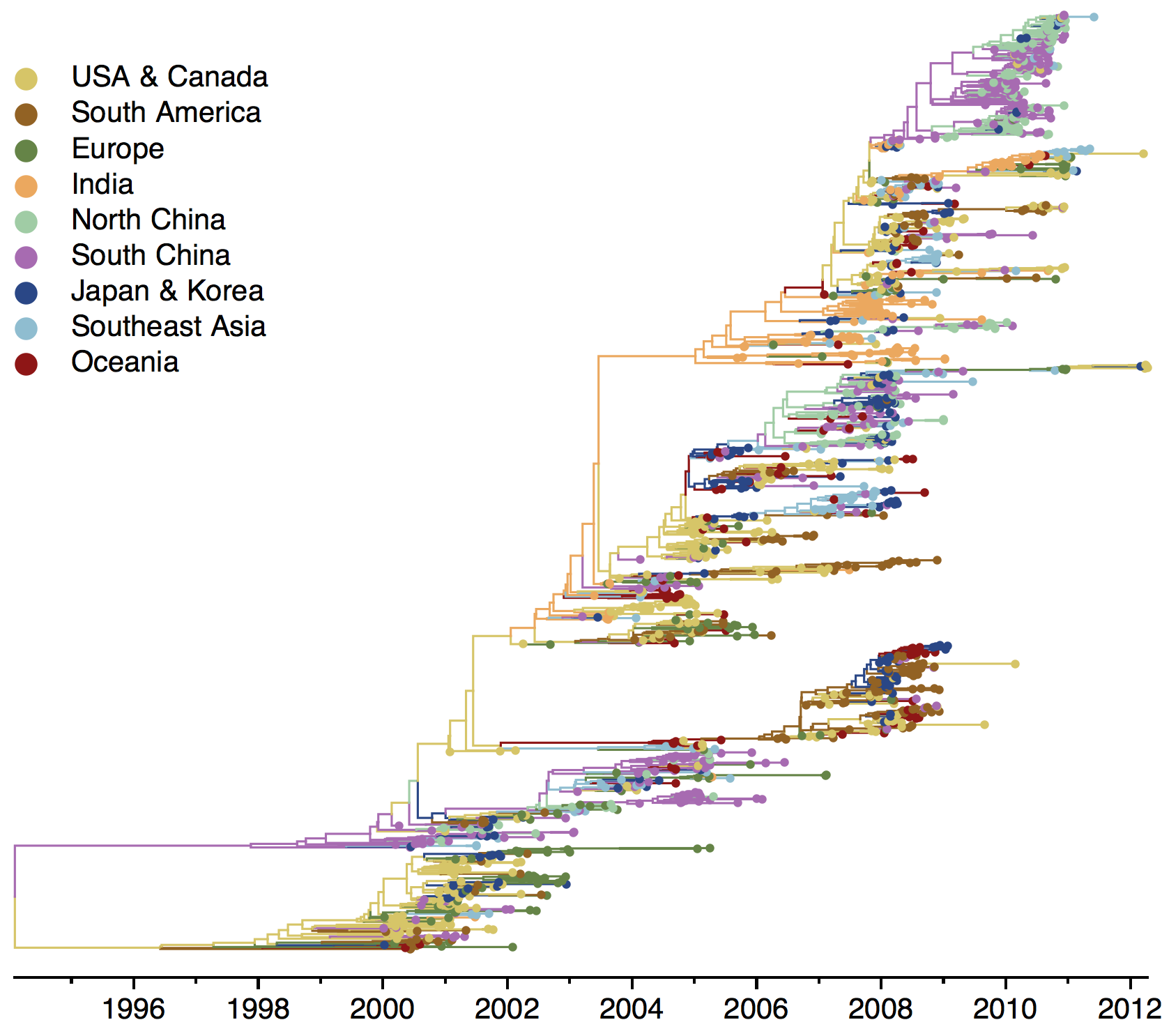
Ancestry patterns across lineages
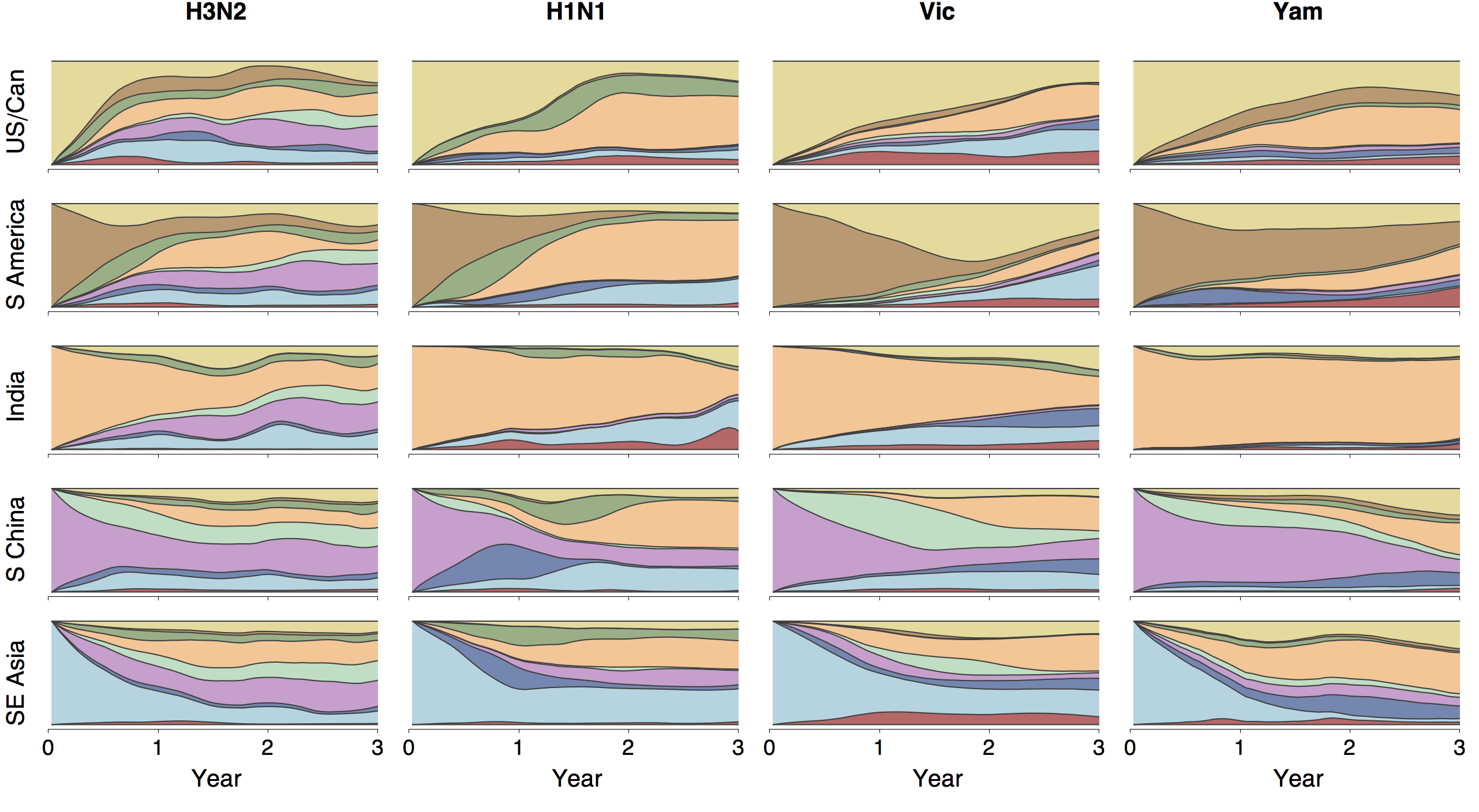
Regional persistence patterns
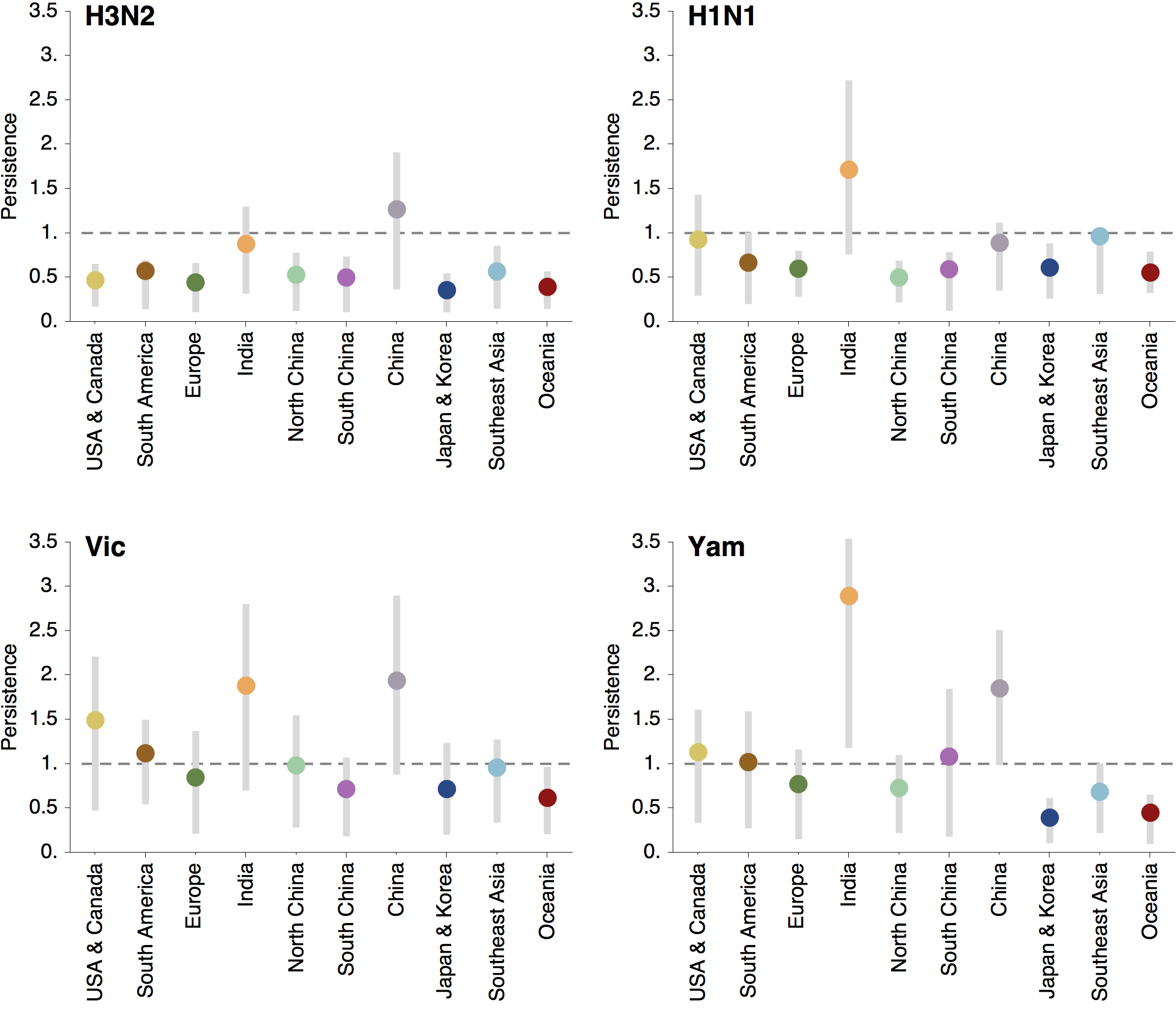
How to explain these differences?
