Real-time tracking of virus evolution
Trevor Bedford (@trvrb)
20 Feb 2018
EPI 583 Seminar
University of Washington
This talk
We work at the interface of virology, evolution and epidemiology
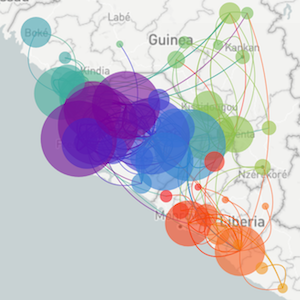

Methods focus on sequencing to reconstruct pathogen spread
Epidemic process
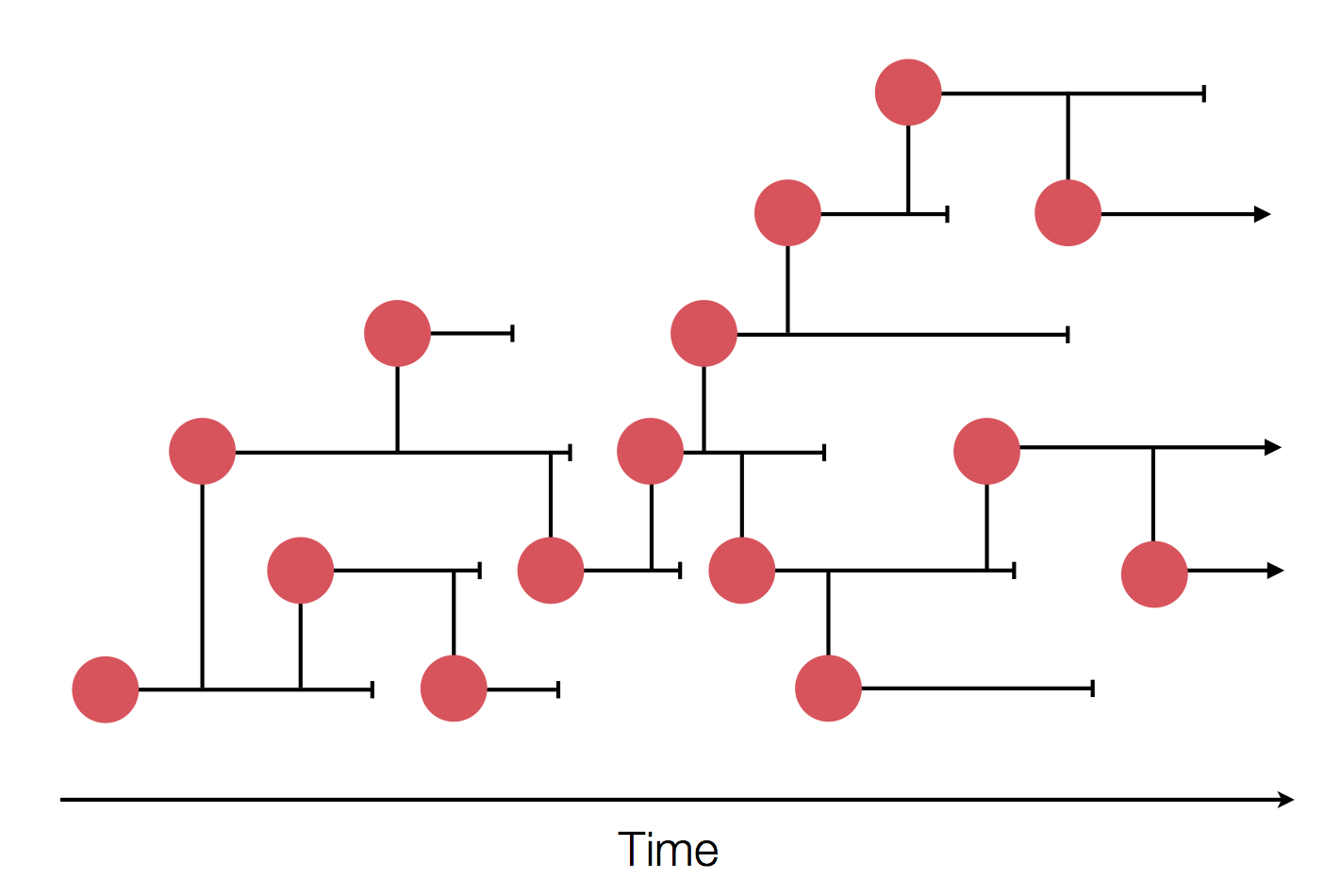
Sample some individuals
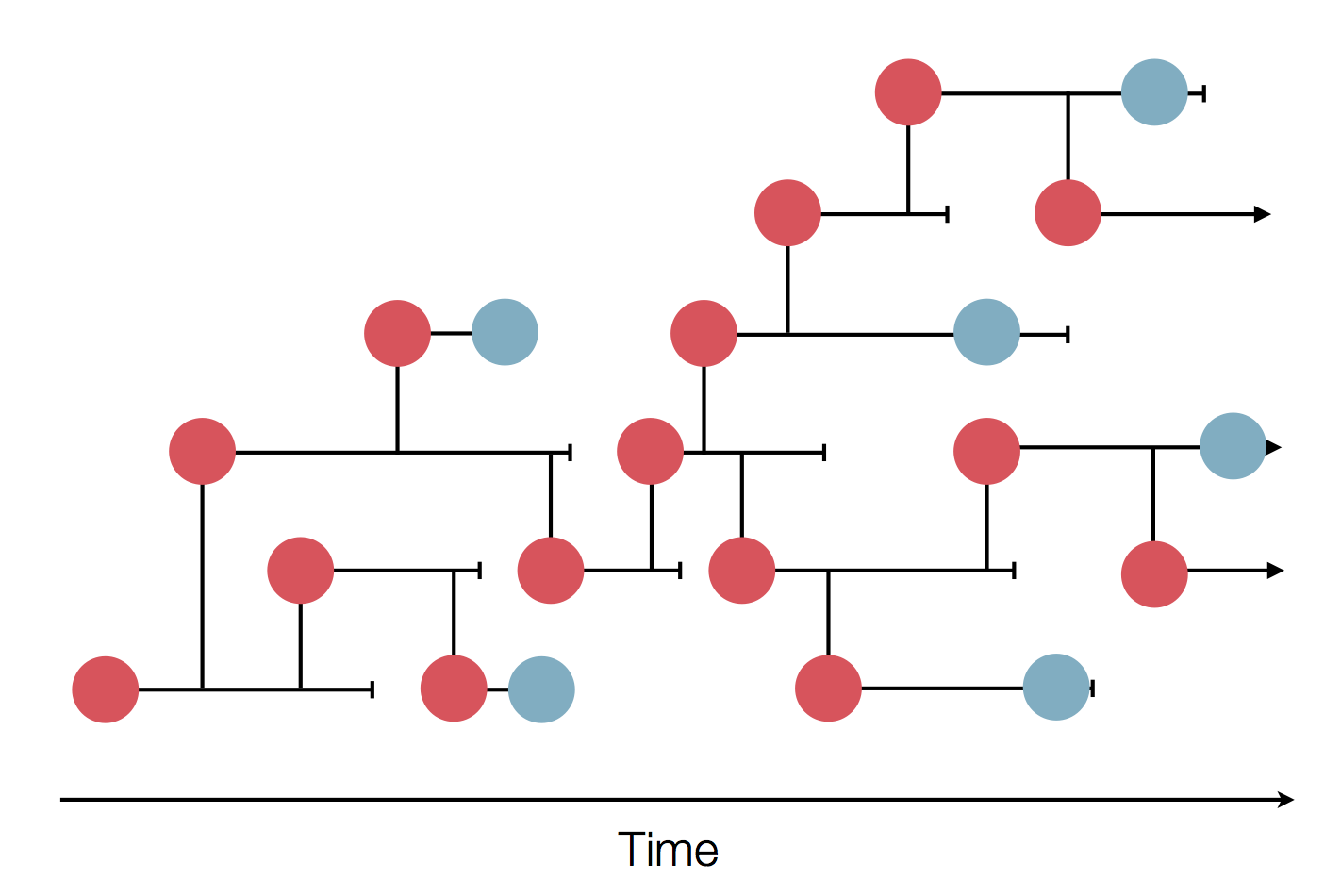
Sequence and determine phylogeny
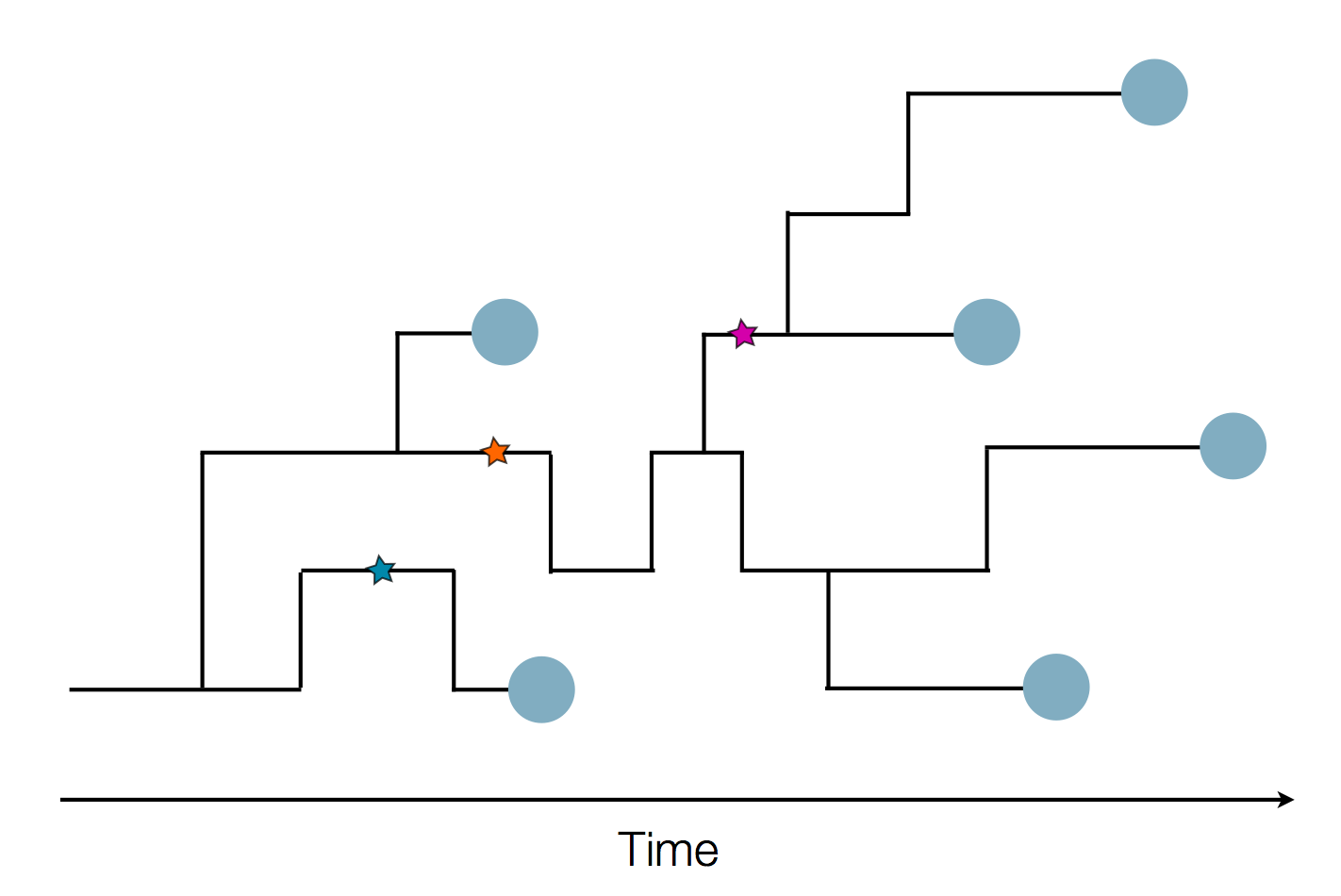
Sequence and determine phylogeny
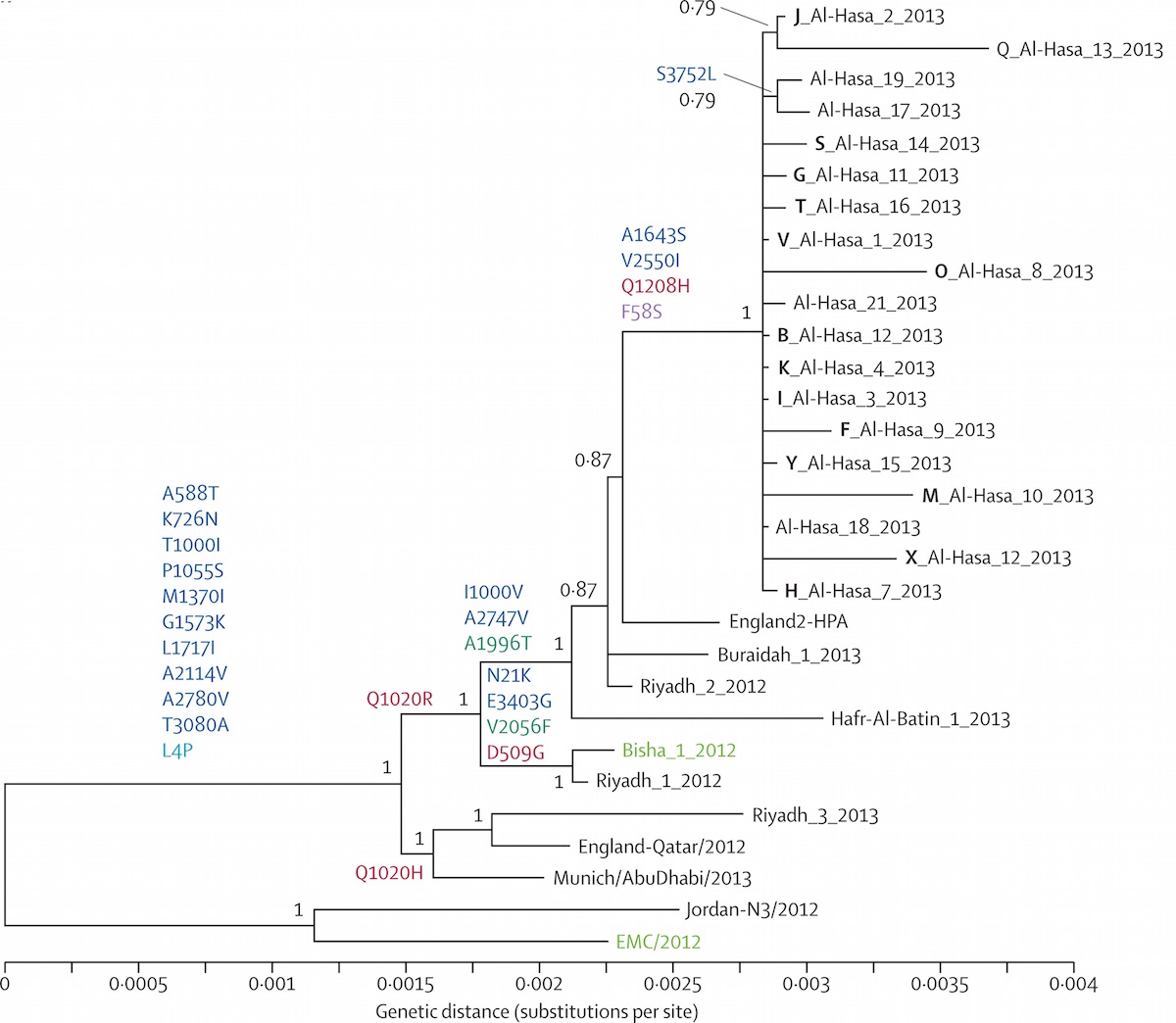
Localized Middle Eastern MERS-CoV phylogeny
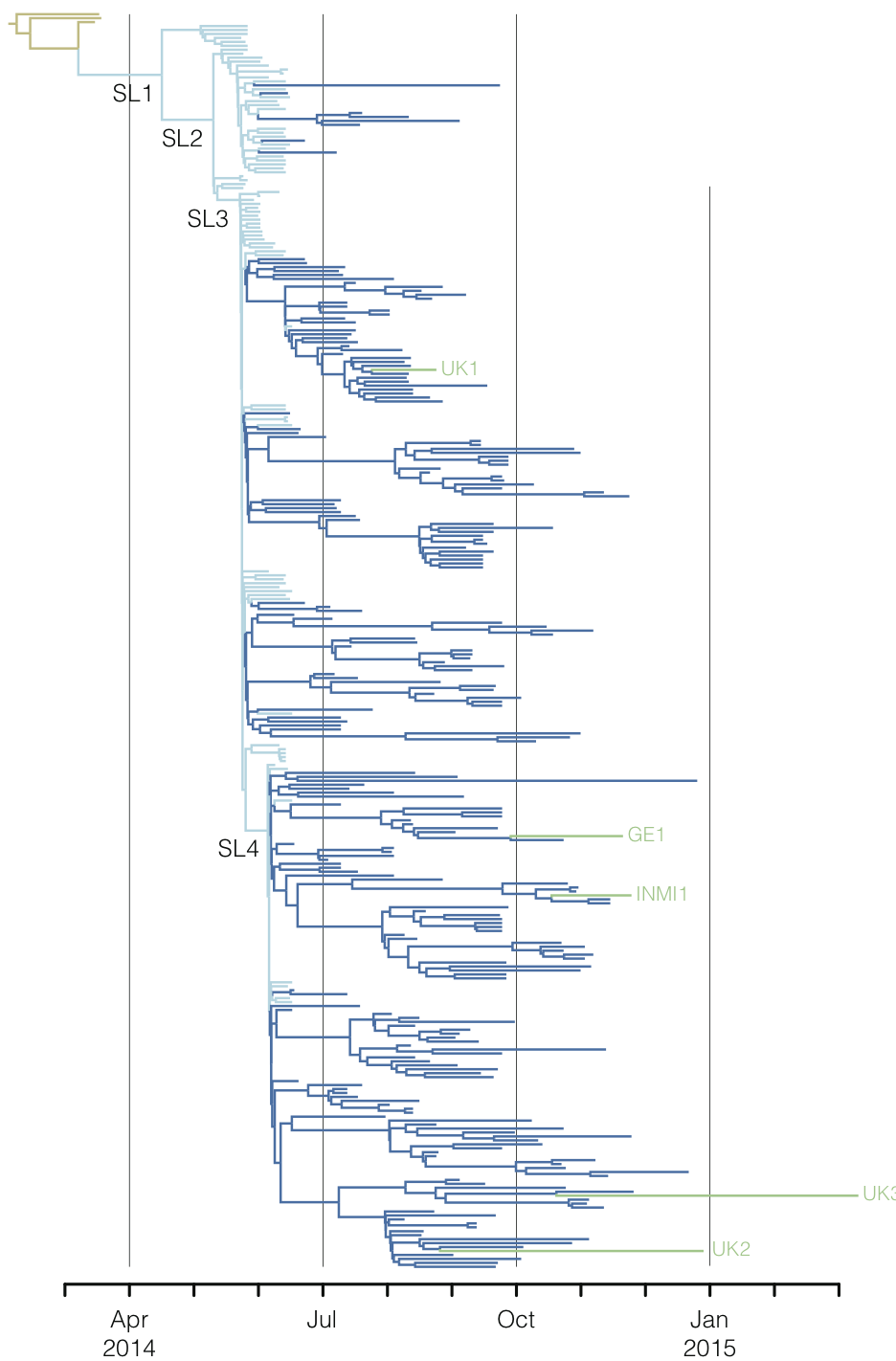
Regional West African Ebola phylogeny
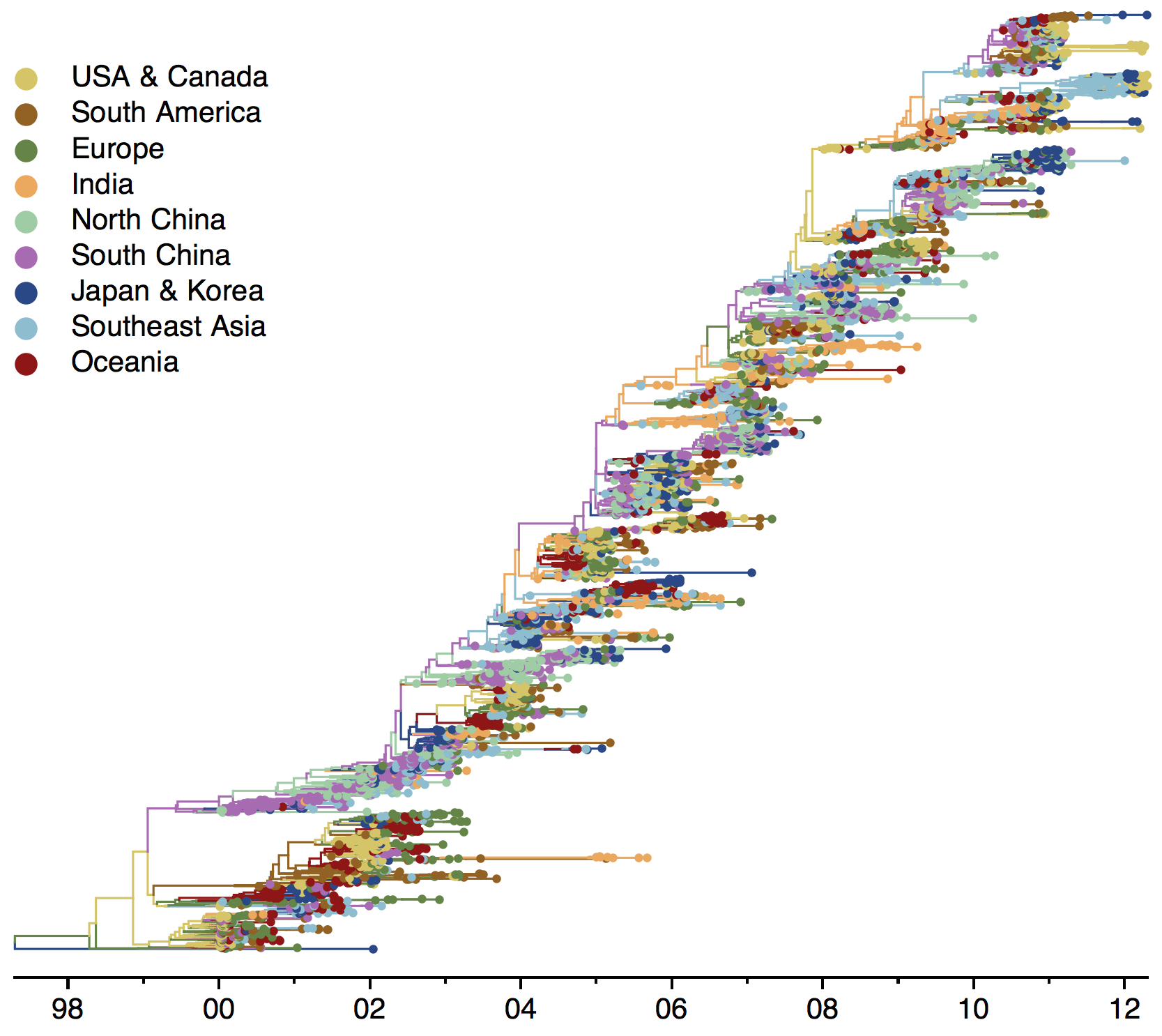
Global influenza phylogeny
Phylogenetic tracking has the capacity to revolutionize epidemiology
Outline
- Influenza circulation and antigenic drift
- Ebola spread in West Africa