Age distribution across viruses
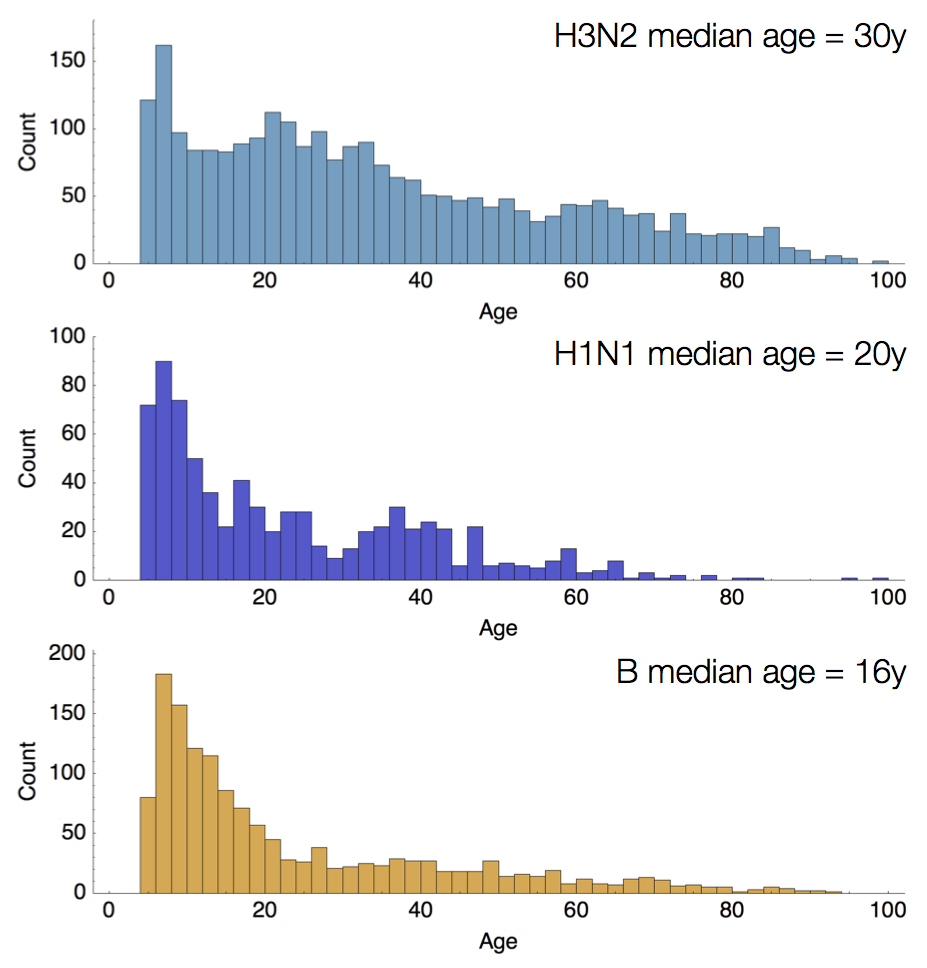
Air travel differences between adults and children
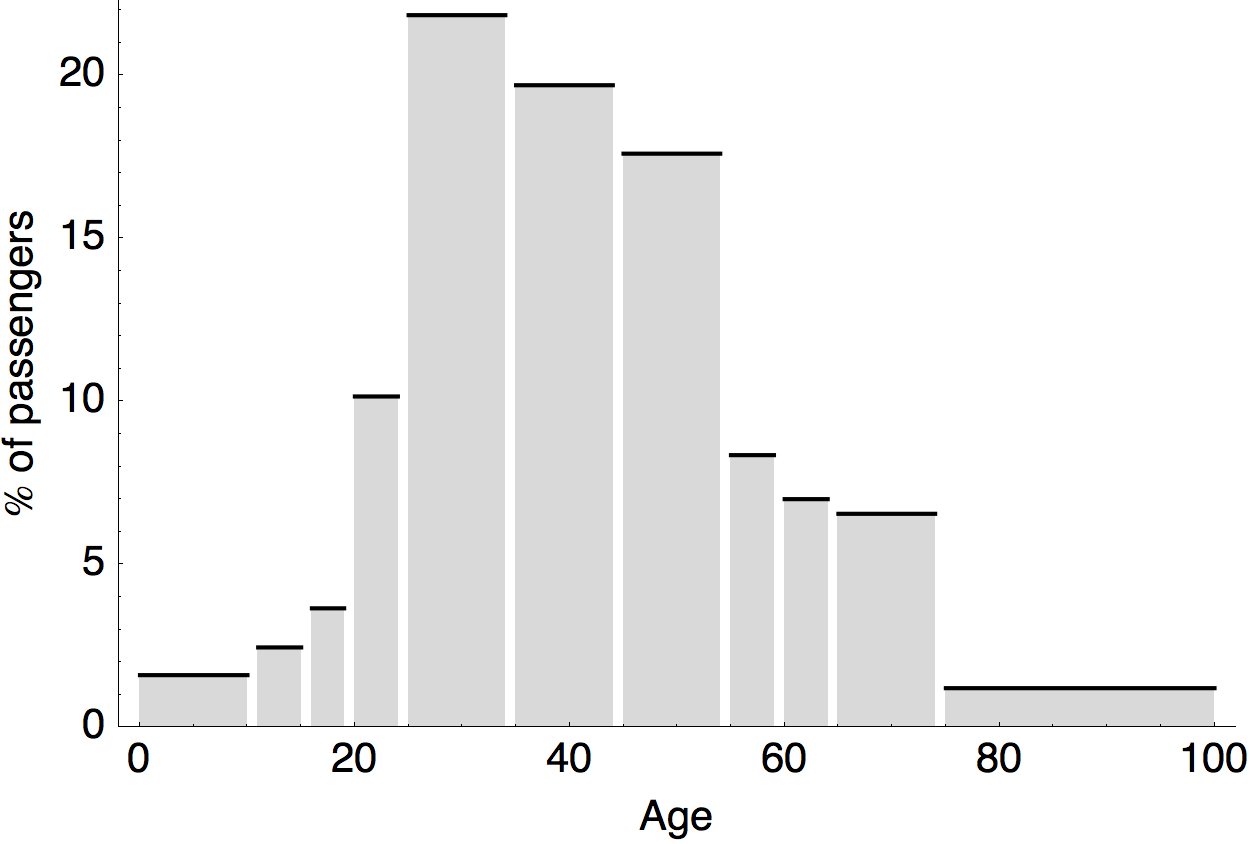
Epidemiological model of varying rates of antigenic drift
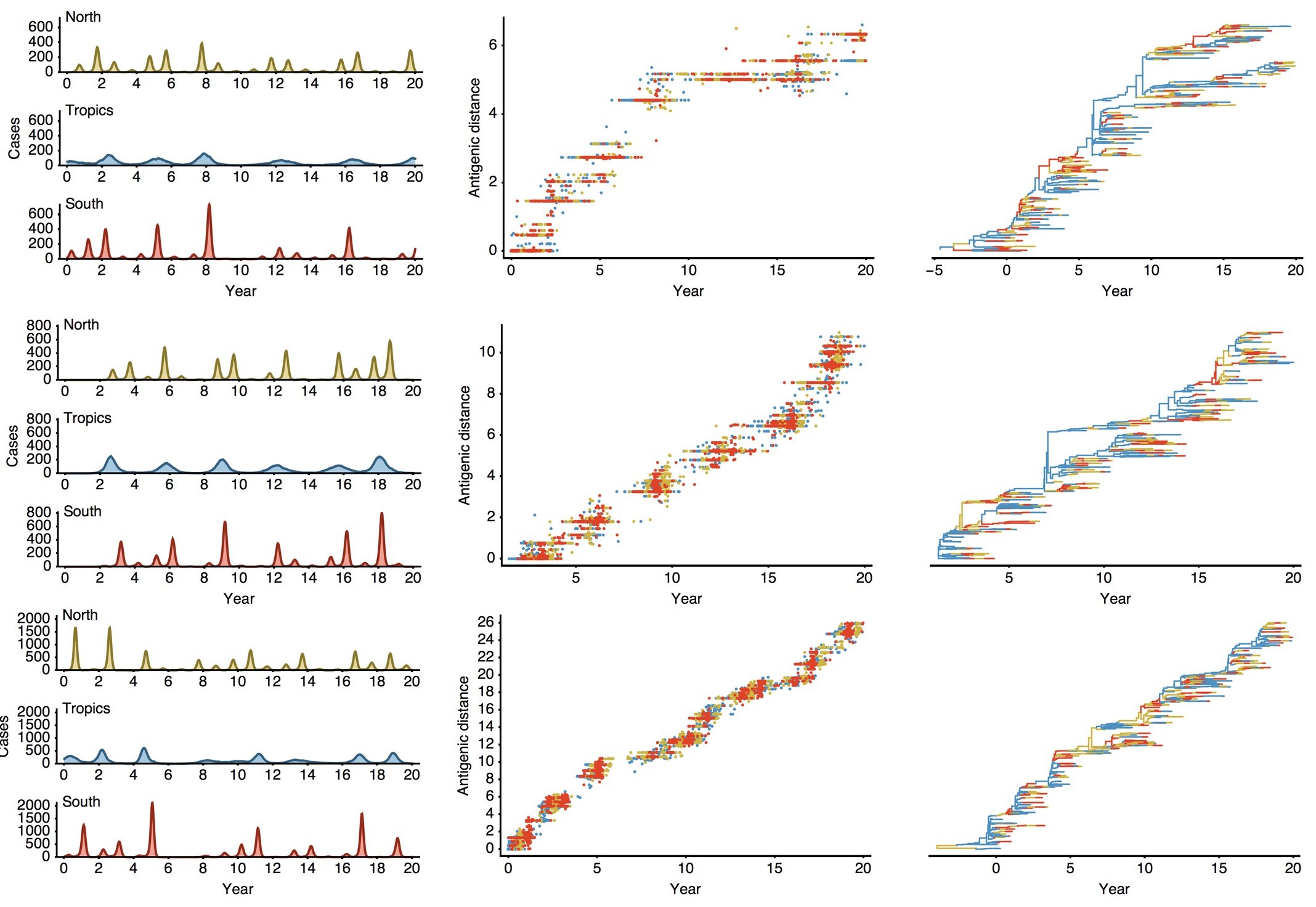
Results of varying antigenic drift
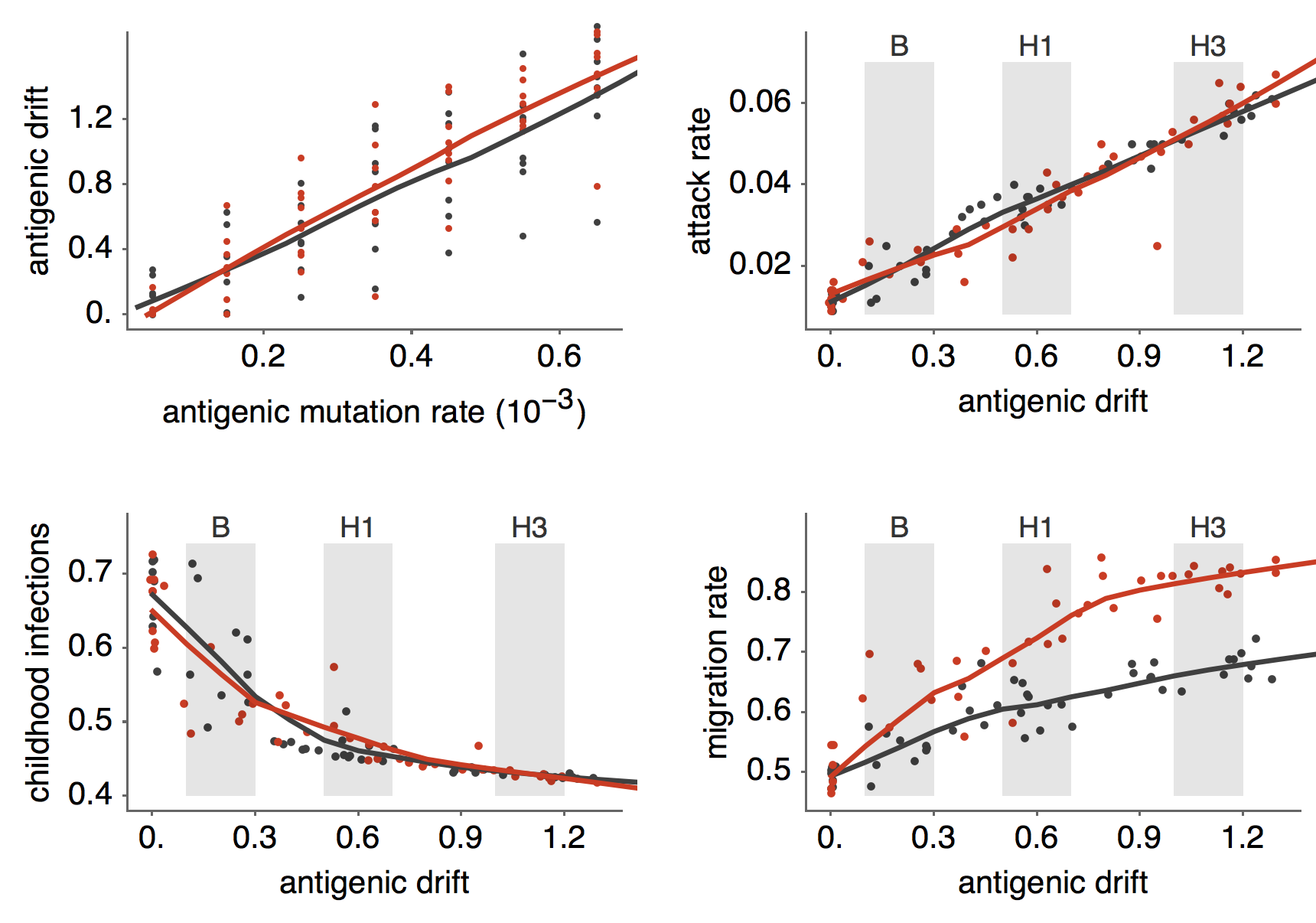
Interaction between virus evolution, epidemiology and human behavior drives migration rate differences
Static vs dynamic inferences and the living paper
Influenza H3N2 vaccine updates
nextflu
Project to provide a real-time view of the evolving influenza population
All in collaboration with Richard Neher

nextflu pipeline
- Download all recent HA sequences from GISAID
- Filter to remove outliers
- Subsample across time and space
- Align sequences