- Zika spread in the Americas
- "Real-time" analyses
Influenza
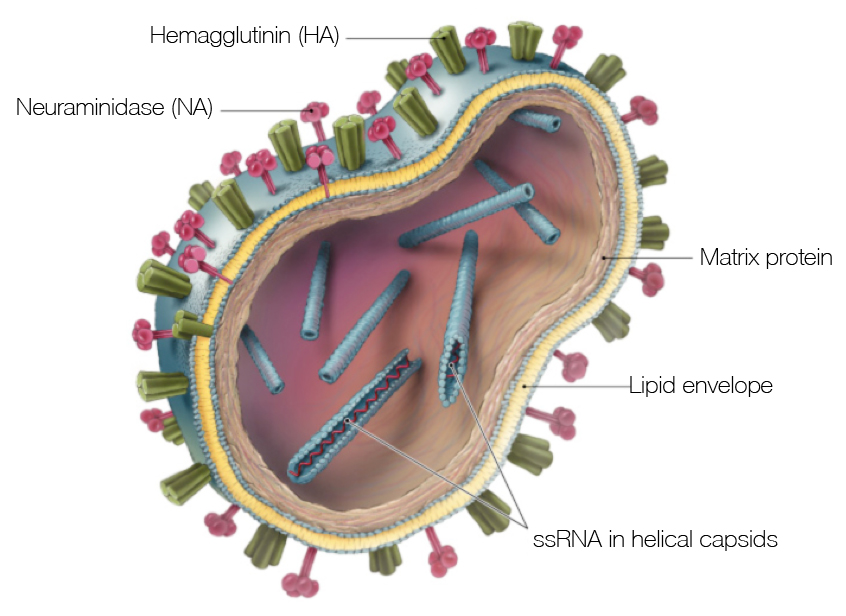
Influenza virion
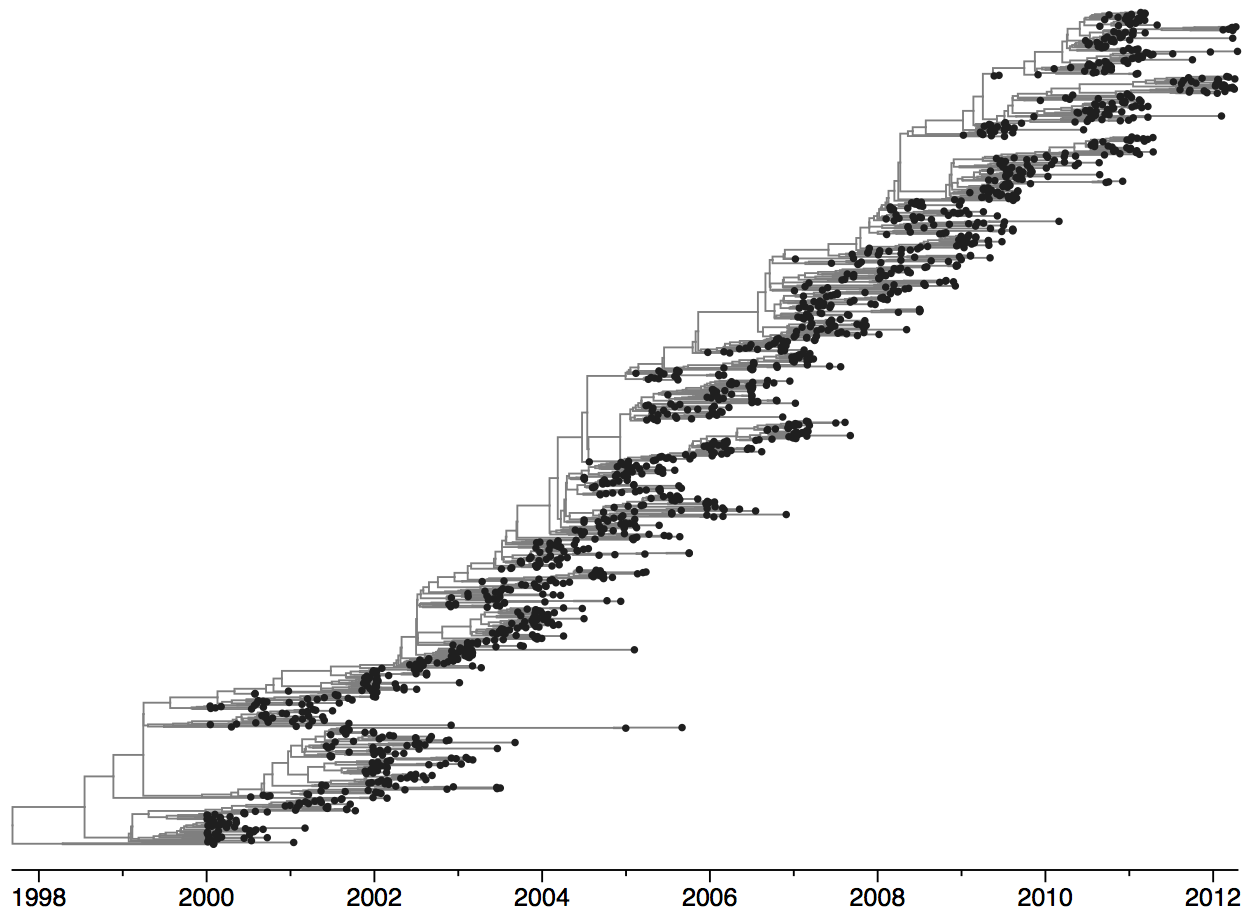
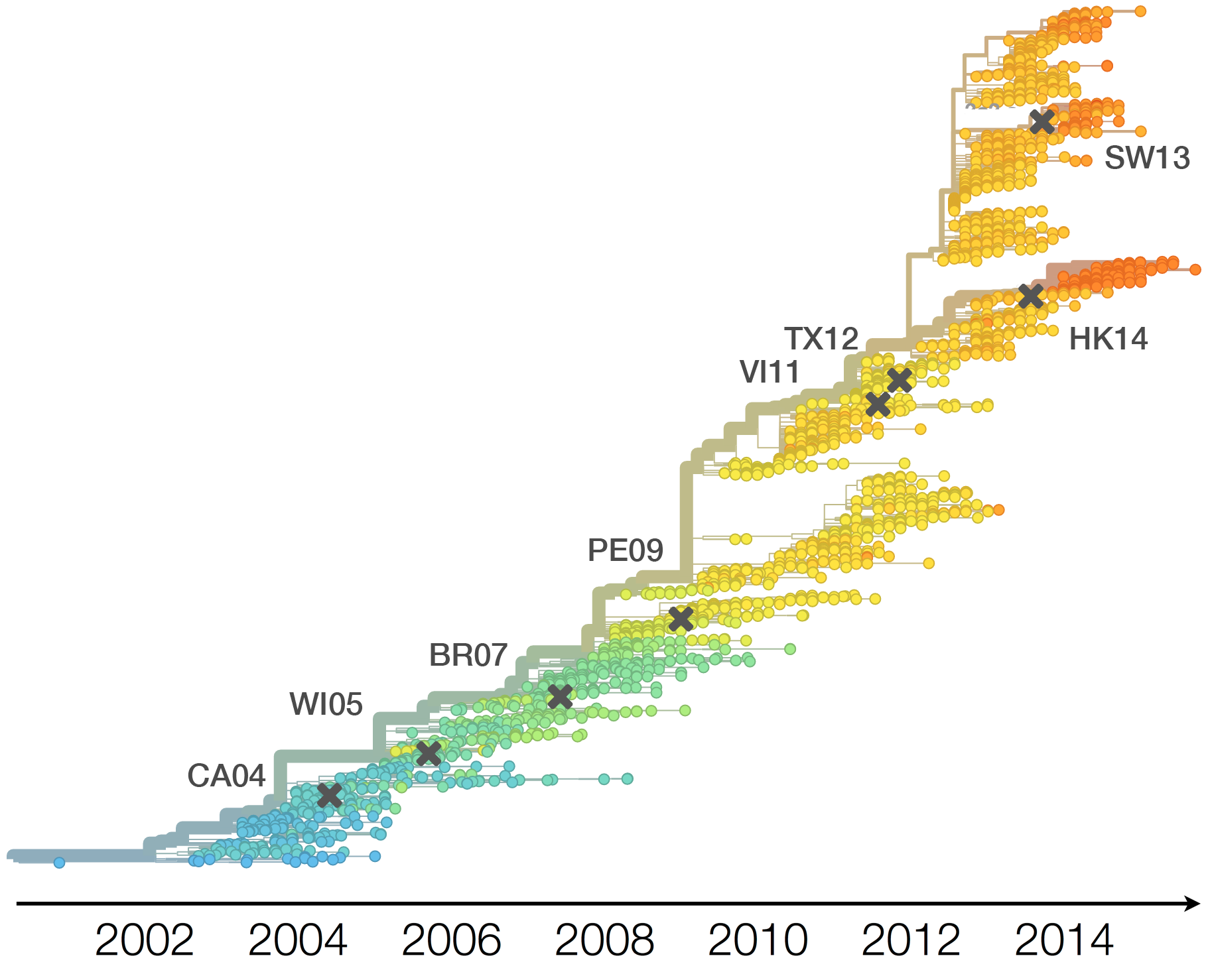
Population turnover (in H3N2) is extremely rapid
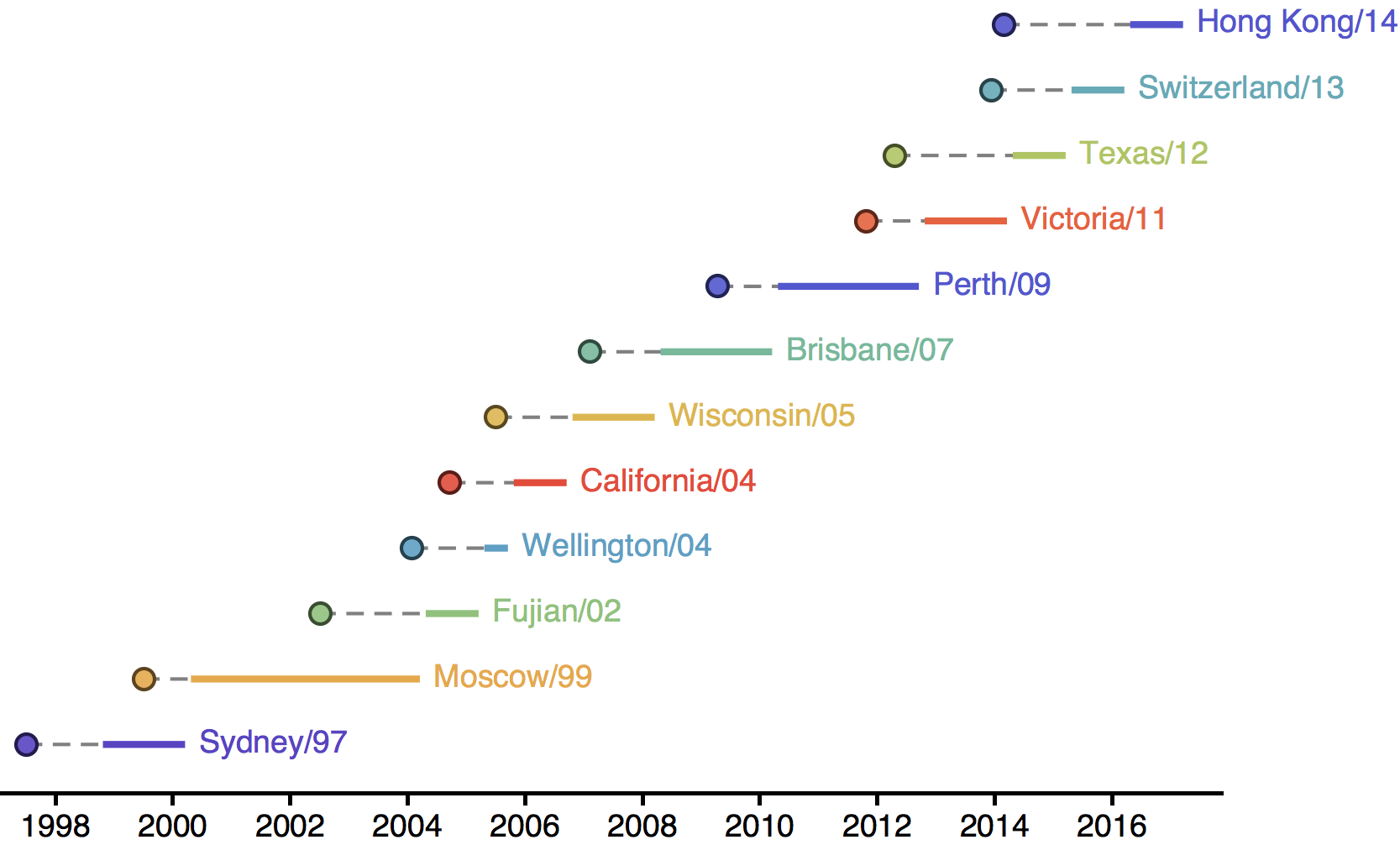
Antigenic drift necessitates frequent H3N2 vaccine updates
Integrating influenza antigenic dynamics with molecular evolution
with Andrew Rambaut, Marc Suchard, Philippe Lemey and others
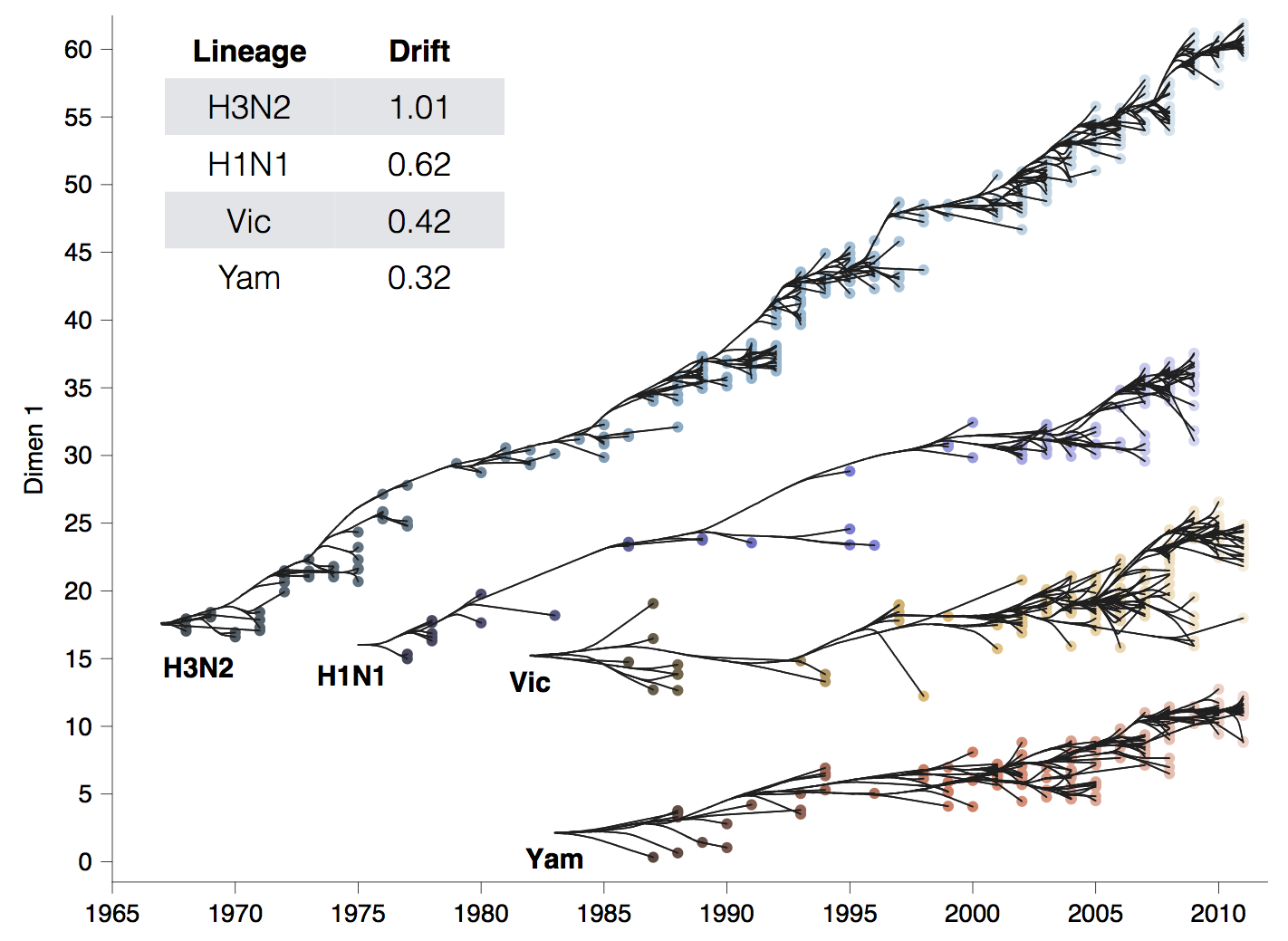
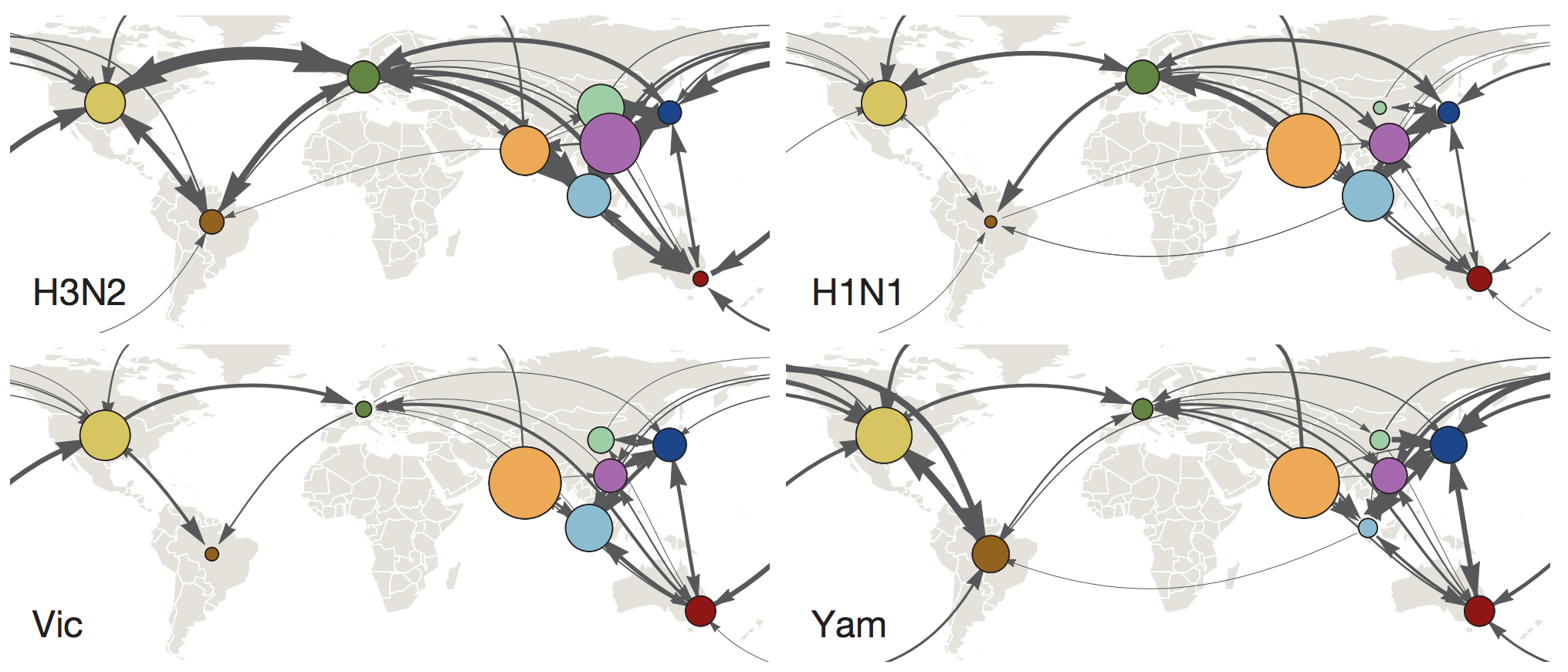
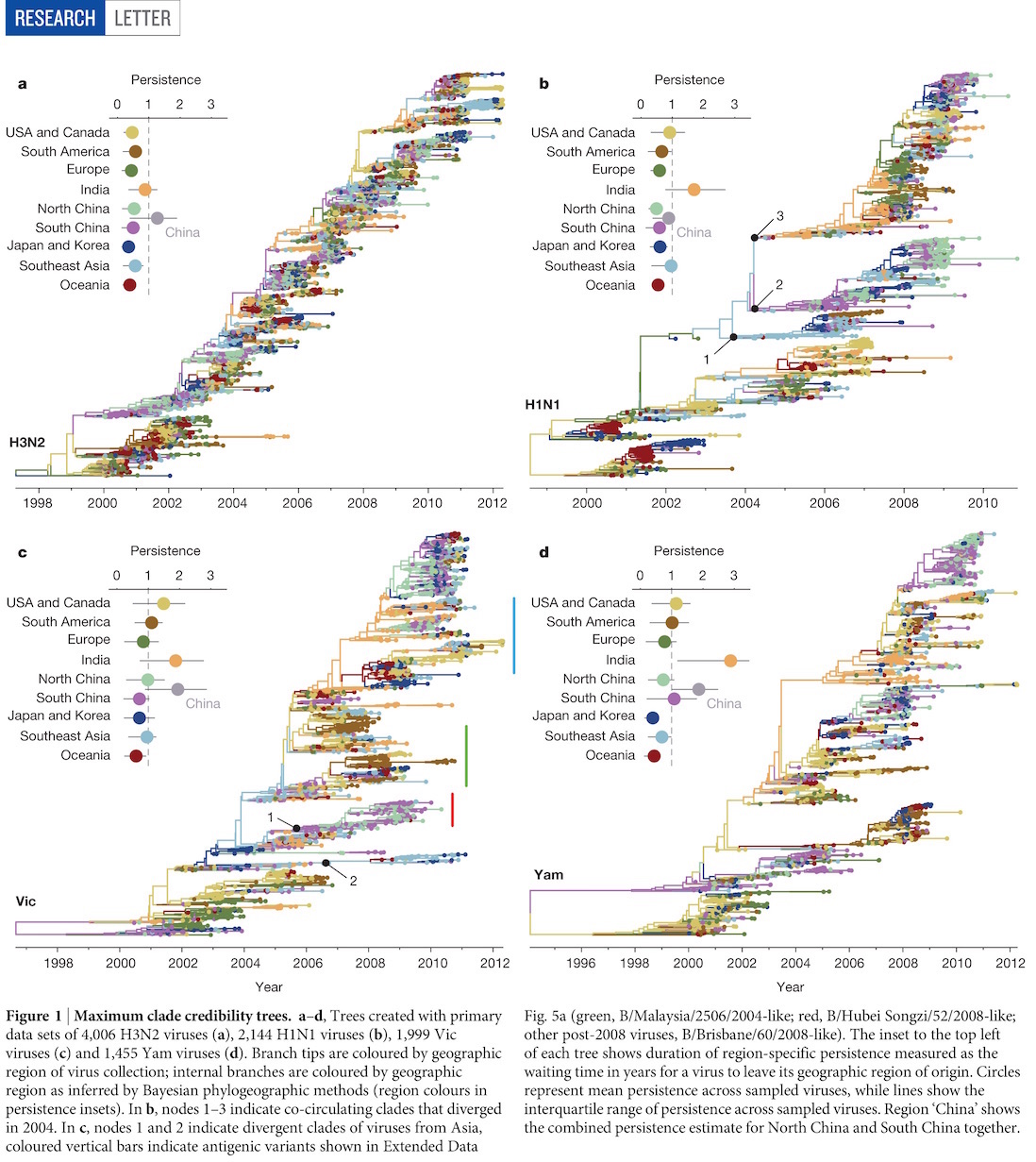
Global circulation patterns of seasonal influenza viruses vary with rates of antigenic drift
with Colin Russell, Philippe Lemey, Steven Riley and many others
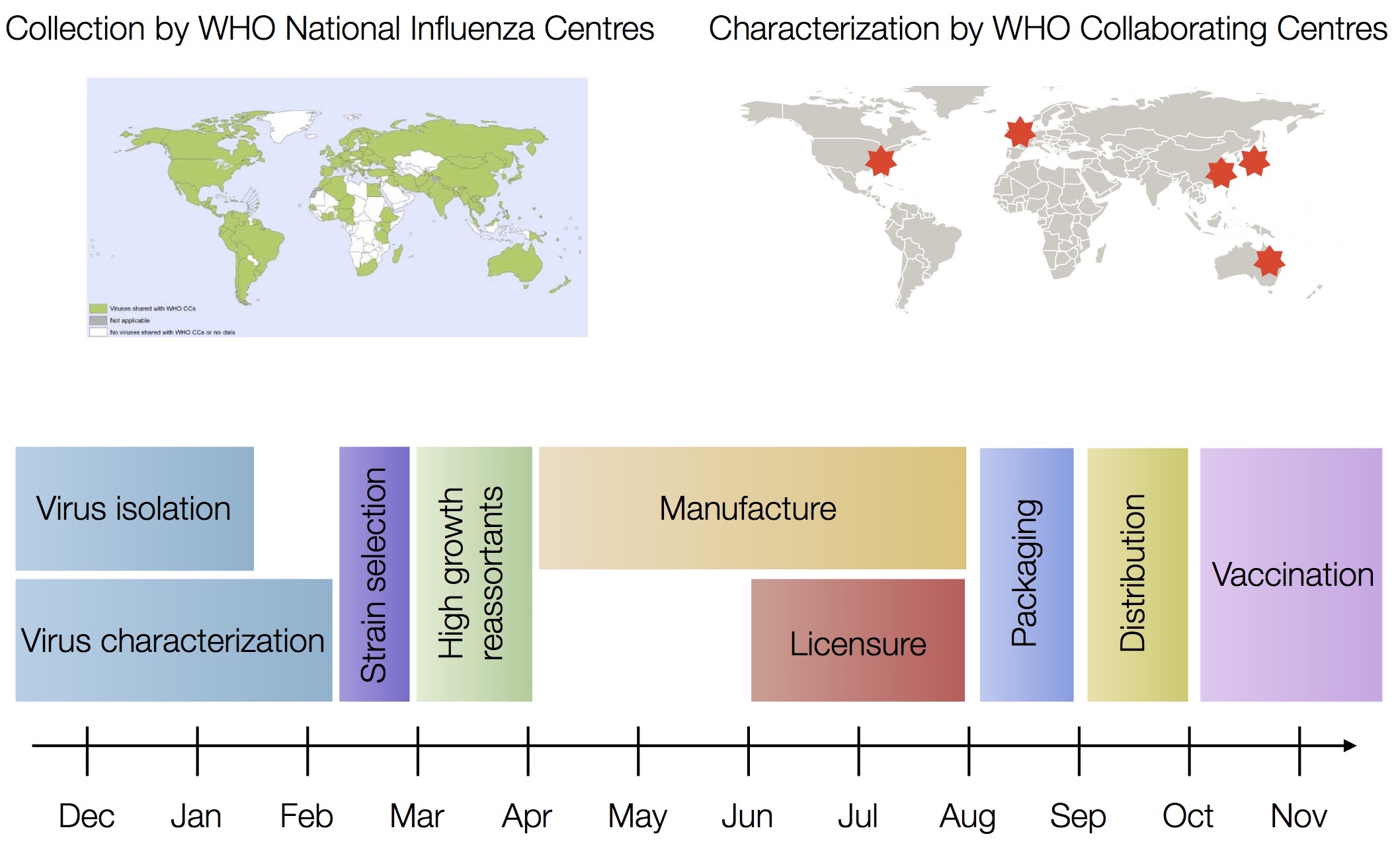
Scientific publishing practices vs
a fast evolving virus
Vaccine strain selection timeline
nextflu