- Build tree
- Estimate frequencies
- Export for visualization
Up-to-date analysis publicly available at:
nextflu.org
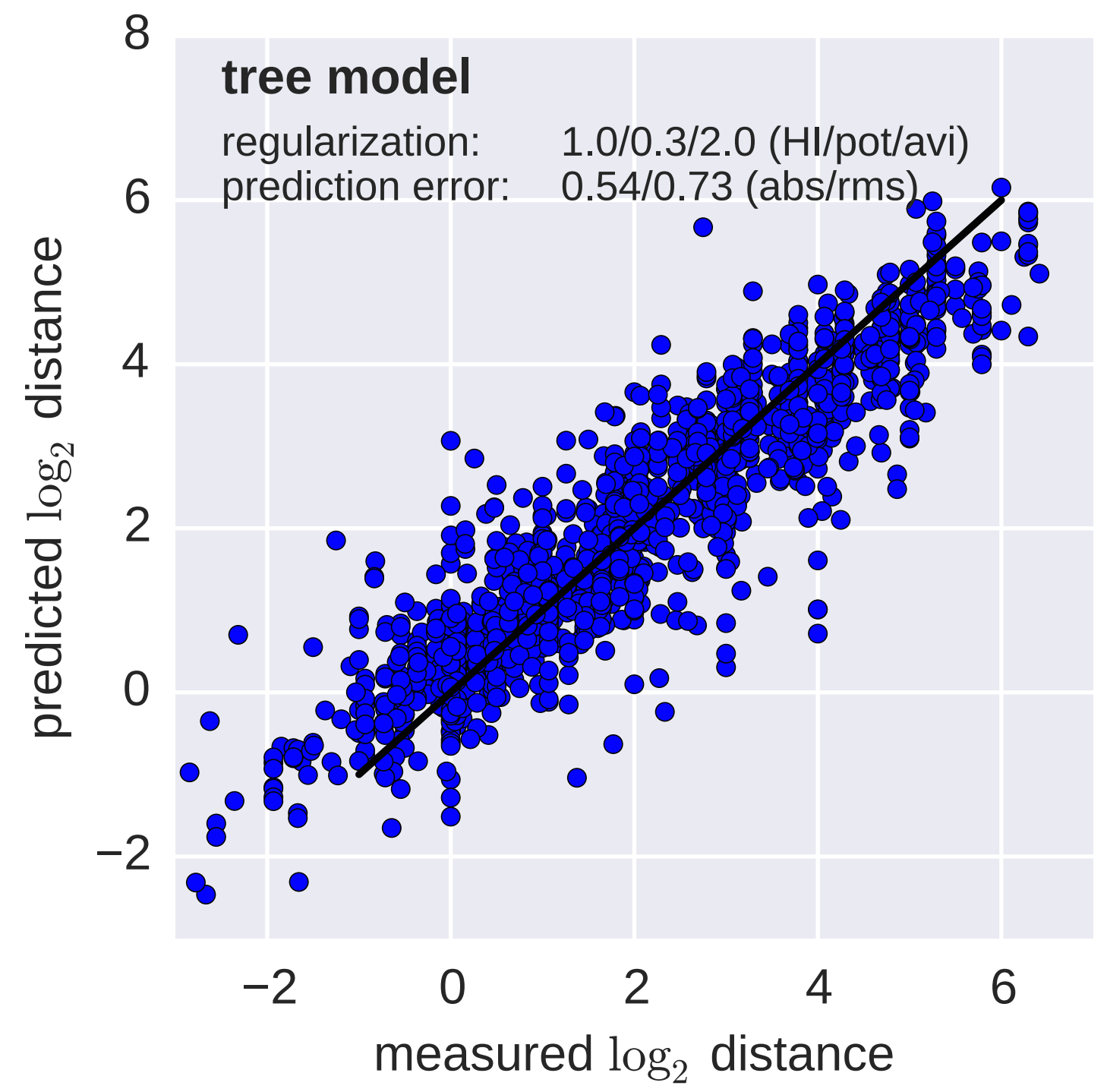
Including HI data, by titer drops to phylogeny branches
Model is highly predictive of missing titer values
Broad patterns agree with cartographic analyses
Recent HI data from WHO CC London annual and interim reports
Up-to-date analysis at:
nextflu.org
Forecasting
The future is here, it's just not evenly distributed yet
— William Gibson
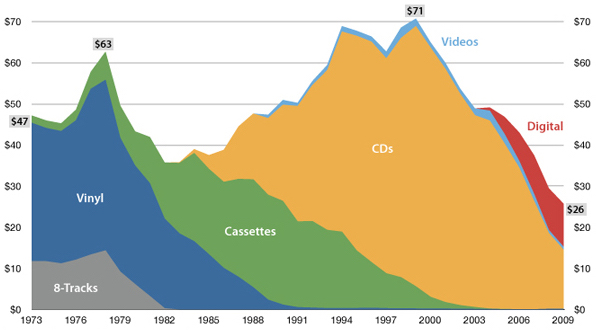
USA music industry, 2011 dollars per capita
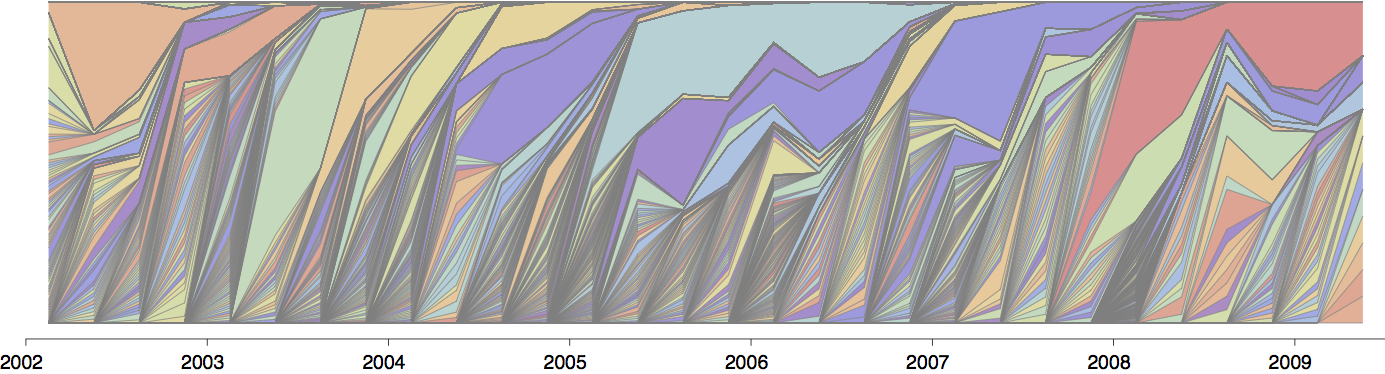
Influenza population turnover
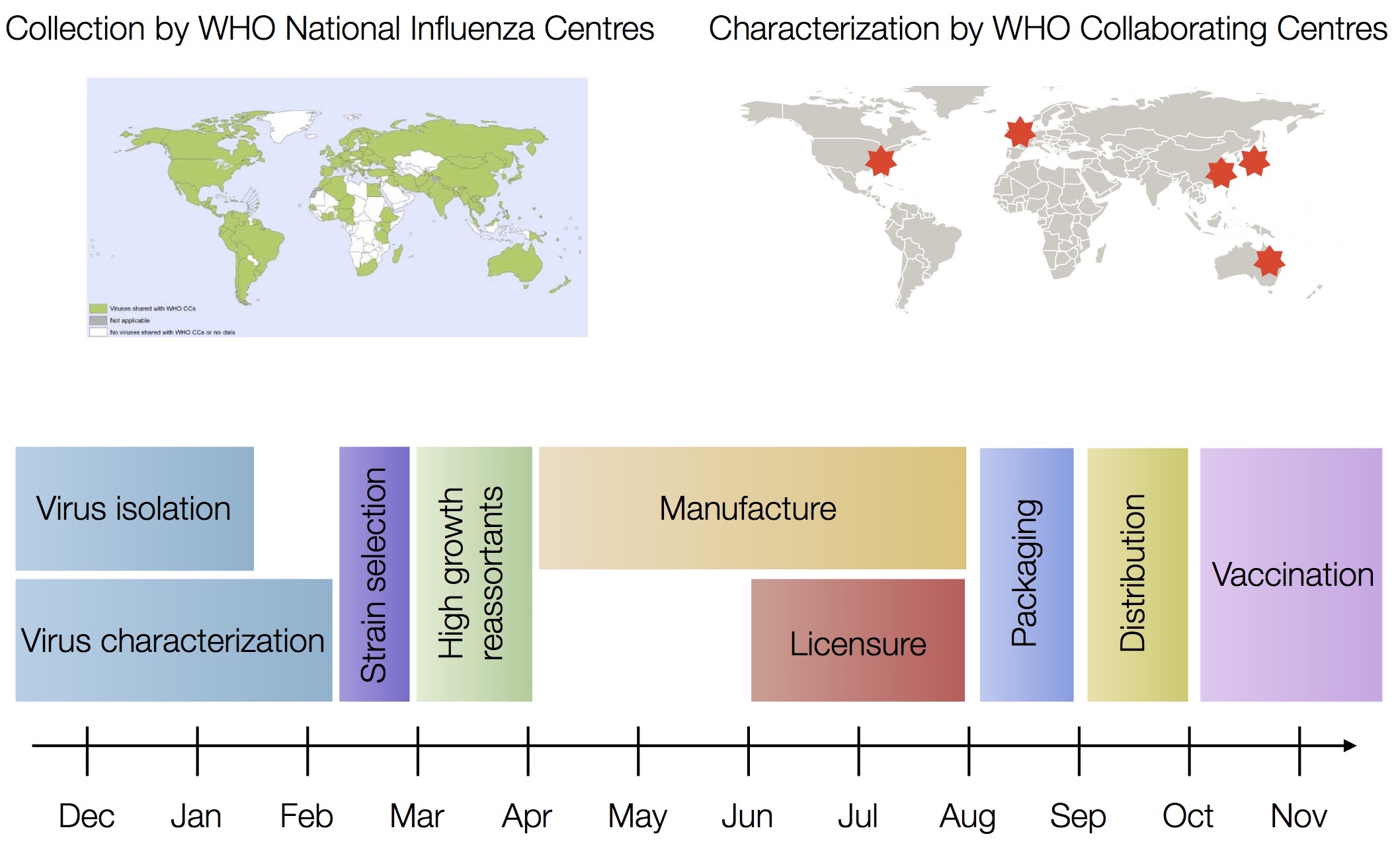
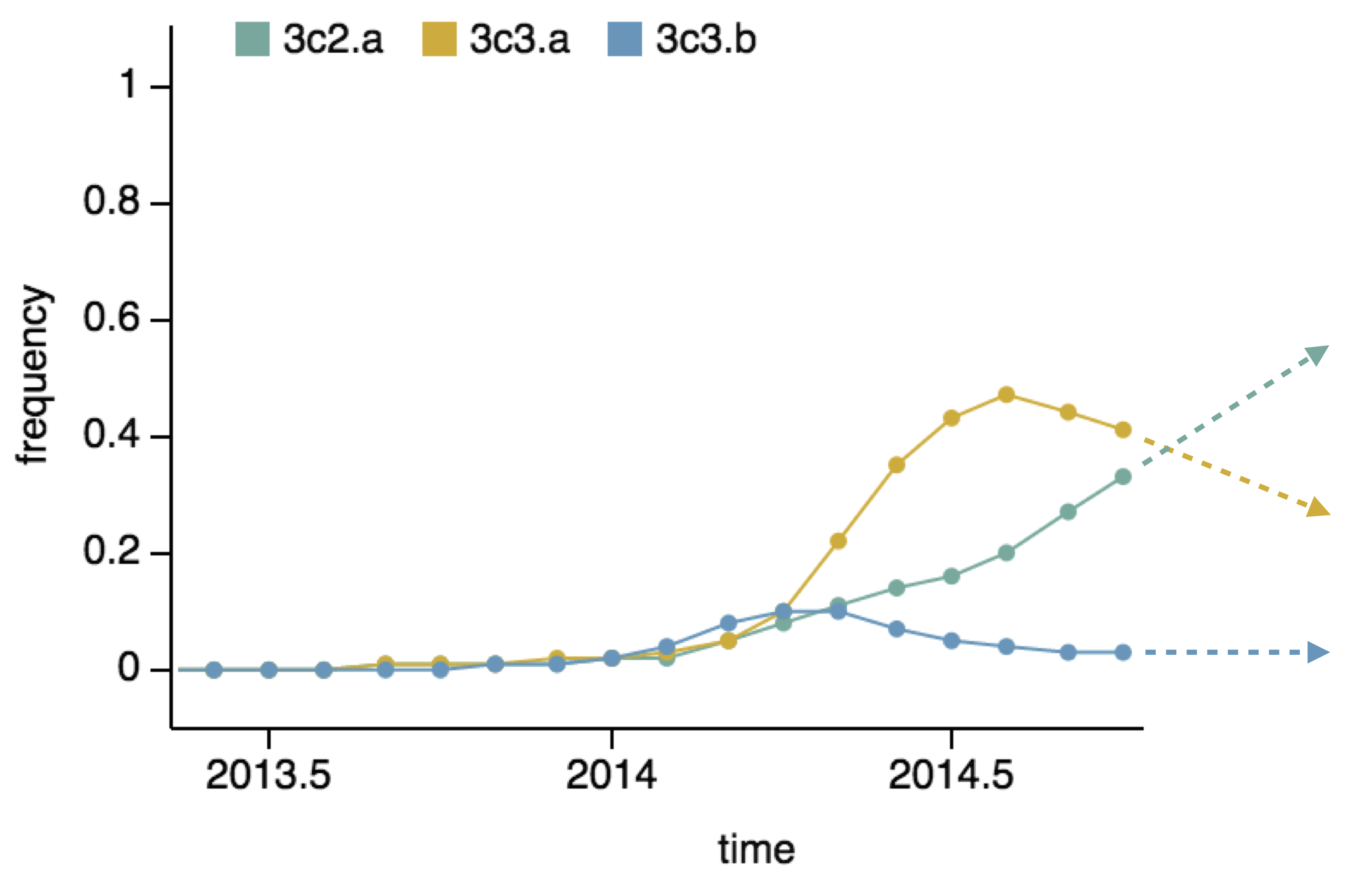
Vaccine strain selection timeline