Project to provide a real-time view of the evolving influenza population
nextflu
Project to provide a real-time view of the evolving influenza population
All in collaboration with Richard Neher

nextflu pipeline
- Download all recent HA sequences from GISAID
- Filter to remove outliers
- Subsample across time and space
- Align sequences
- Build tree
- Estimate clade frequencies
- Infer antigenic phenotypes
- Export for visualization
Phenotypic assay data used to directly infer titer drops on the phylogeny
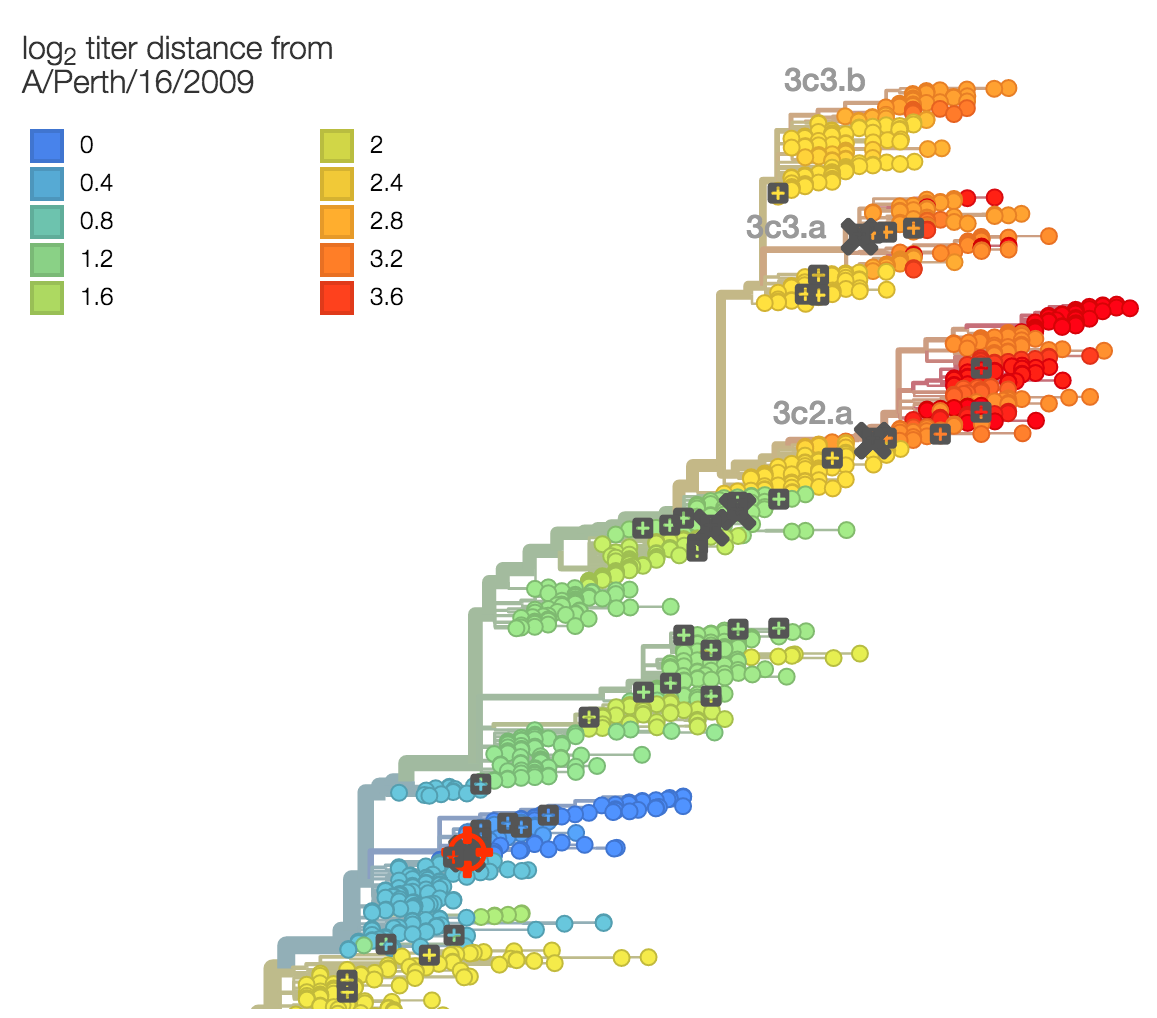
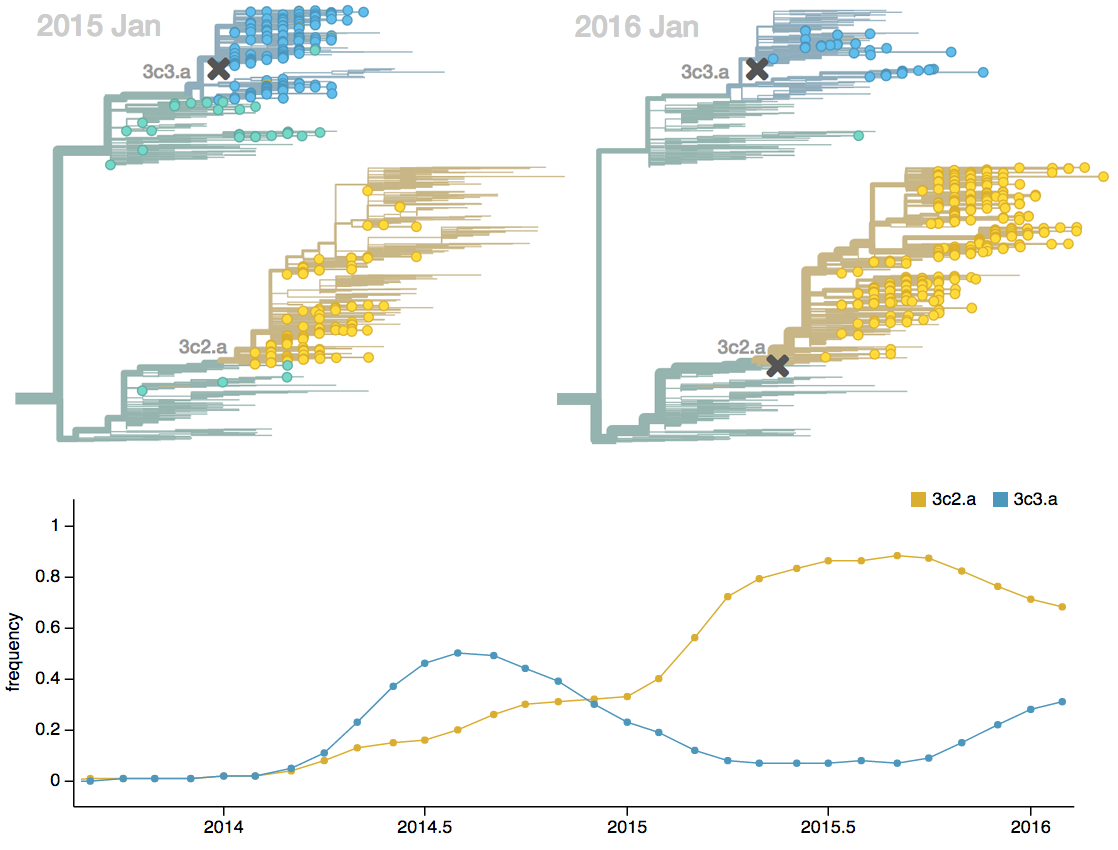
Antigenic drift drives population turnover
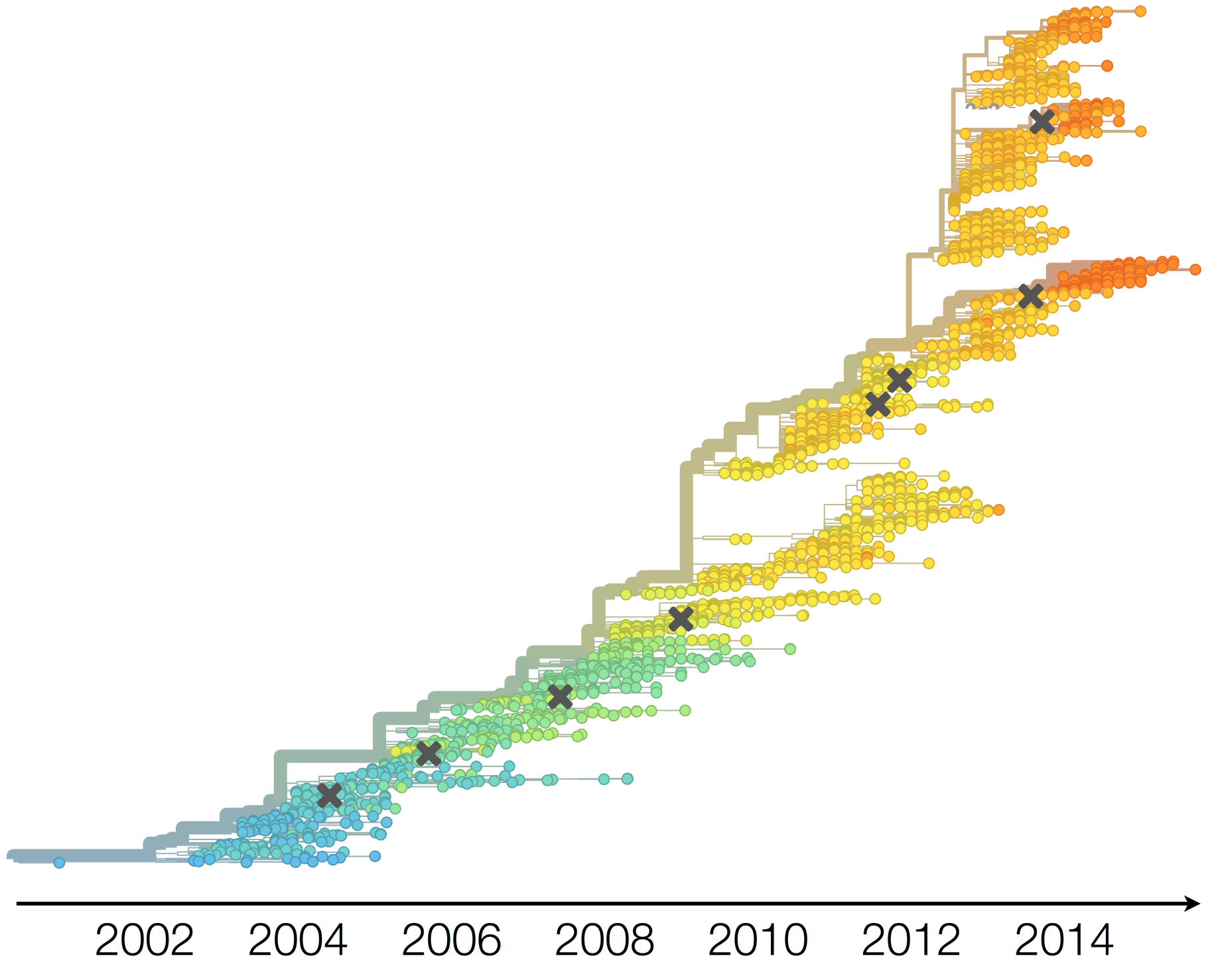
Antigenic drift drives population turnover
nextflu.org
Forecasting
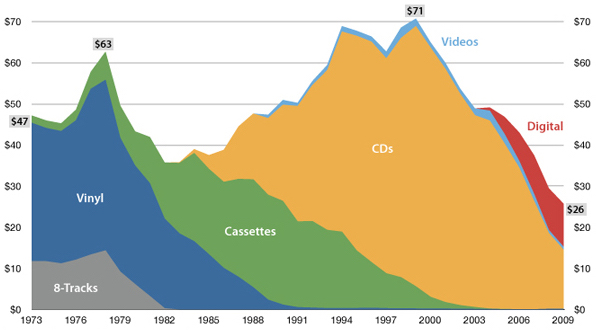
"The future is here, it's just not evenly distributed yet"
— William Gibson
USA music industry, 2011 dollars per capita
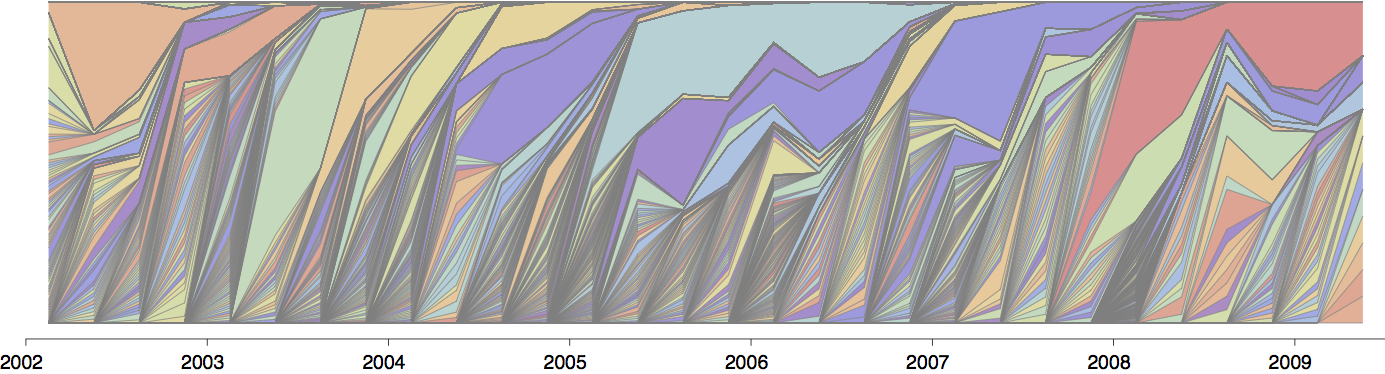
Influenza population turnover
Vaccine strain selection timeline
Seek to explain change in clade frequencies over 1 year
Fitness models can project clade frequencies
Clade frequencies $X$ derive from the fitnesses $f$ and frequencies $x$ of constituent viruses, such that
$$\hat{X}_v(t+\Delta t) = \sum_{i:v} x_i(t) \, \mathrm{exp}(f_i \, \Delta t)$$
This captures clonal interference between competing lineages
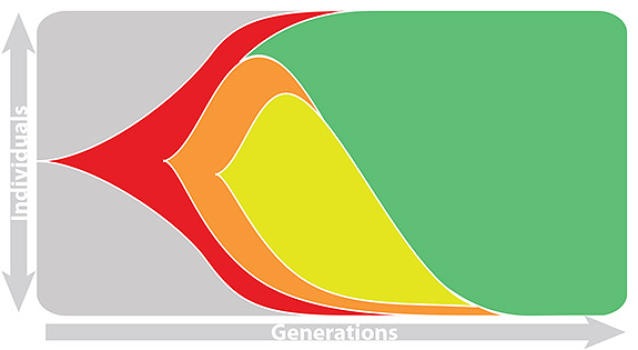
The question of forecasting becomes: how do we accurately estimate fitnesses of circulating viruses?
Fortunately, there's lots of training data and previously successful strains have had:
- Amino acid changes at epitope sites
- Antigenic novelty based on HI
- Rapid phylogenetic growth
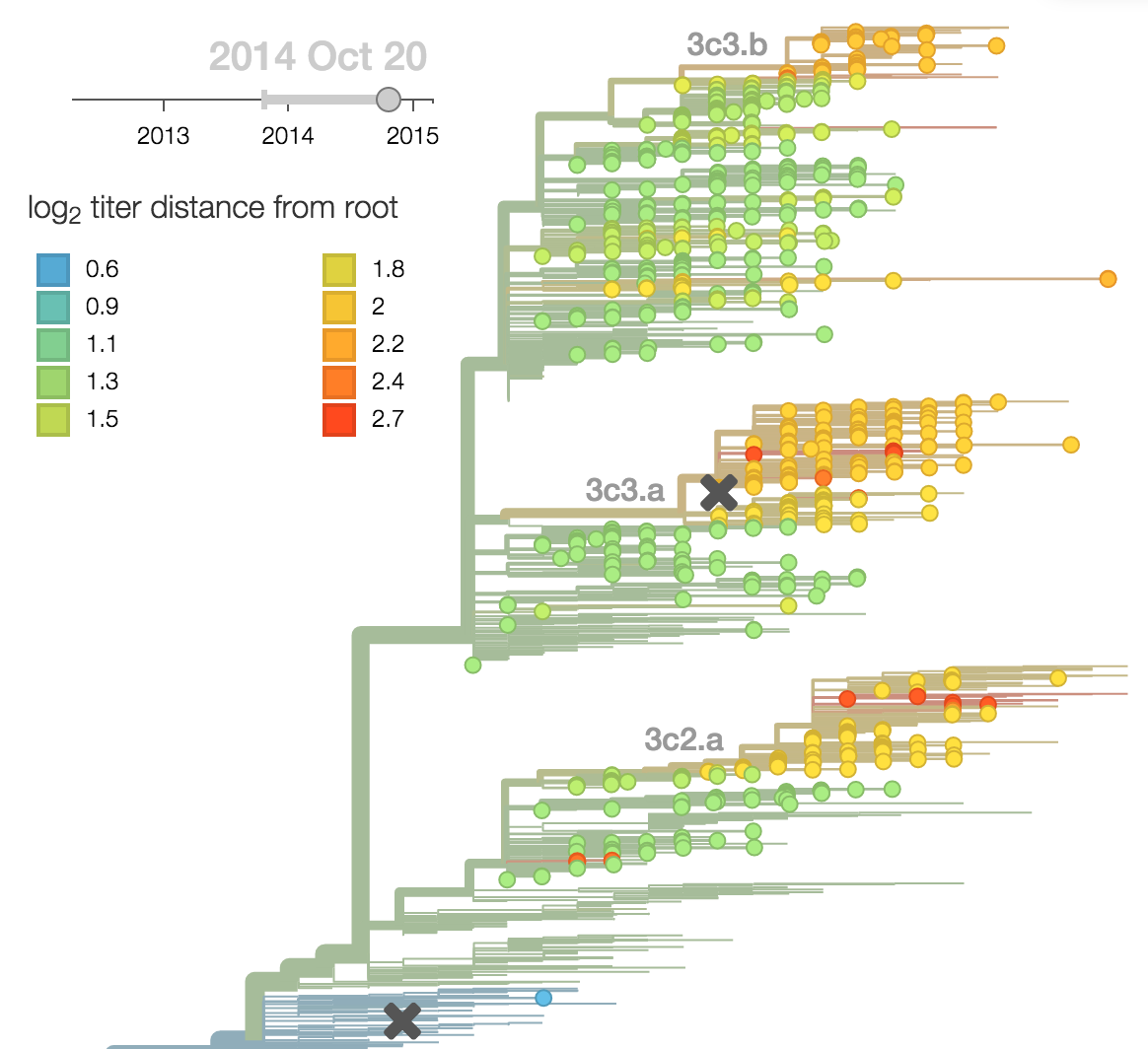
Predictor: calculate HI drop from ancestor,
drifted clades have high fitness
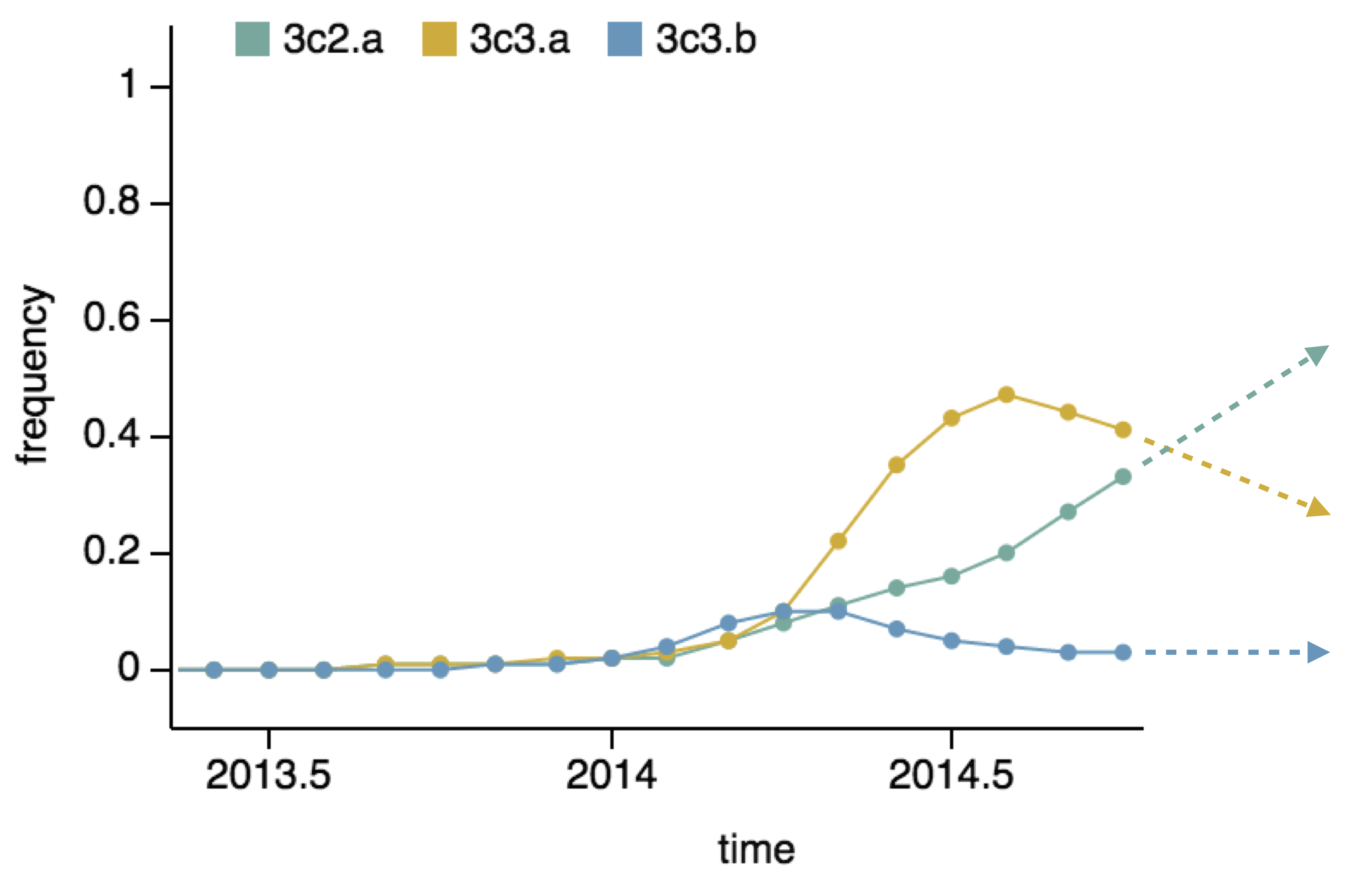