Seek to explain change in clade frequencies over 1 year
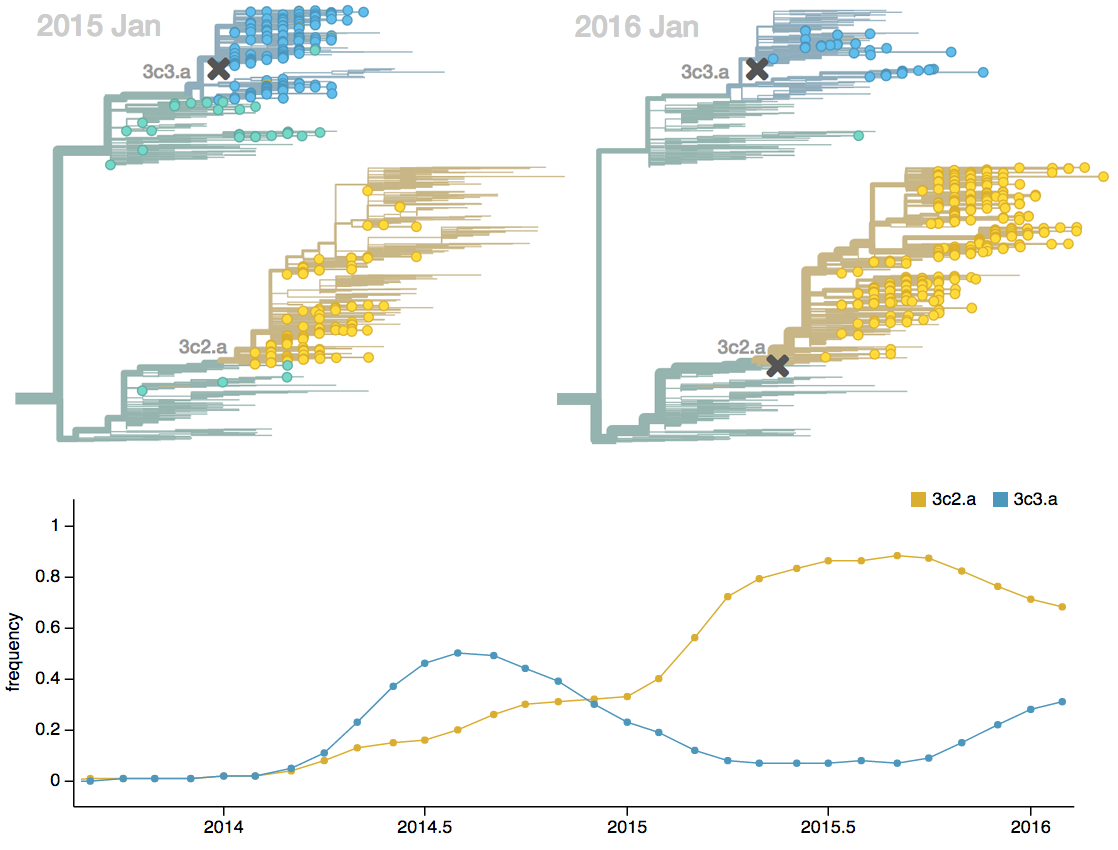
Fitness models can project clade frequencies
Clade frequencies $X$ derive from the fitnesses $f$ and frequencies $x$ of constituent viruses, such that
$$\hat{X}_v(t+\Delta t) = \sum_{i:v} x_i(t) \, \mathrm{exp}(f_i \, \Delta t)$$
This captures clonal interference between competing lineages
Predictive fitness models
A simple predictive model estimates the fitness $f$ of virus $i$ as
$$\hat{f}_i = \beta^\mathrm{ep} \, f_i^\mathrm{ep} + \beta^\mathrm{ne} \, f_i^\mathrm{ne}$$
where $f_i^\mathrm{ep}$ measures cross-immunity via substitutions at epitope sites and $f_i^\mathrm{ep}$ measures mutational load via substitutions at non-epitope sites
We implement a similar model based on two predictors
- Clade frequency change