Predictor: project frequencies forward,
growing clades have high fitness
We predict fitness based on a simple formula
where the fitness $f$ of virus $i$ is estimated as
$$\hat{f}_i = \beta^\mathrm{HI} \, f_i^\mathrm{HI} + \beta^\mathrm{freq} \, f_i^\mathrm{freq}$$
where $f_i^\mathrm{HI}$ measures antigenic drift via HI and $f_i^\mathrm{freq}$ measures clade growth/decline
We learn coefficients and validate model based on previous 15 H3N2 seasons
Clade growth rate is well predicted (ρ = 0.66)
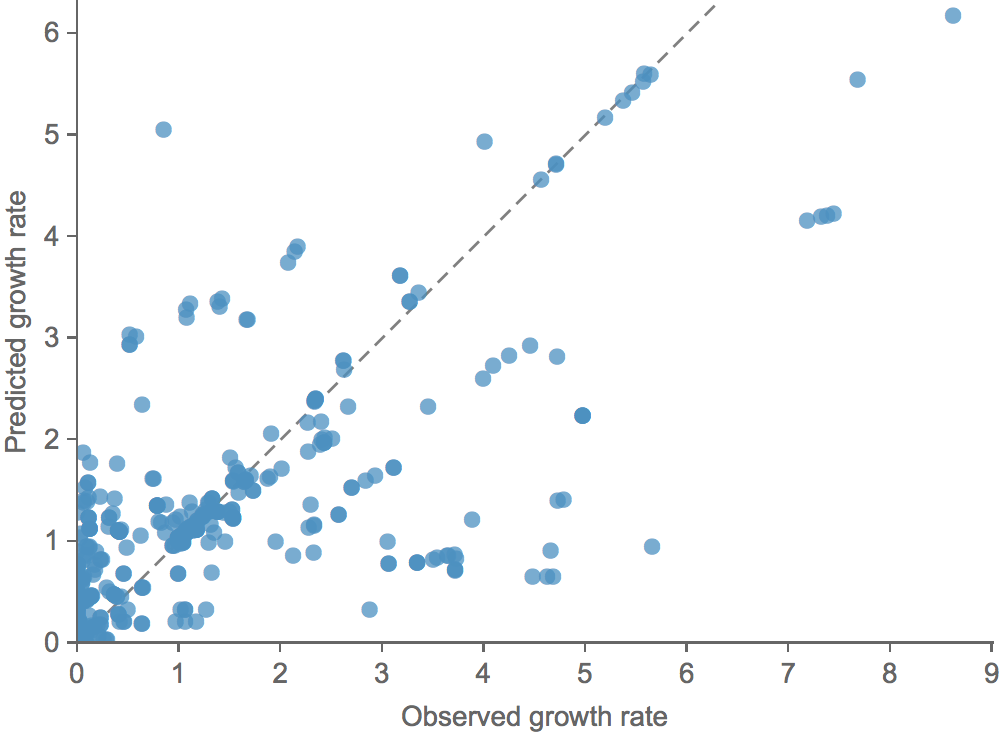
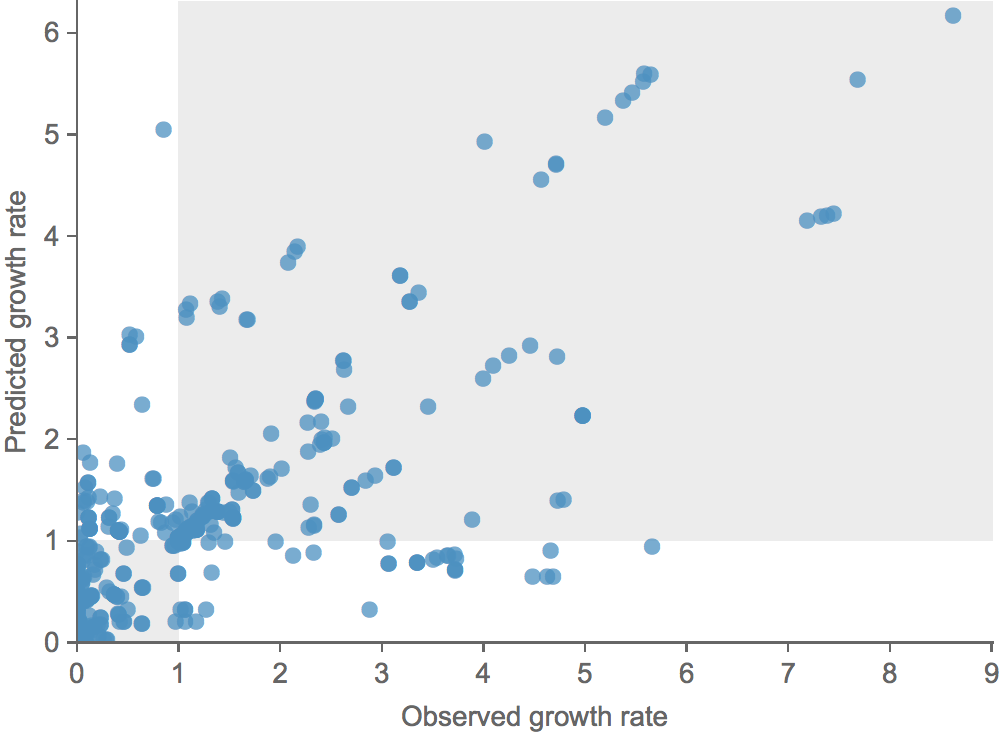
Growth vs decline correct in 84% of cases
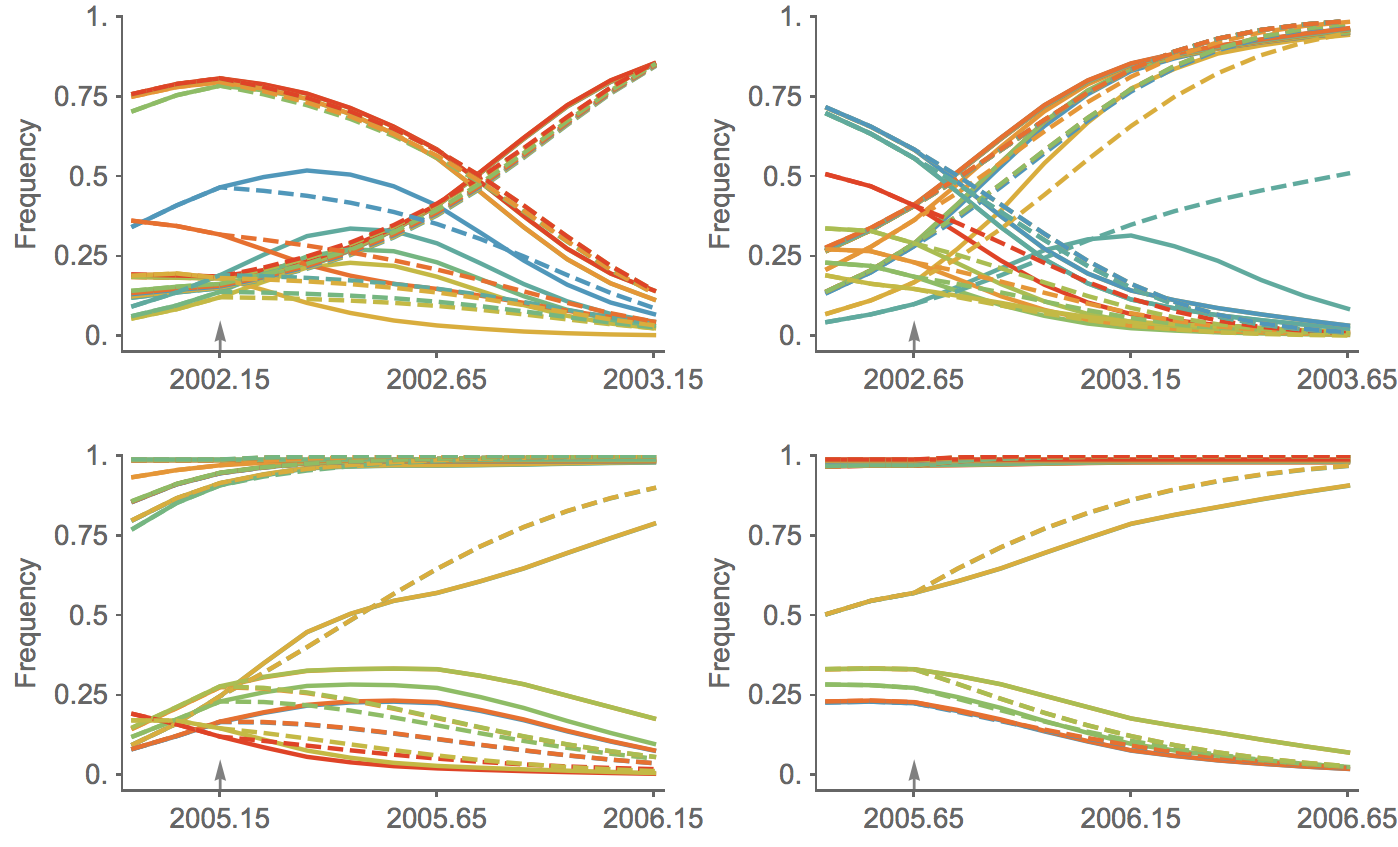
Trajectories show more detailed congruence
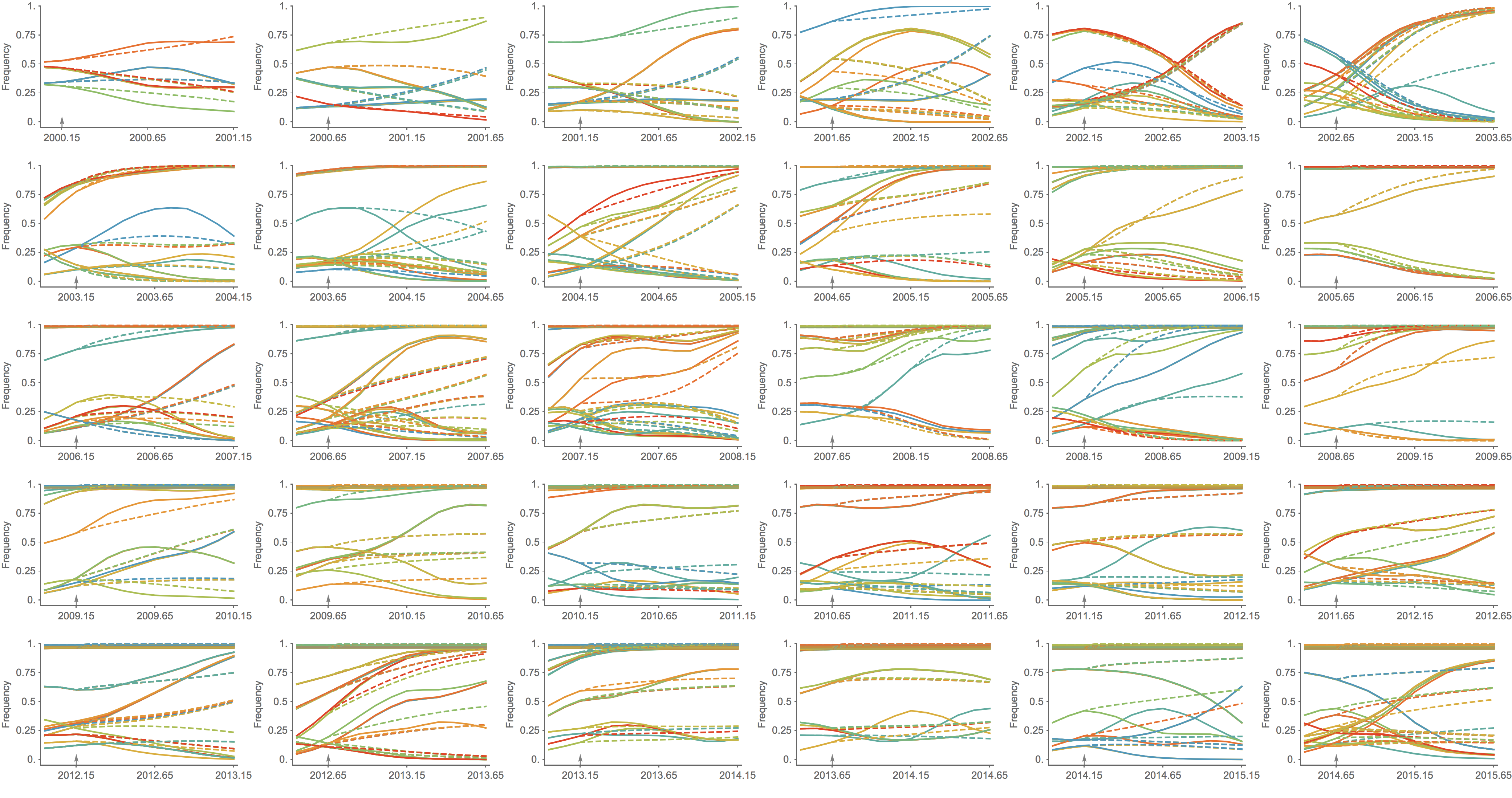
Trajectories show more detailed congruence
Working directly with CDC to provide analytics and the WHO to provide technical reports
Ebola
Virus genomes reveal factors that spread and sustained the Ebola epidemic