- Antigenic advancement
Project frequencies forward,
growing clades have high fitness
Calculate HI drop from ancestor,
drifted clades have high fitness
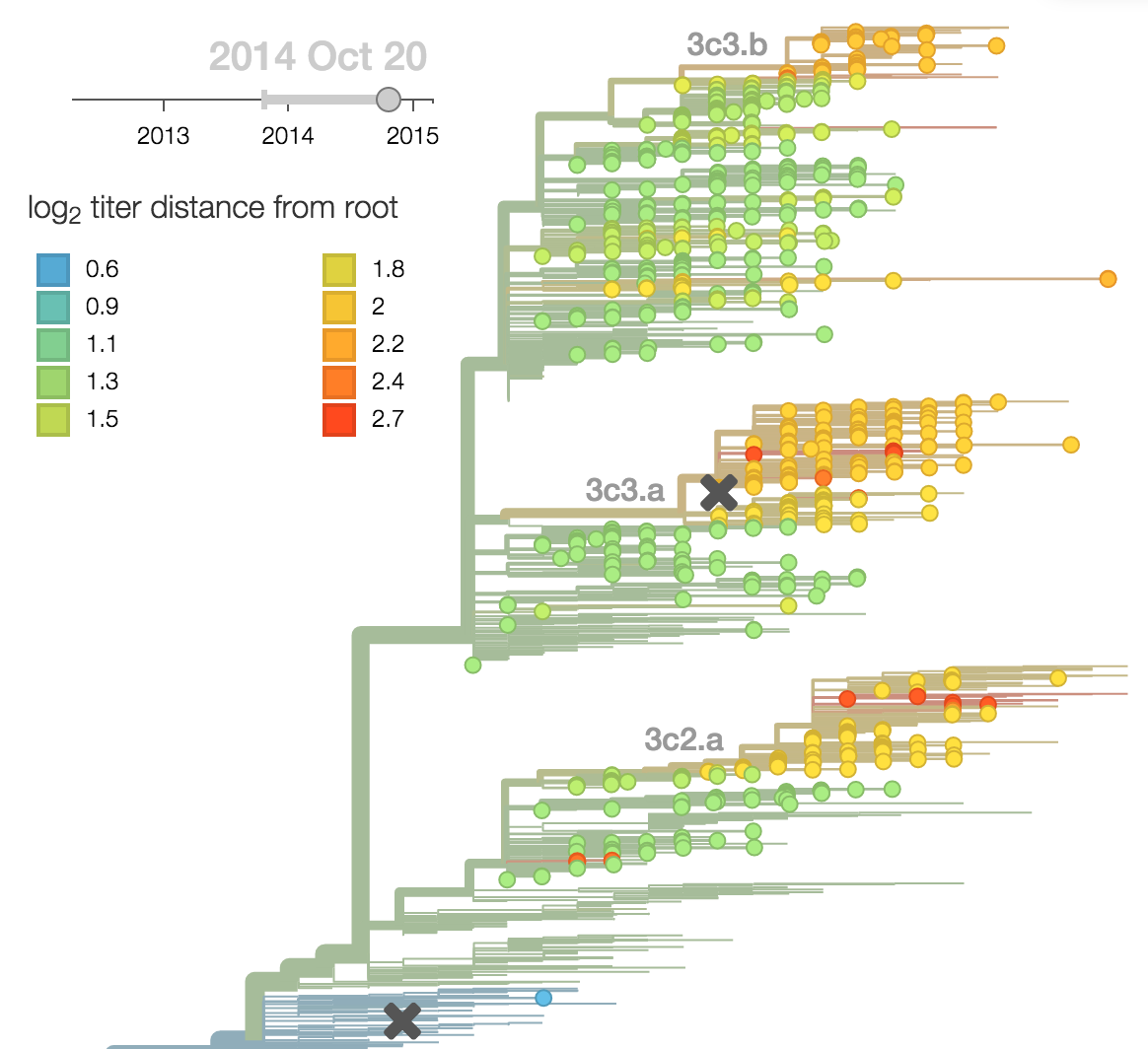
Fitness model parameterization
Our predictive model estimates the fitness $f$ of virus $i$ as
$$\hat{f}_i = \beta^\mathrm{freq} \, f_i^\mathrm{freq} + \beta^\mathrm{HI} \, f_i^\mathrm{HI}$$
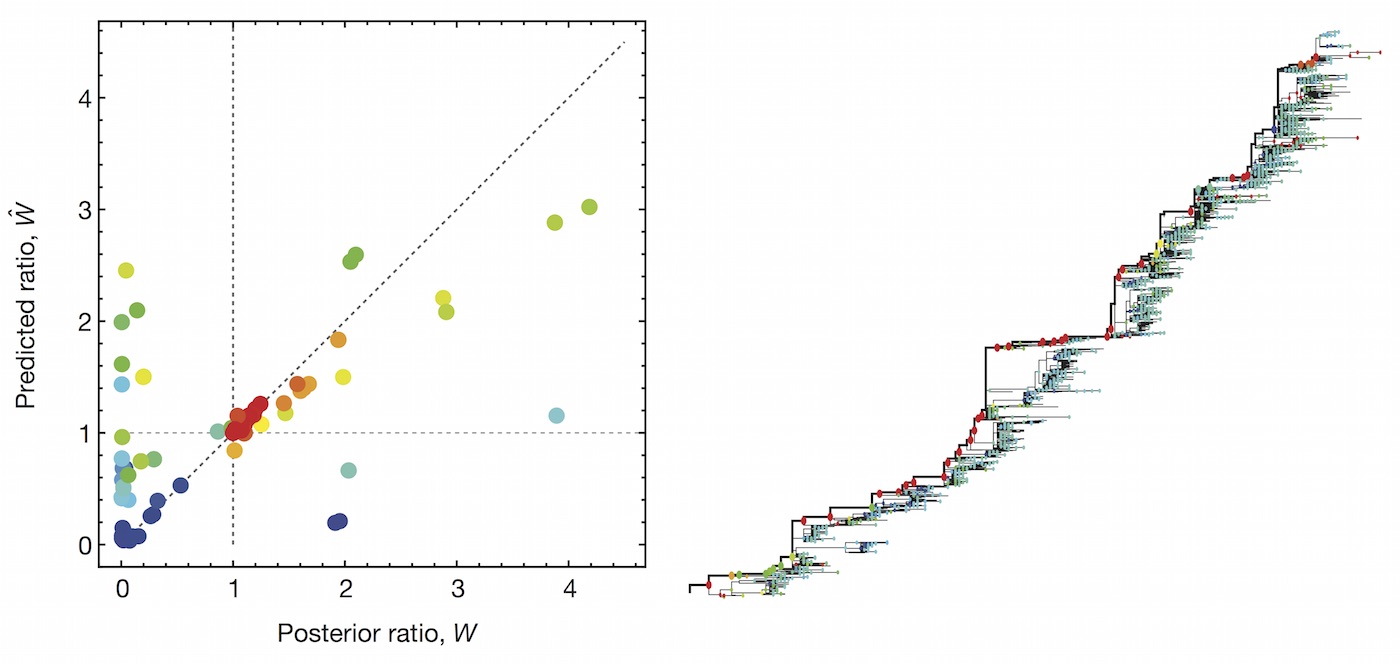
We learn coefficients and validate model based on previous 15 H3N2 seasons
Clade growth rate is well correlated (ρ = 0.66)
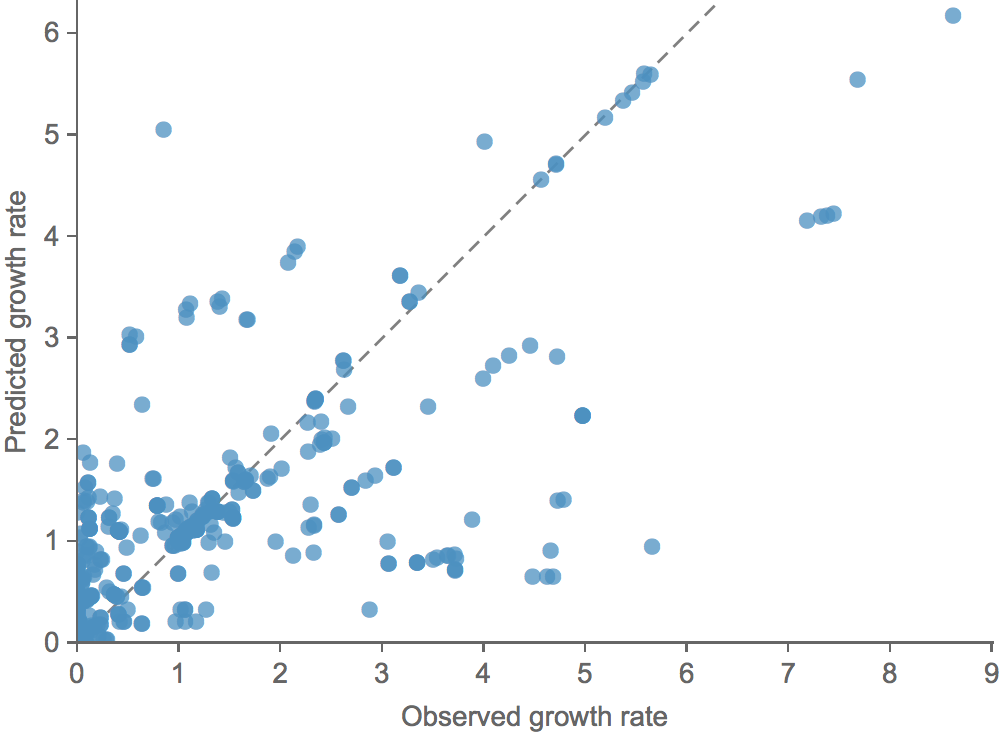
Growth vs decline correct in 84% of cases
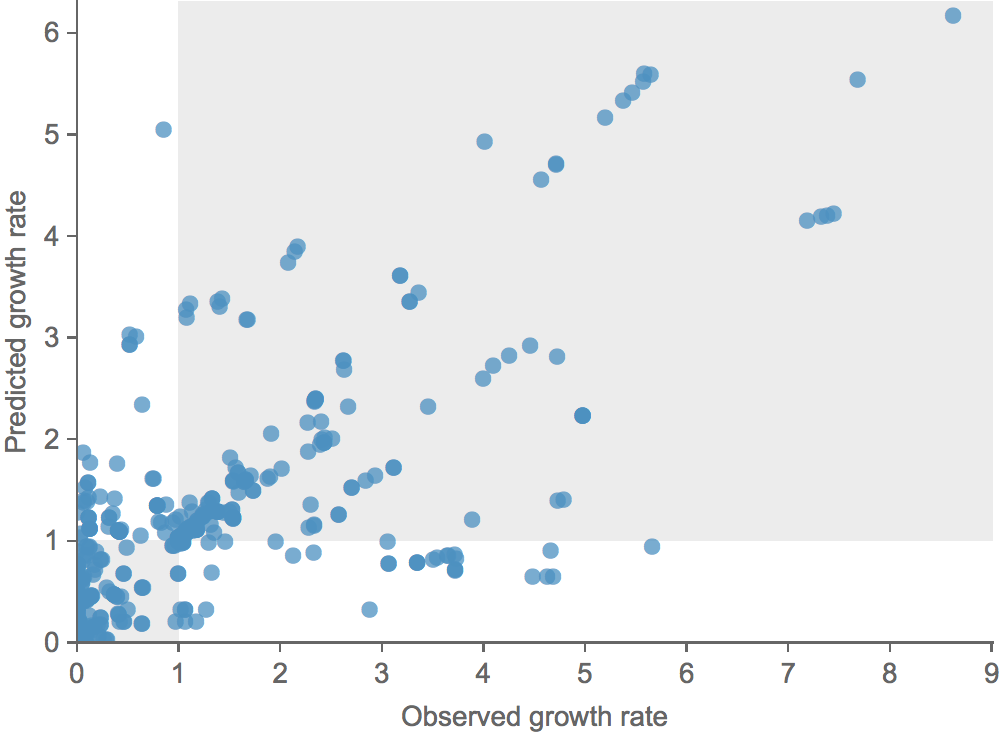
Trajectories show more detailed congruence
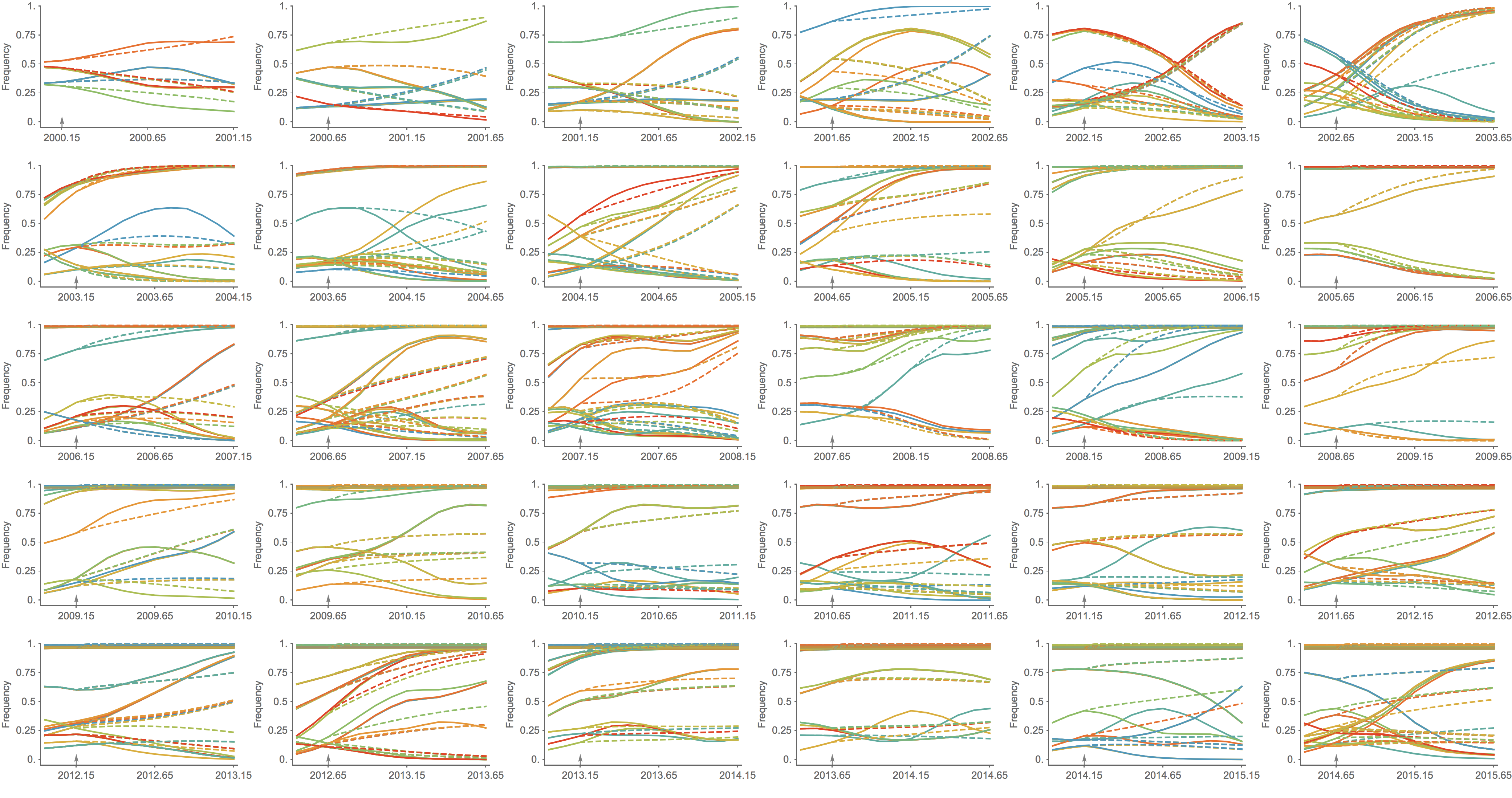