with ![]() Gytis Dudas, Andrew Rambaut, Luiz Carvalho, Marc Suchard, Philippe Lemey,
Gytis Dudas, Andrew Rambaut, Luiz Carvalho, Marc Suchard, Philippe Lemey,
and many others
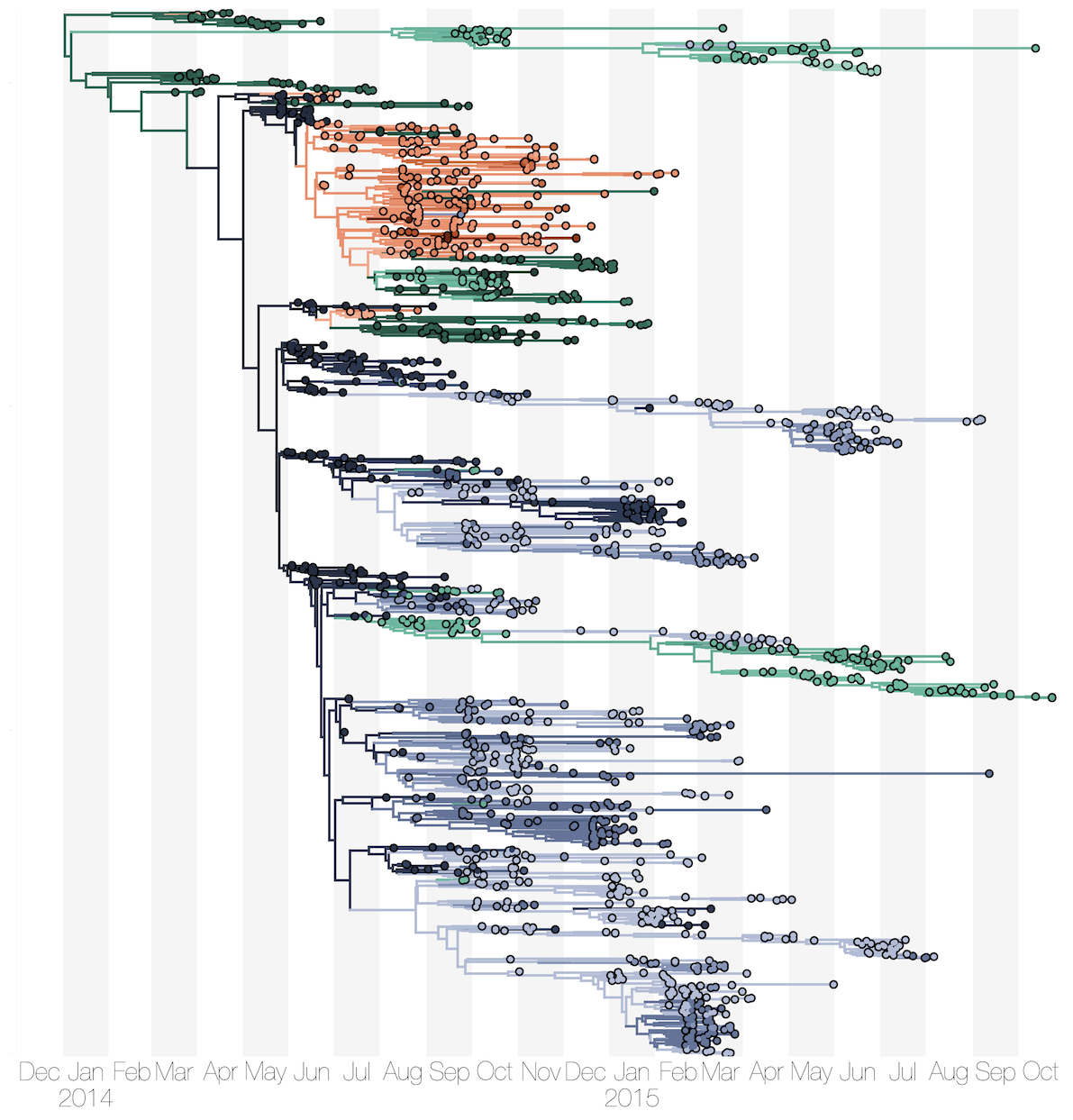
Sequencing of 1610 Ebola virus genomes collected during the 2013-2016 West African epidemic
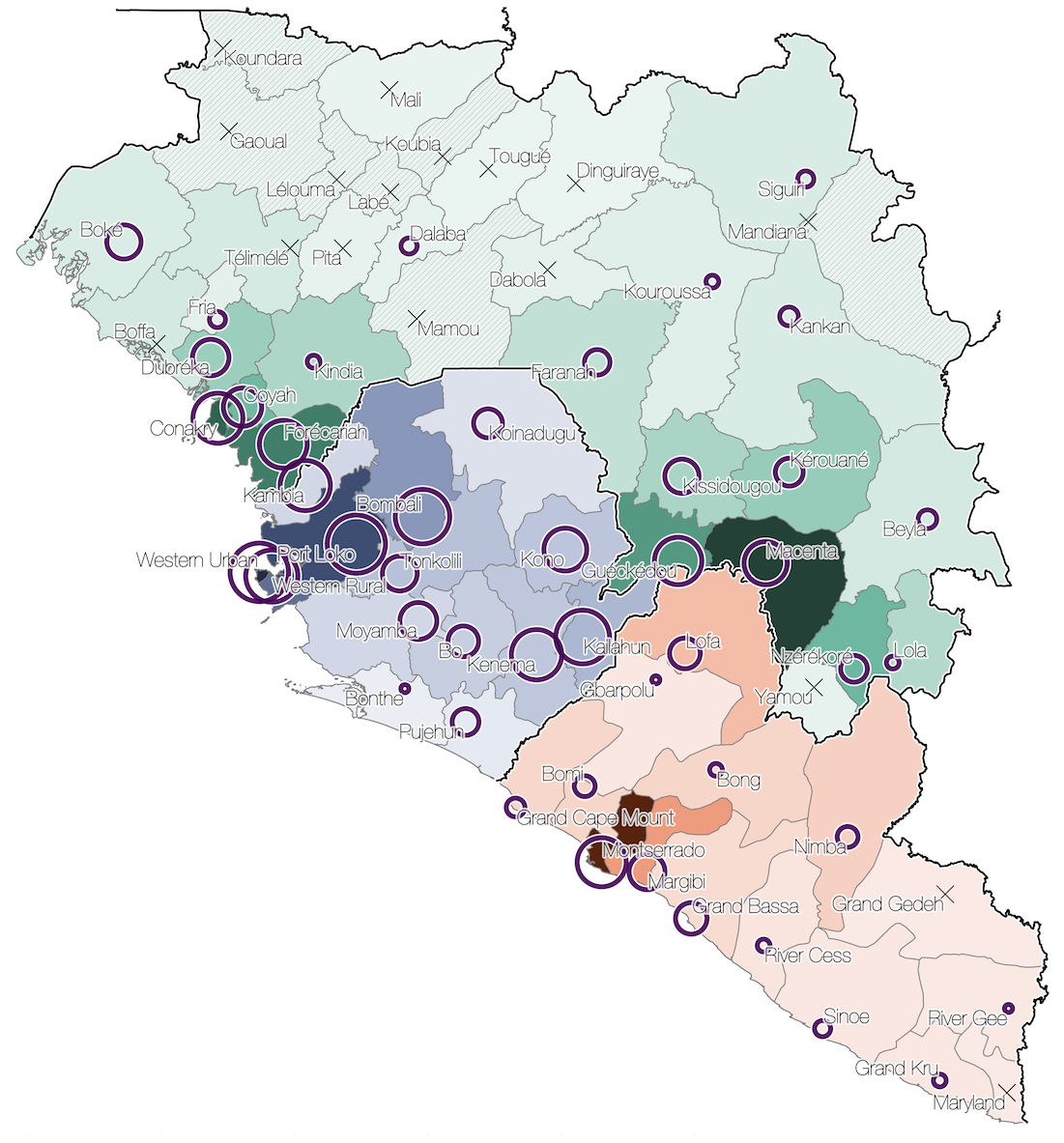
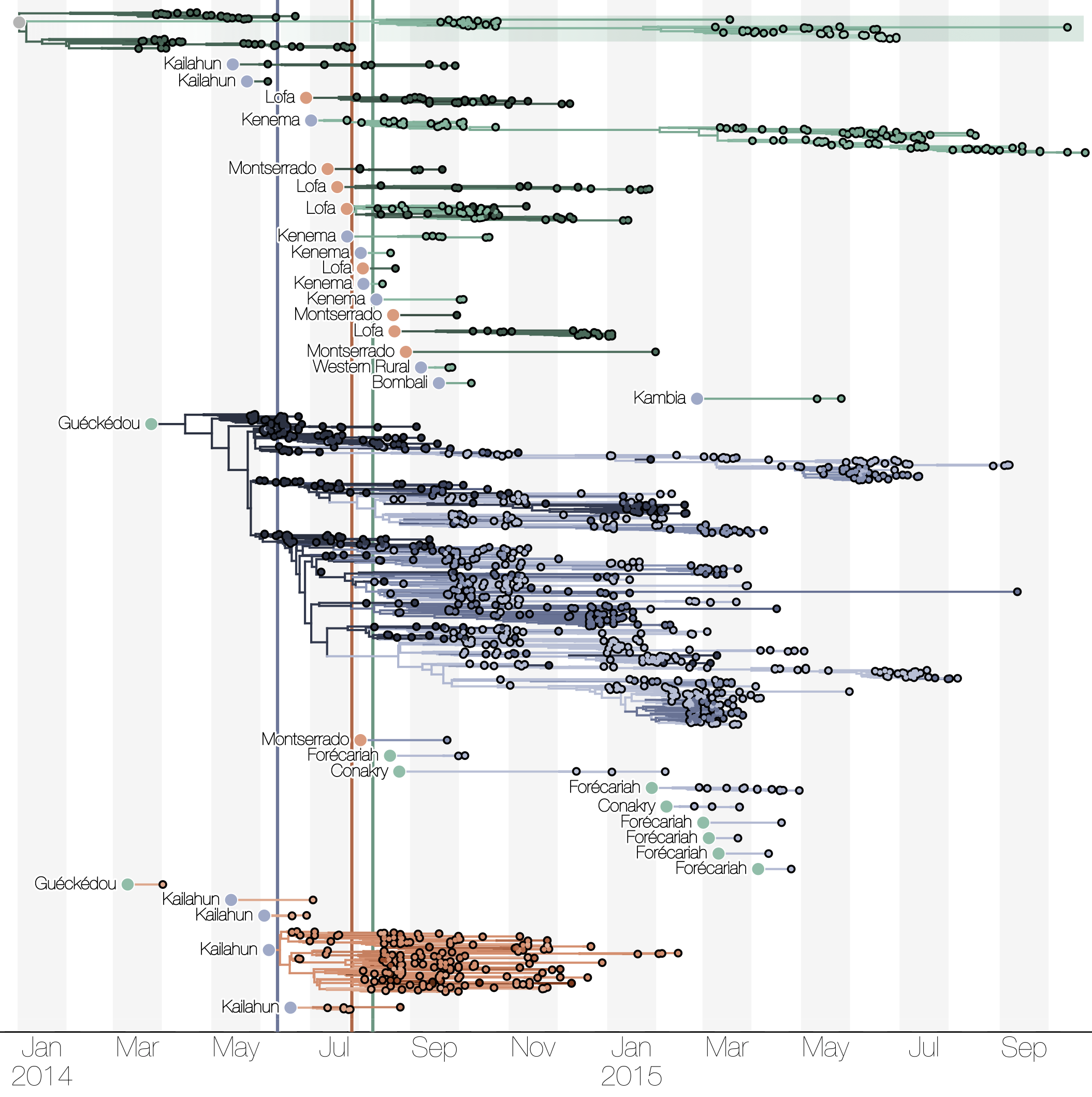
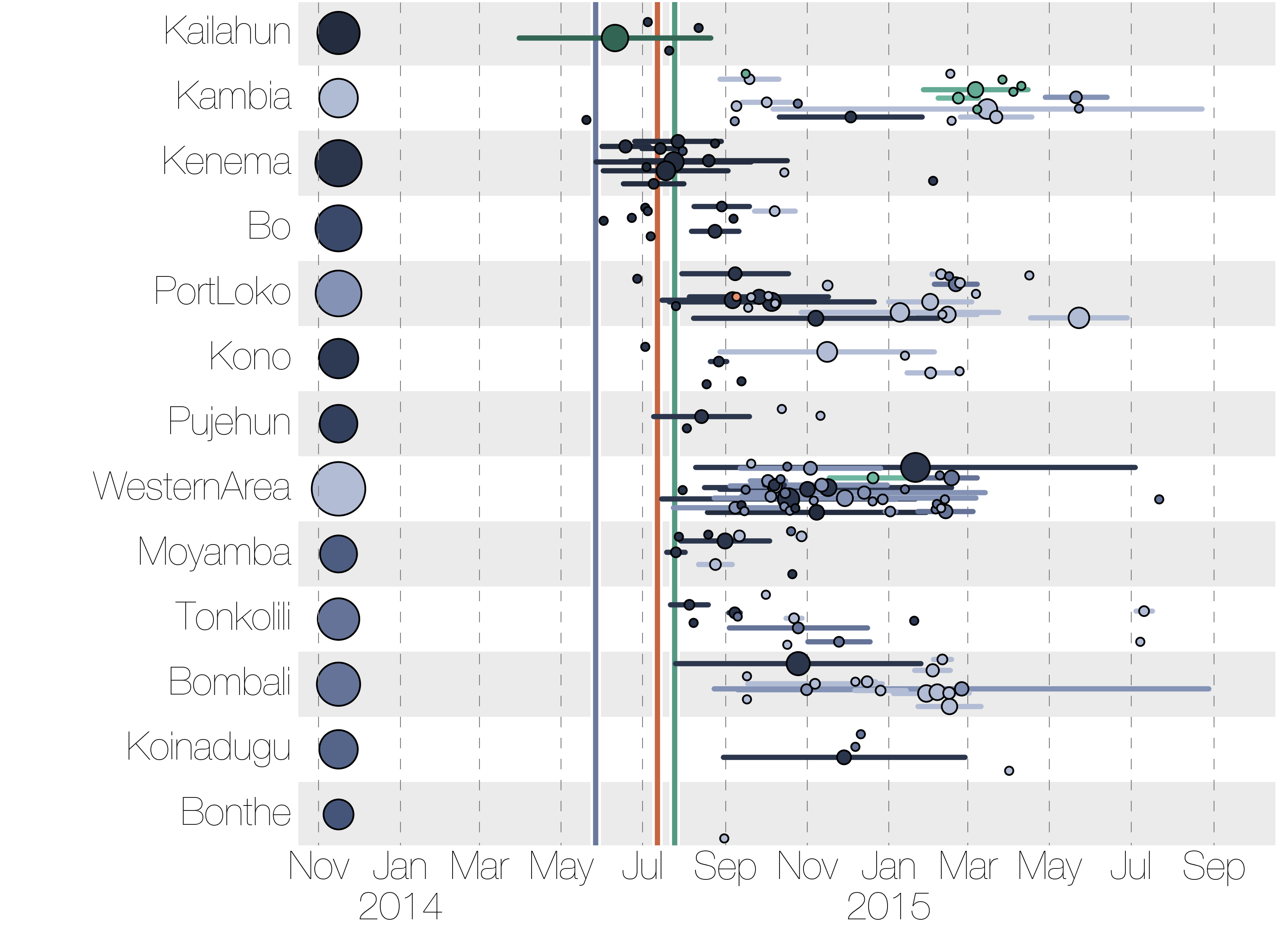
Phylogenetic reconstruction of evolution and spread
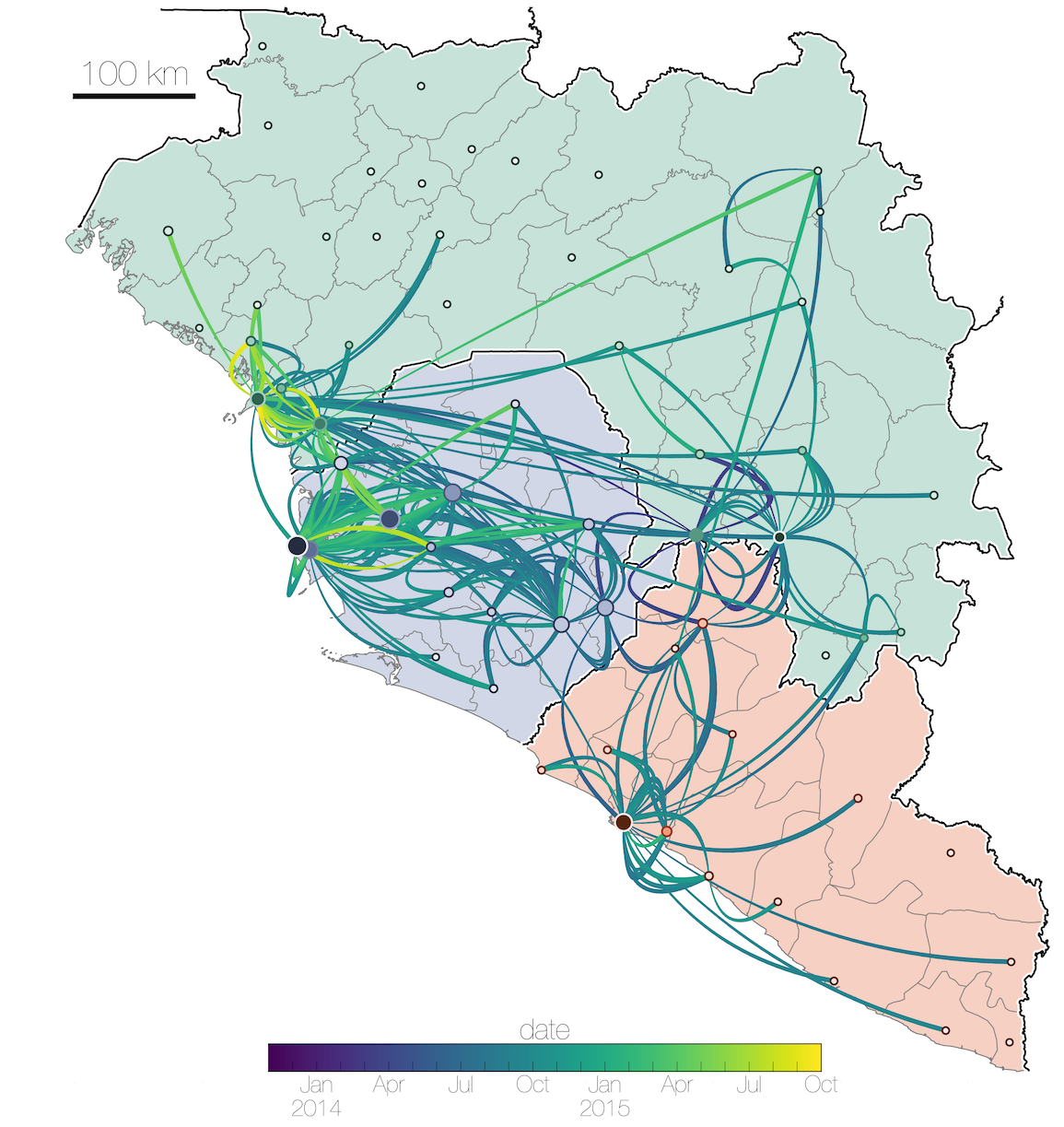
Tracking migration events
Factors influencing migration rates
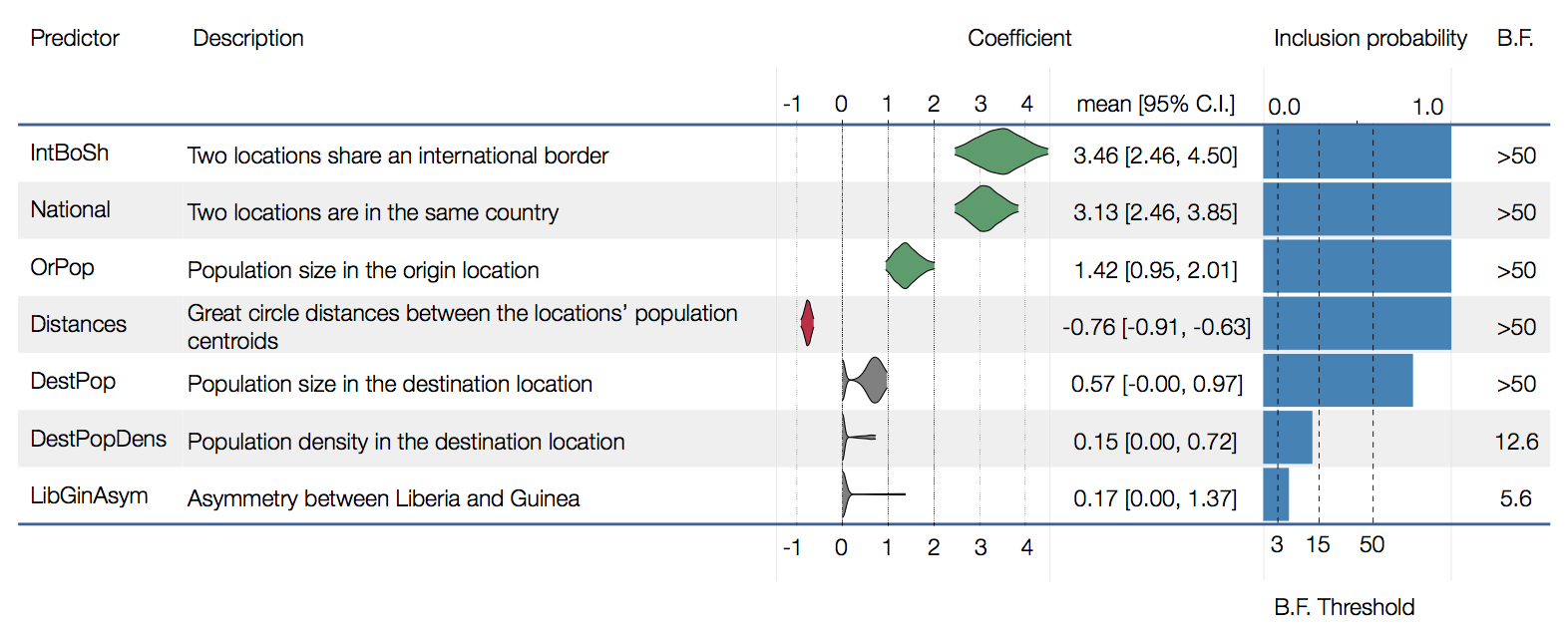
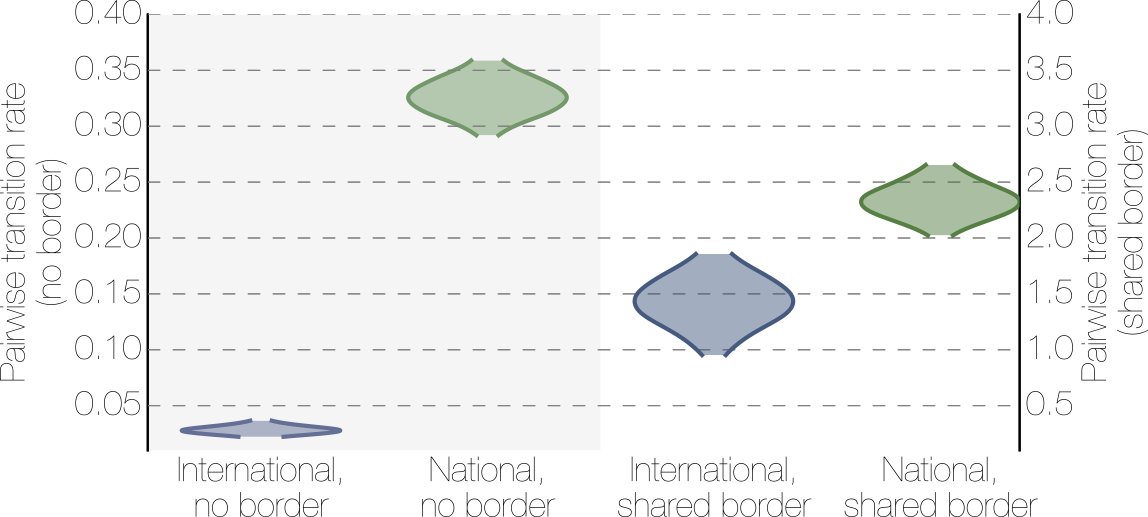
Effect of borders on migration rates
Spatial structure at the country level
Substantial mixing at the regional level
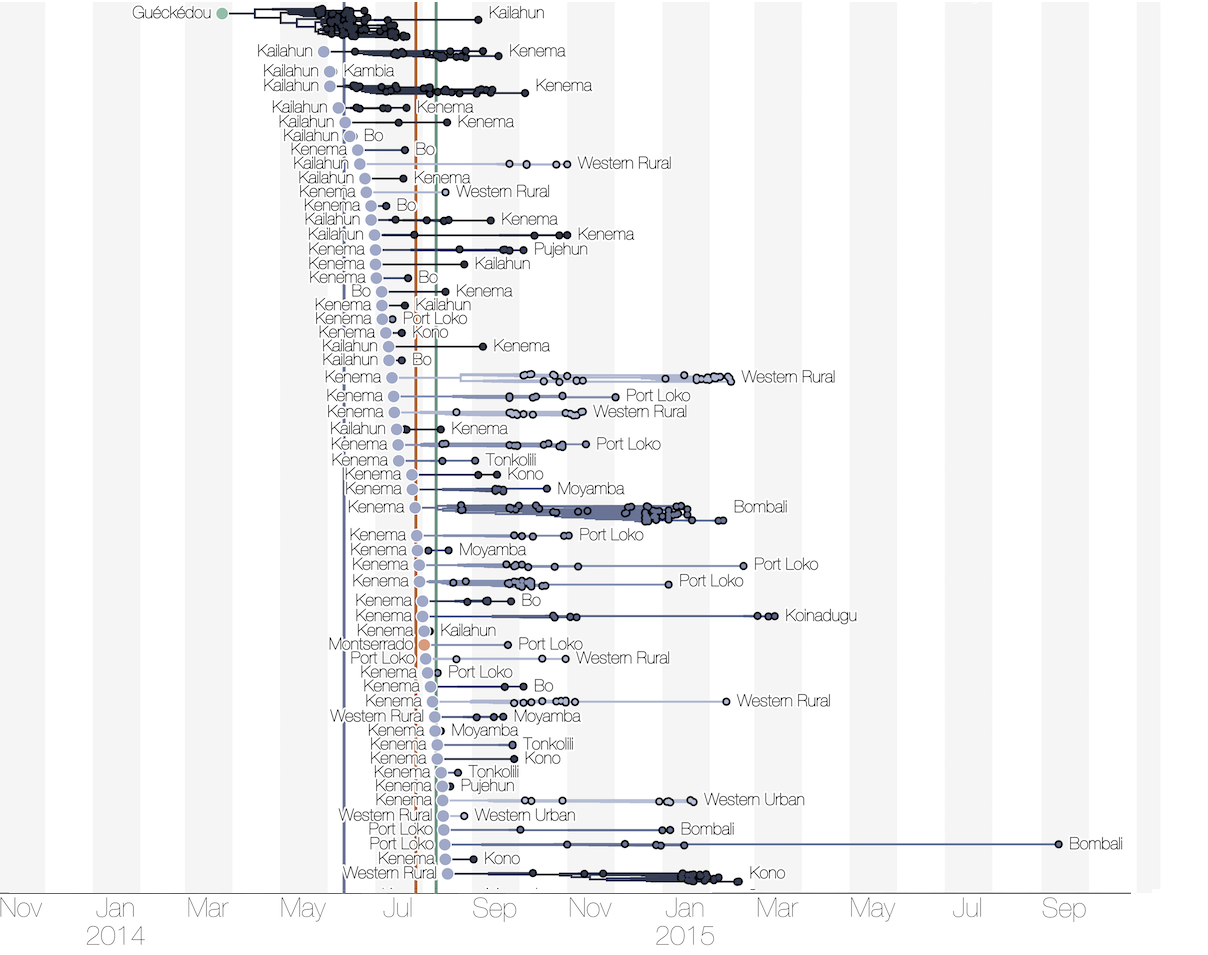
Regional outbreaks due to multiple introductions
Each introduction results in a minor outbreak
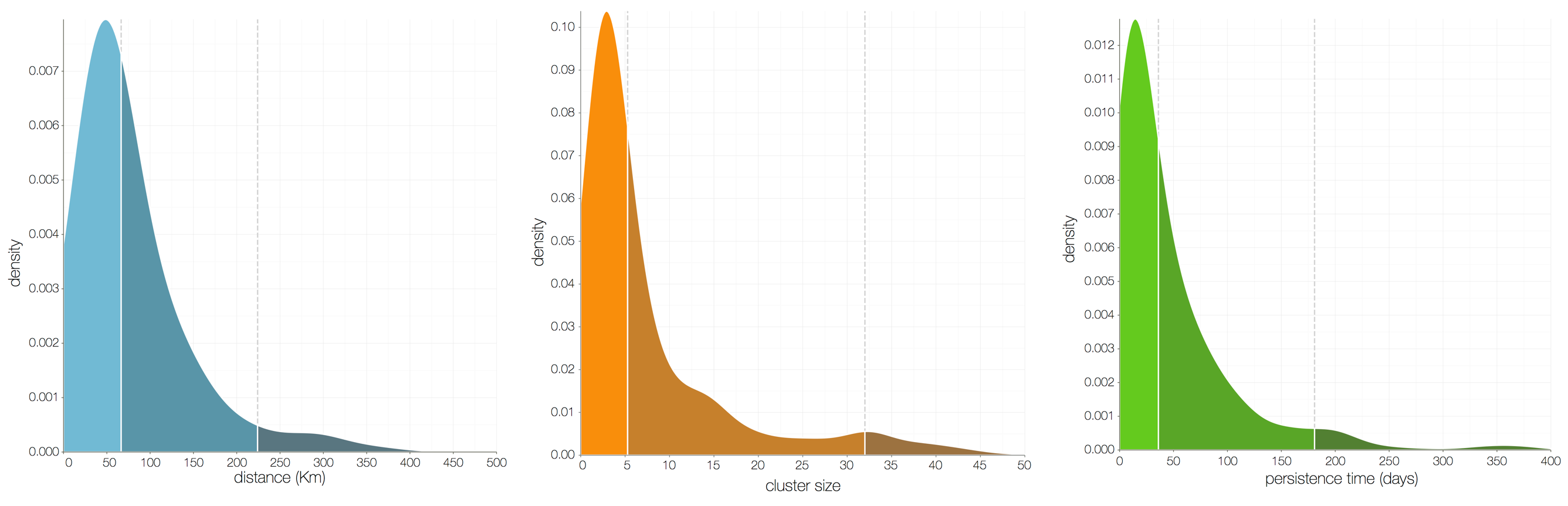
Ebola spread in West Africa followed a gravity model with moderate slowing by international borders, in which spread is driven by short-lived migratory clusters
Zika
Zika's arrival and spread in the Americas
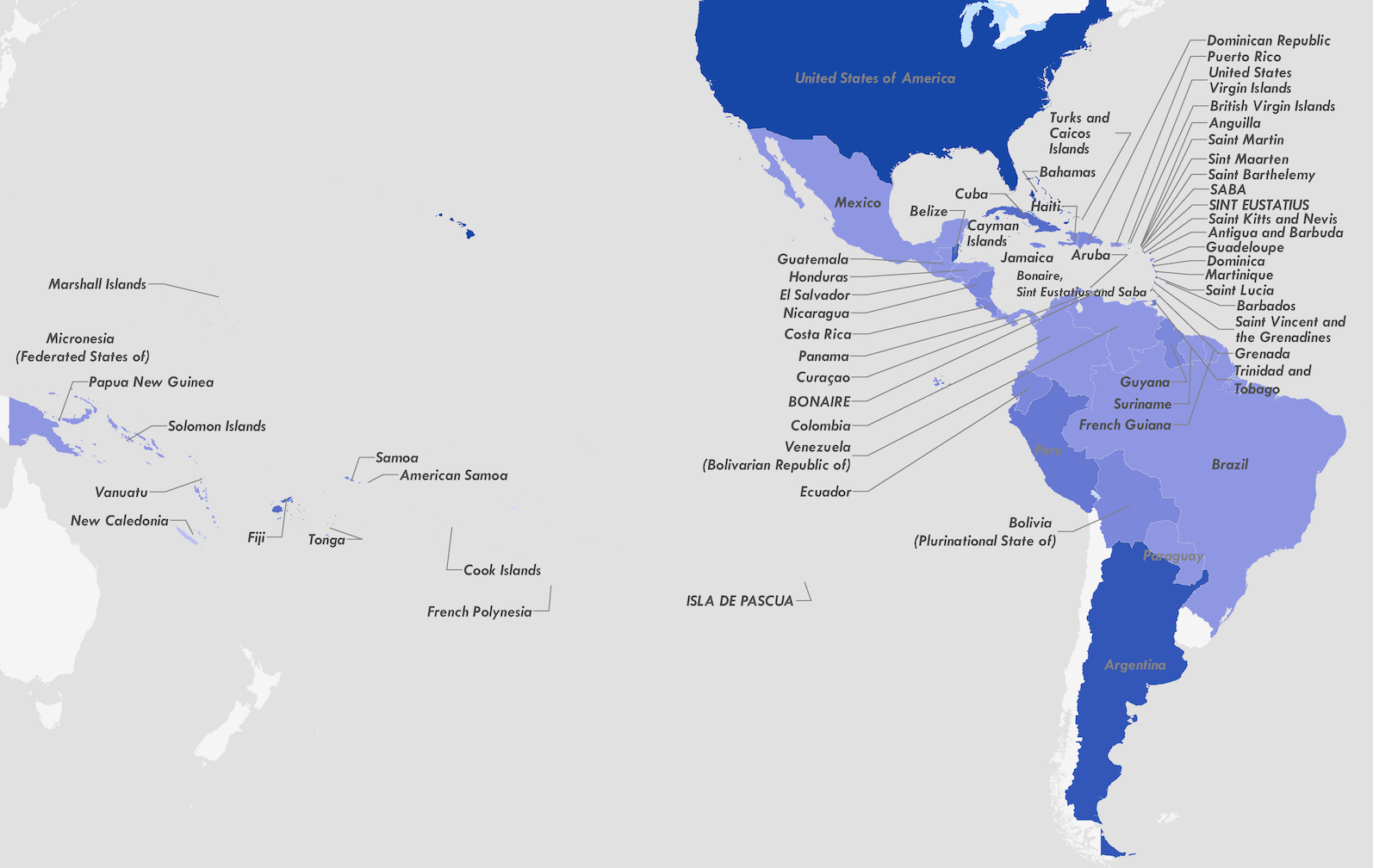
Establishment and cryptic transmission of Zika virus in Brazil and the Americas
with Nuno Faria, Nick Loman, Oli Pybus, Luiz Alcantara, Ester Sabino, Josh Quick,
![]() Alli Black,
Ingra Morales, Julien Thézé, Marcio Nunes, Jacqueline de Jesus,
Alli Black,
Ingra Morales, Julien Thézé, Marcio Nunes, Jacqueline de Jesus,
Marta Giovanetti, Moritz Kraemer, Sarah Hill and many others

Road trip through northeast Brazil to collect samples and sequence
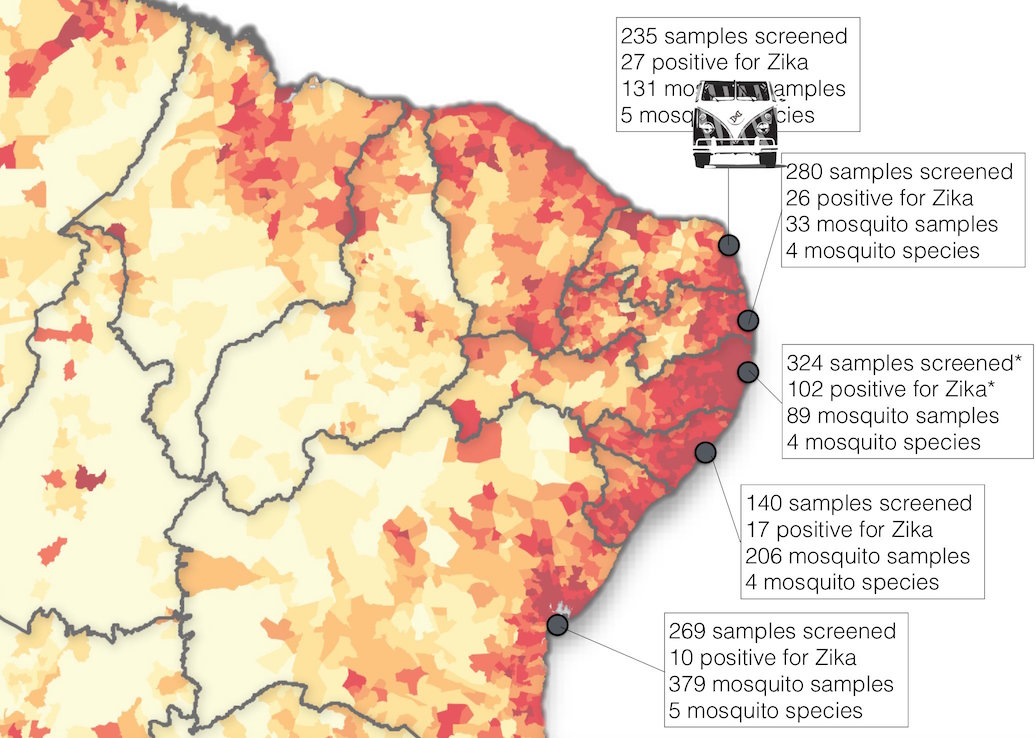
Case reports and diagnostics suggest initiation in northeast Brazil
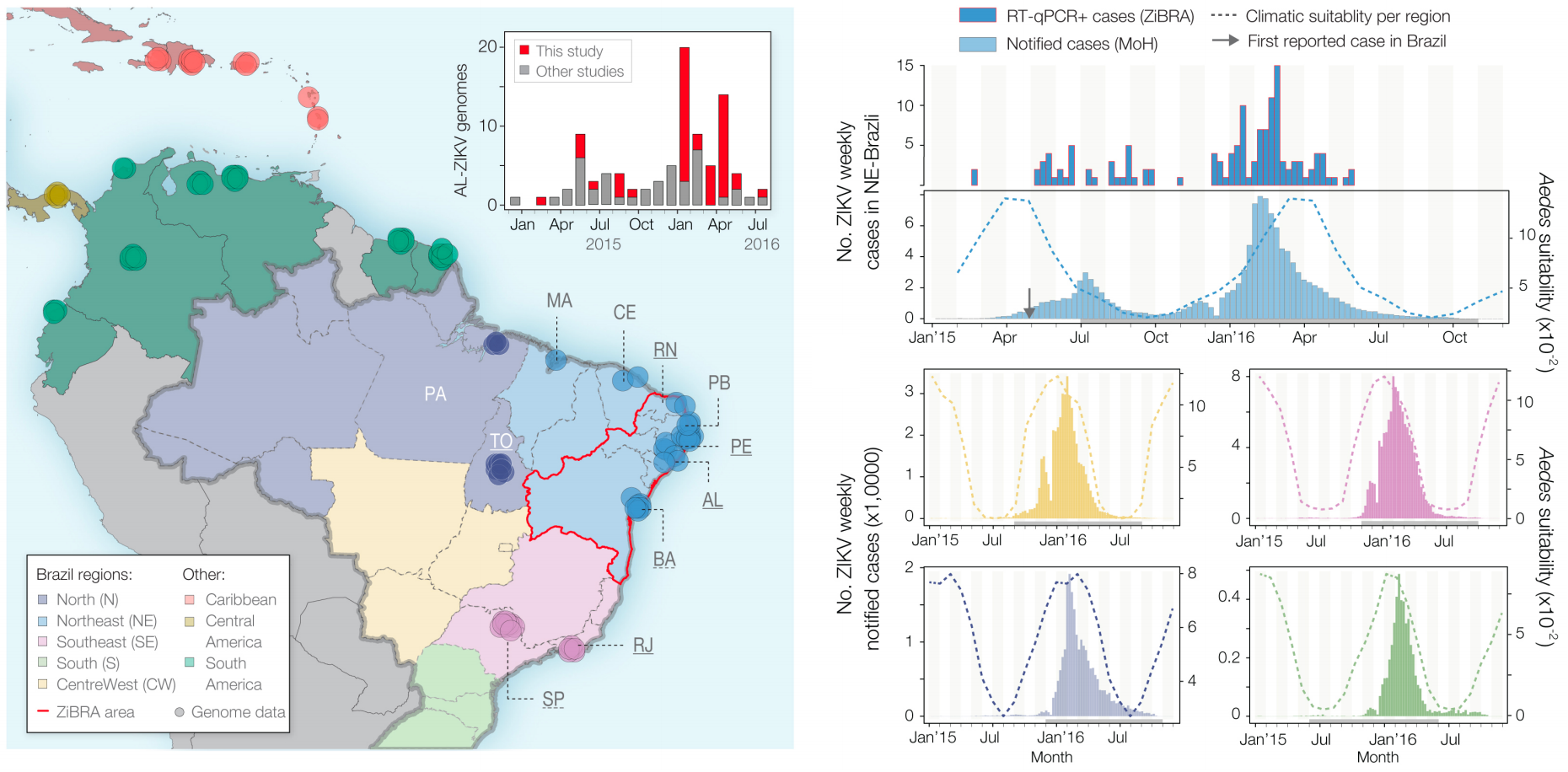