Formalizes intuition about drivers of influenza dynamics
| Model | Ep coefficient | HI coefficient | Freq error | Growth corr |
|---|---|---|---|---|
| Epitope only | 2.36 | -- | 0.10 | 0.57 |
| HI only | -- | 2.05 | 0.08 | 0.63 |
| Epitope + HI | -0.11 | 2.15 | 0.08 | 0.67 |
Further work on predictive modeling
- Integrate data predictors and data sources, e.g. plan to investigate a geographic predictor
- Possible to build predictive models for H1N1 and B and to forecast NA evolution
Real-time analyses are actionable and thus, may inform influenza vaccine strain selection
Outbreak analysis
Ebola
Epidemic nearly contained, but resulted in >28,000 confirmed cases and >11,000 deaths
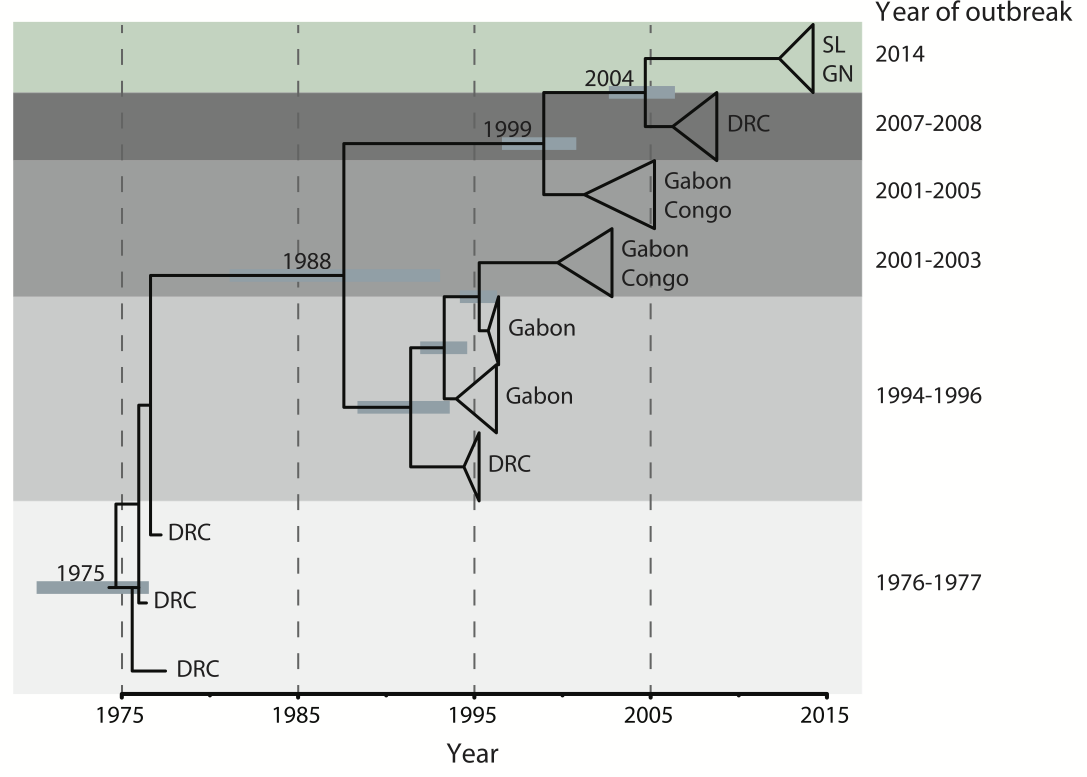
Outbreaks are independent spillovers from the animal reservoir
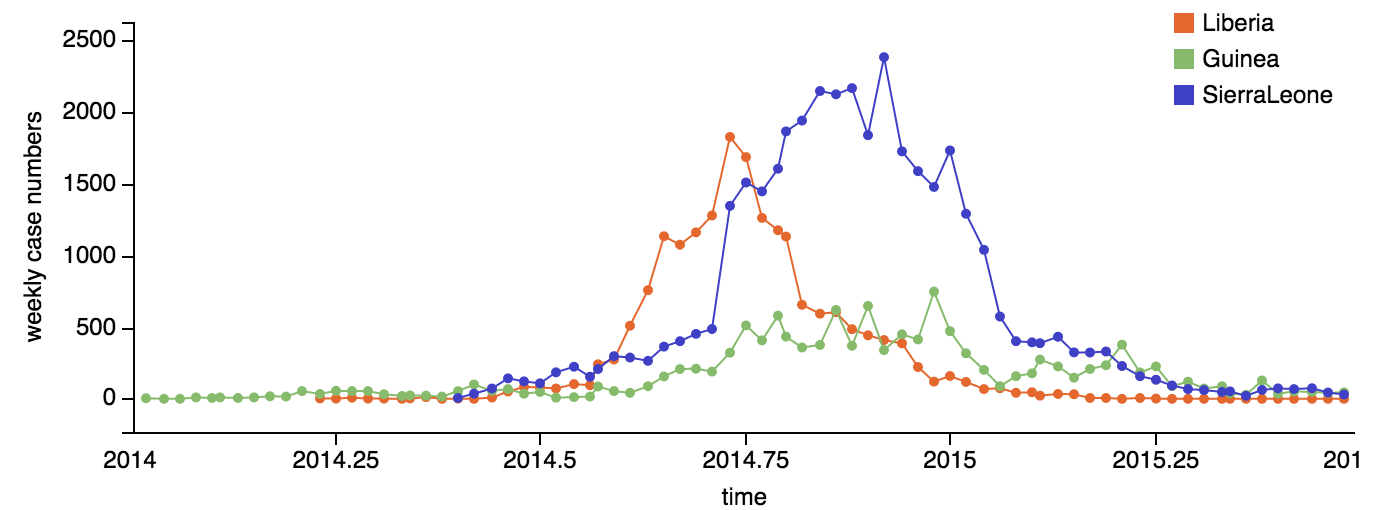
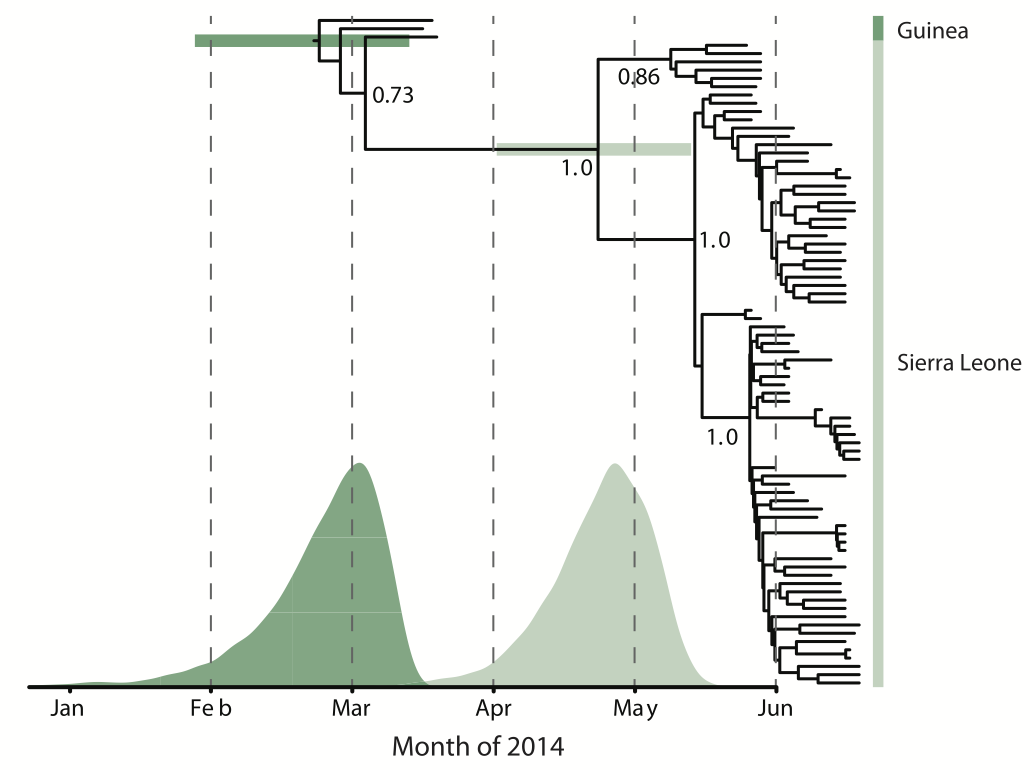
Person-to-person spread in the early West African outbreak
Continued spread through Dec 2014
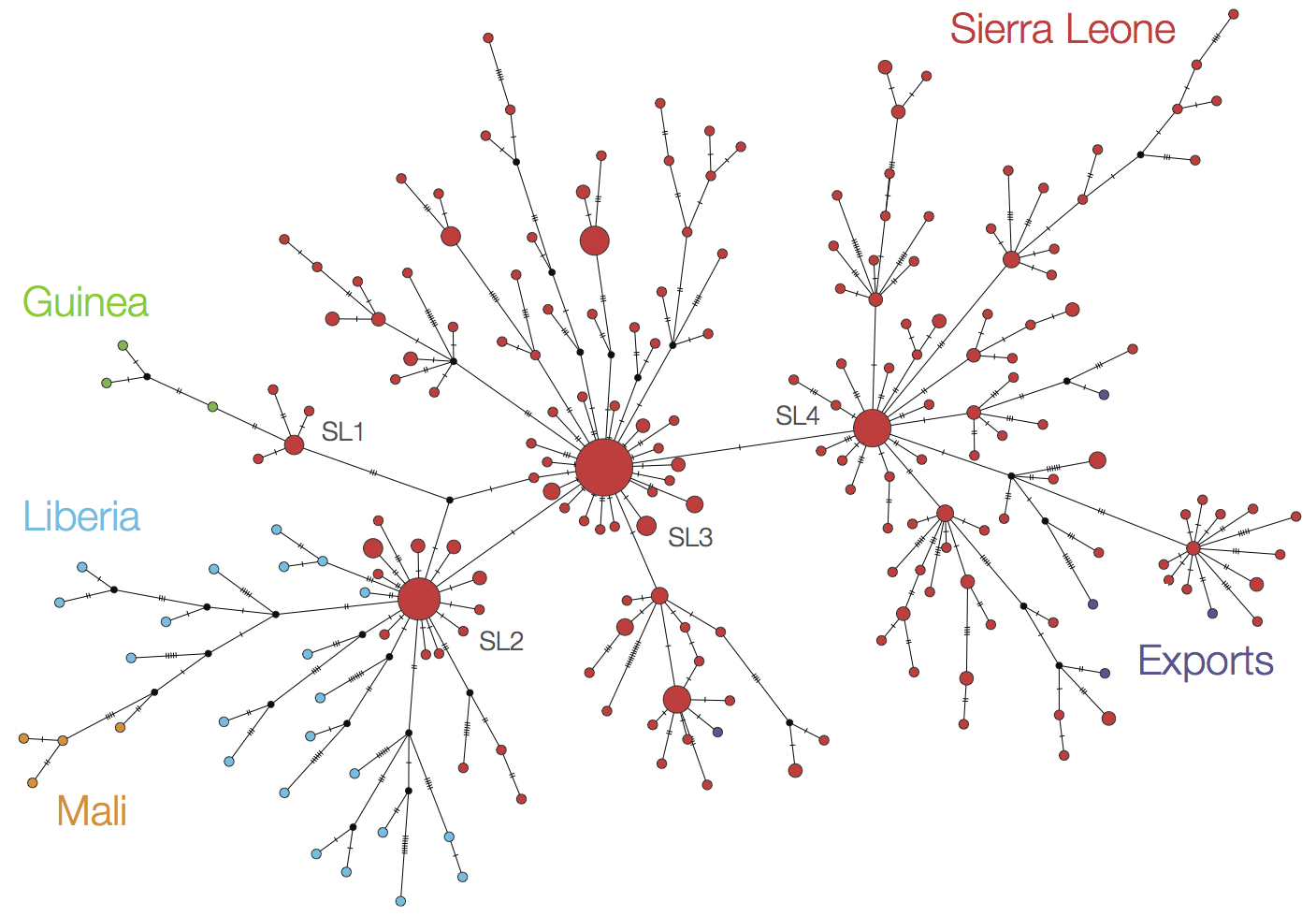
At epidemic height, geographic spread of particular interest
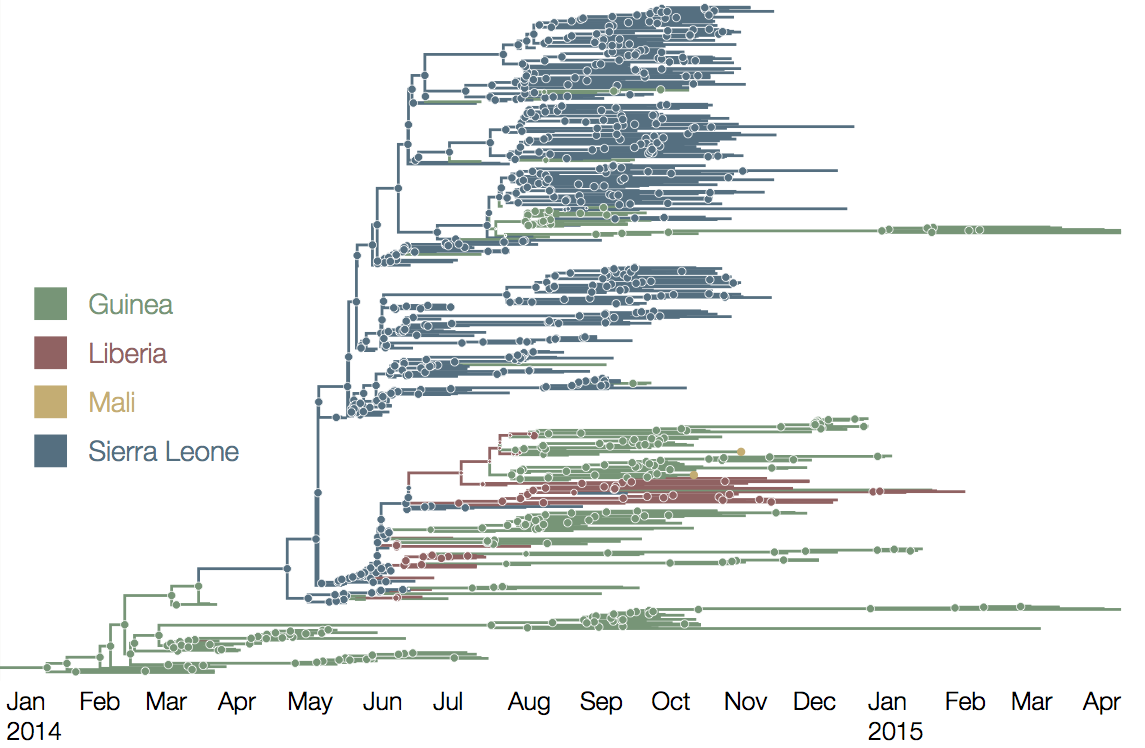
Later on, tracking transmission clusters of primary importance
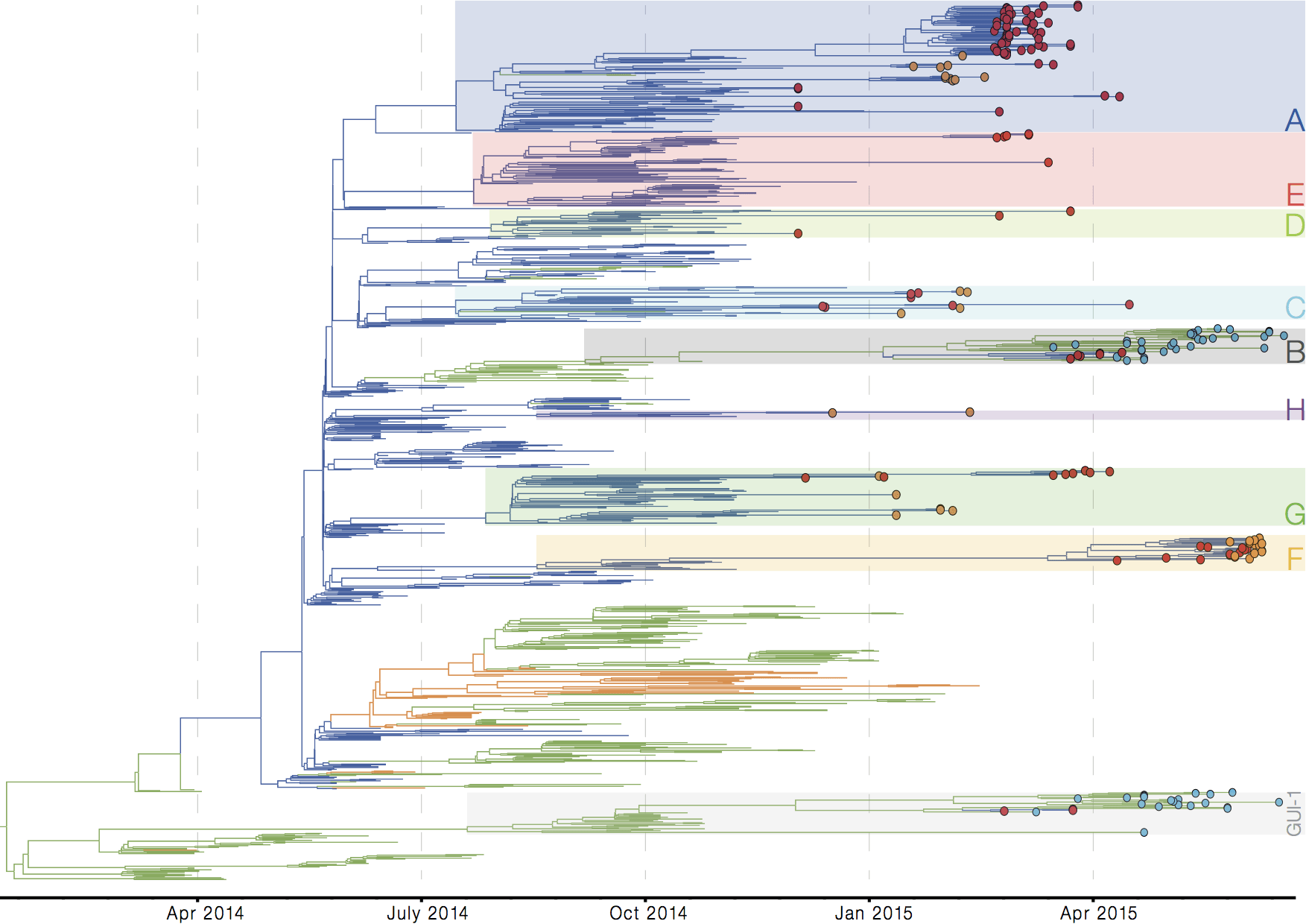
Tracking epidemic spread in real-time:
ebola.nextstrain.org
Zika
Virus source in Africa, spread eastward

Virus source in Africa, spread eastward
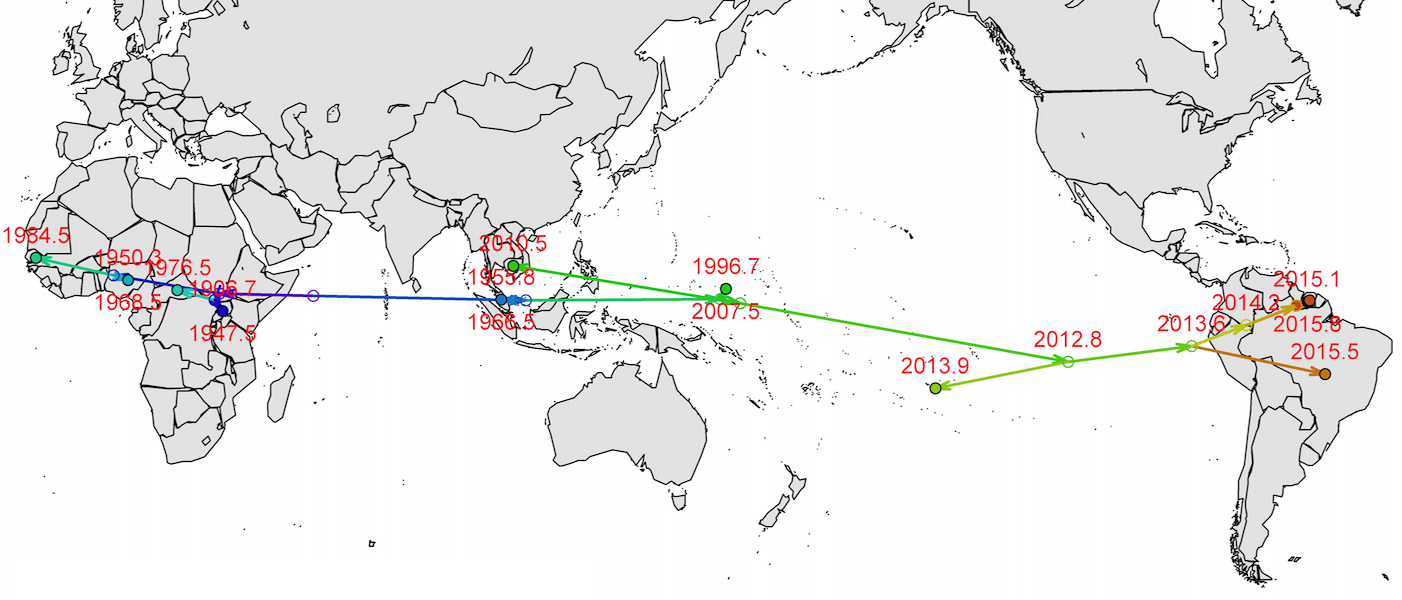
Isolated epidemics in the South Pacific

Single arrival into the Americas, circa mid-2014
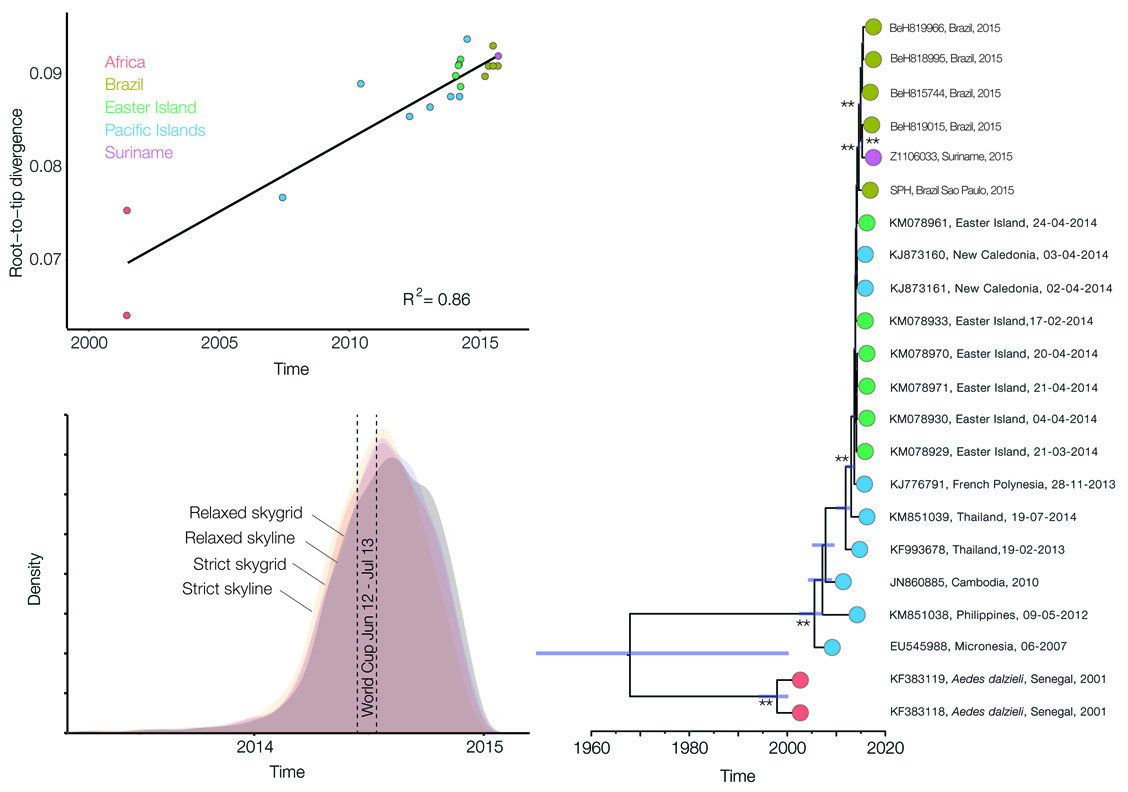
Working on next/zika
Moving forward, genetically-informed outbreak response requires:
- Rapid sharing of sequence data, genetic context critical
- Technologies to rapidly conduct phylogenetic inference
- Technologies to explore genetic relationships and inform epidemiological investigation
Future work

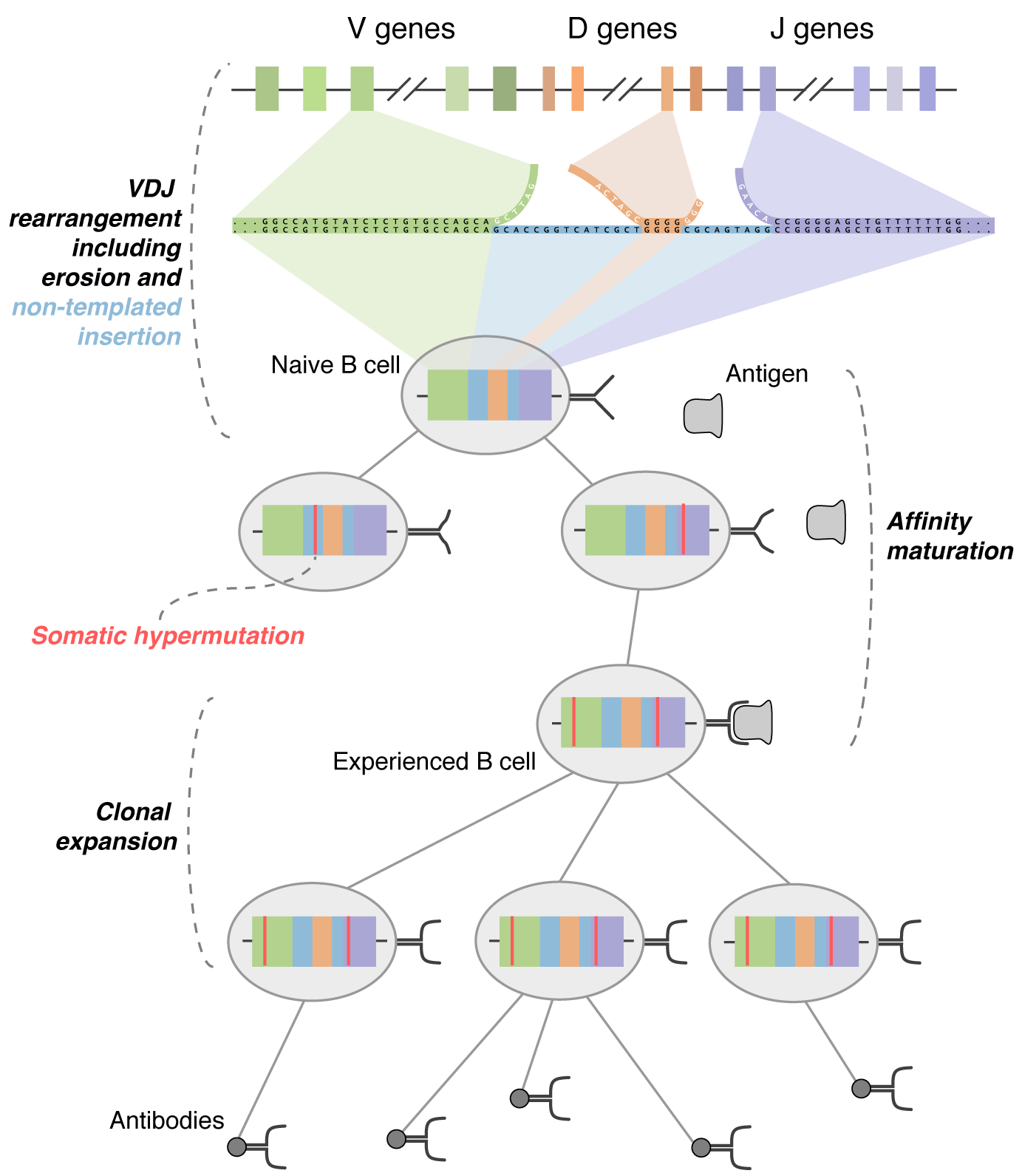
Phylogenetic analysis of B cell affinity maturation
Acknowledgements
WHO Global Influenza Surveillance Network, GISAID, Richard Neher (Max Planck Tübingen), Andrew Rambaut (University of Edinburgh), Colin Russell (Cambridge University), Philipe Lemey (KU Leuven), Marc Suchard (UCLA), Steven Riley (Imperial College), Gytis Dudas (University of Edinburgh), Jesse Bloom (Fred Hutch), Erick Matsen (Fred Hutch), the Lab.
Contact
- Website: bedford.io
- Twitter: @trvrb
- Slides: bedford.io/talks/real-time-tracking-vidd/