Phylogeny infers an origin in northeast Brazil
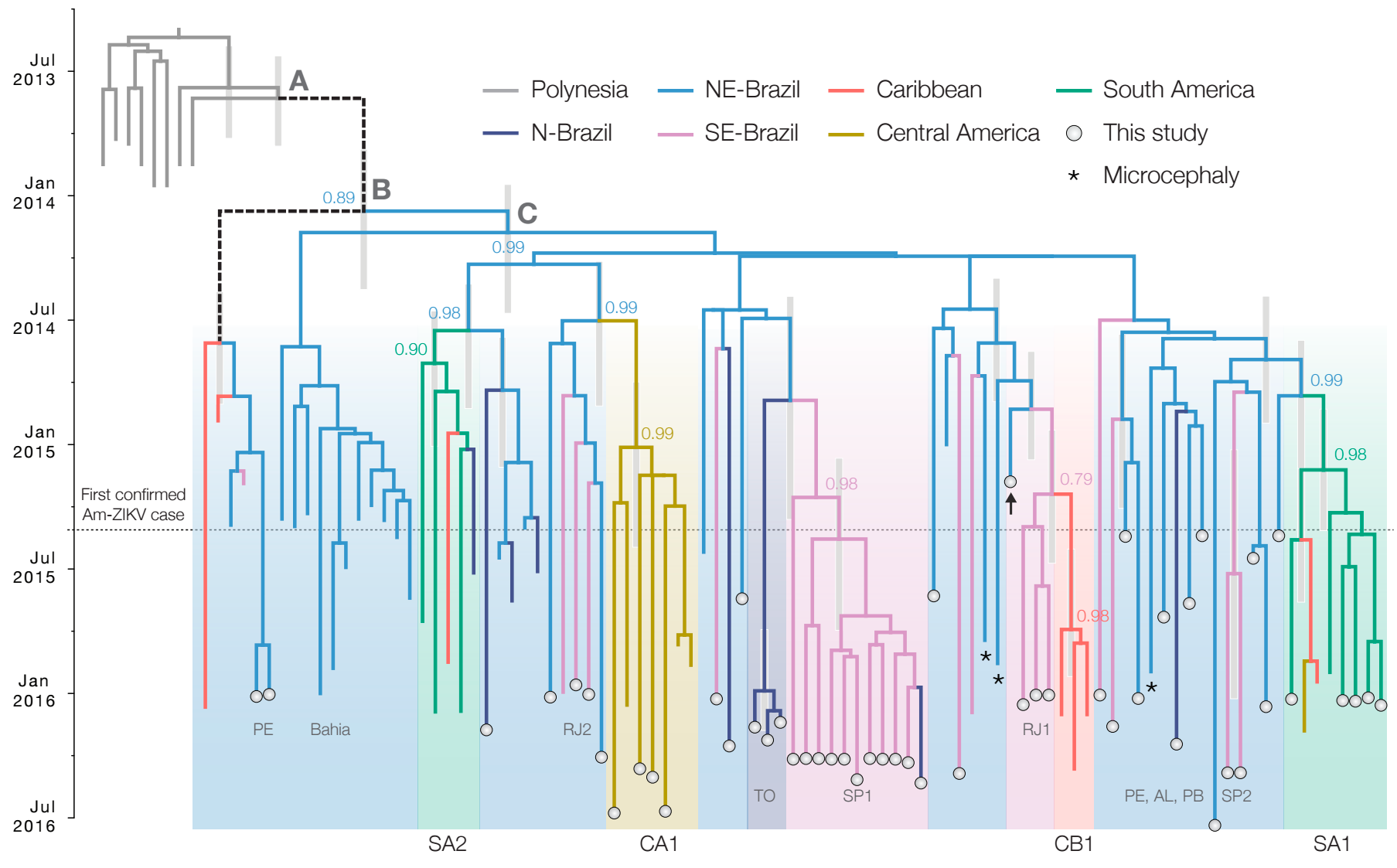
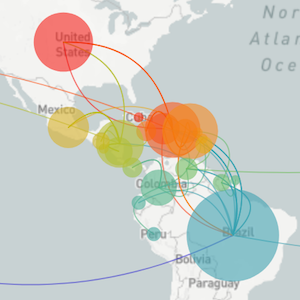
Genomic epidemiology reveals multiple introductions of Zika virus into the United States
with Nathan Grubaugh, Kristian Andersen, Jason Ladner, Gustavo Palacios, Sharon Isern, Oli Pybus,
Moritz Kraemer, ![]() Gytis Dudas,
Amanda Tan, Karthik Gangavarapu, Michael Wiley, Stephen White,
Julien Thézé, Scott Michael, Leah Gillis, Pardis Sabeti, and many others
Gytis Dudas,
Amanda Tan, Karthik Gangavarapu, Michael Wiley, Stephen White,
Julien Thézé, Scott Michael, Leah Gillis, Pardis Sabeti, and many others
Outbreak of locally-acquired infections focused in Miami-Dade county
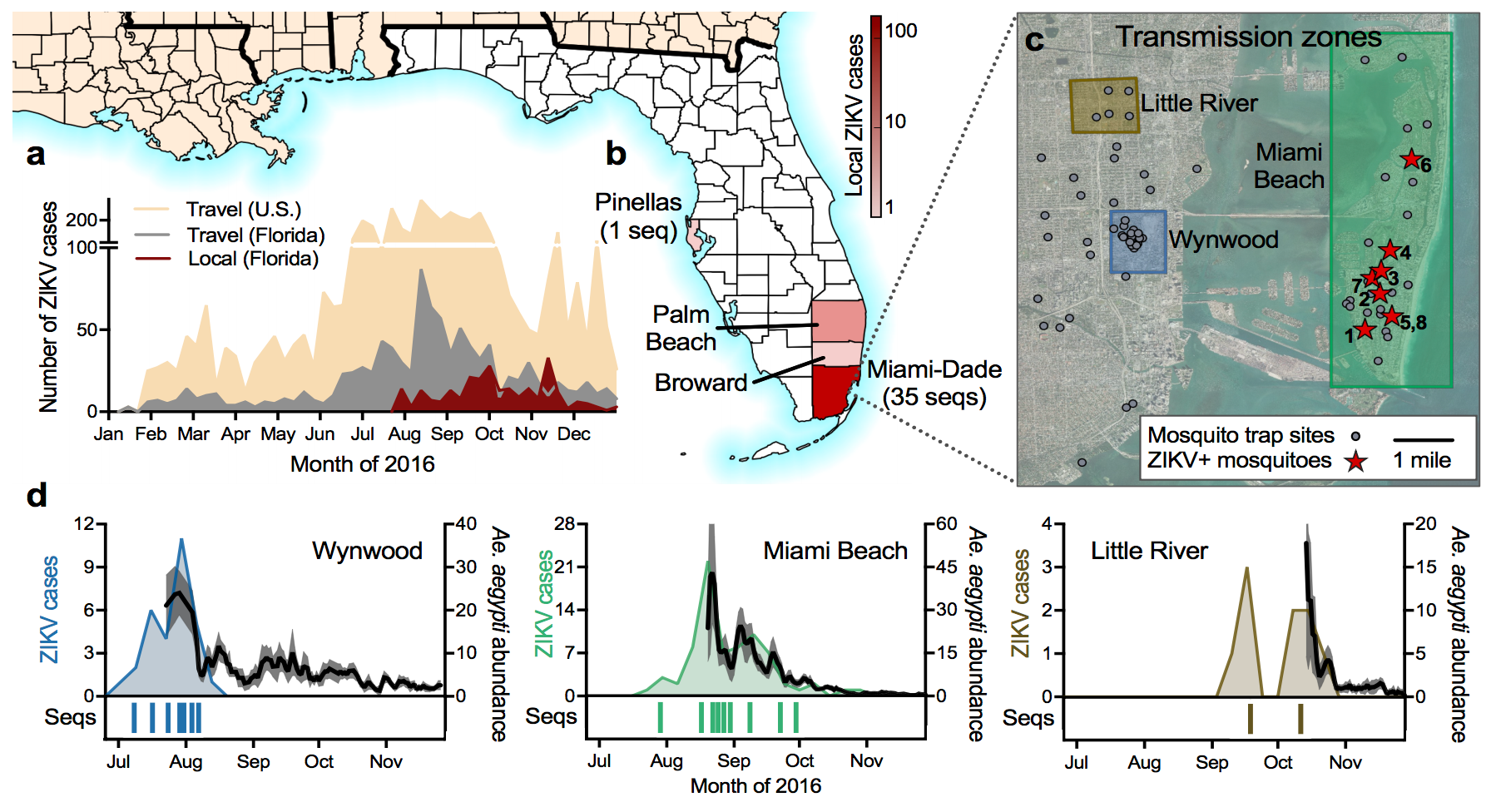
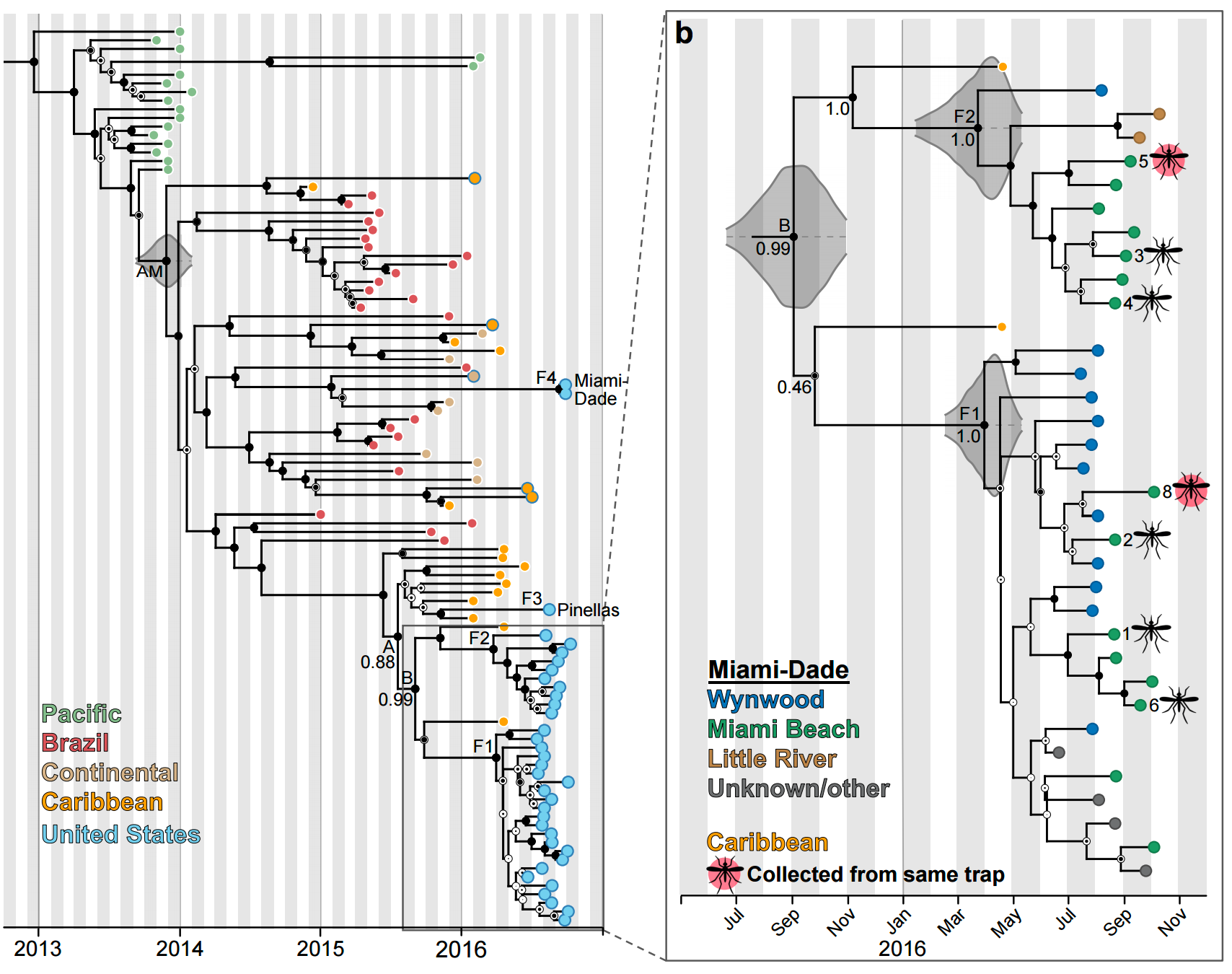
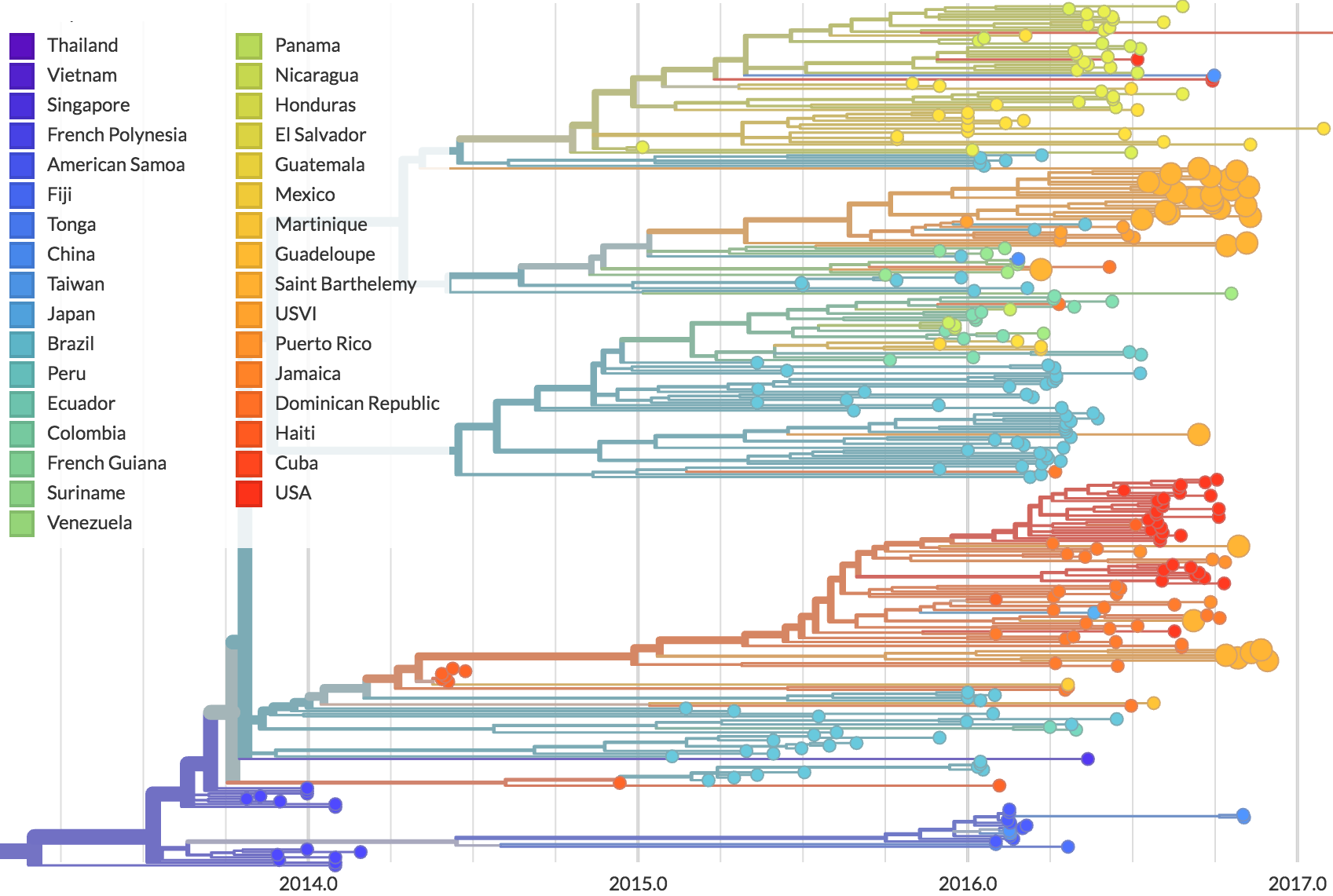
Phylogeny shows introductions from the Caribbean and a surprising degree of clustering
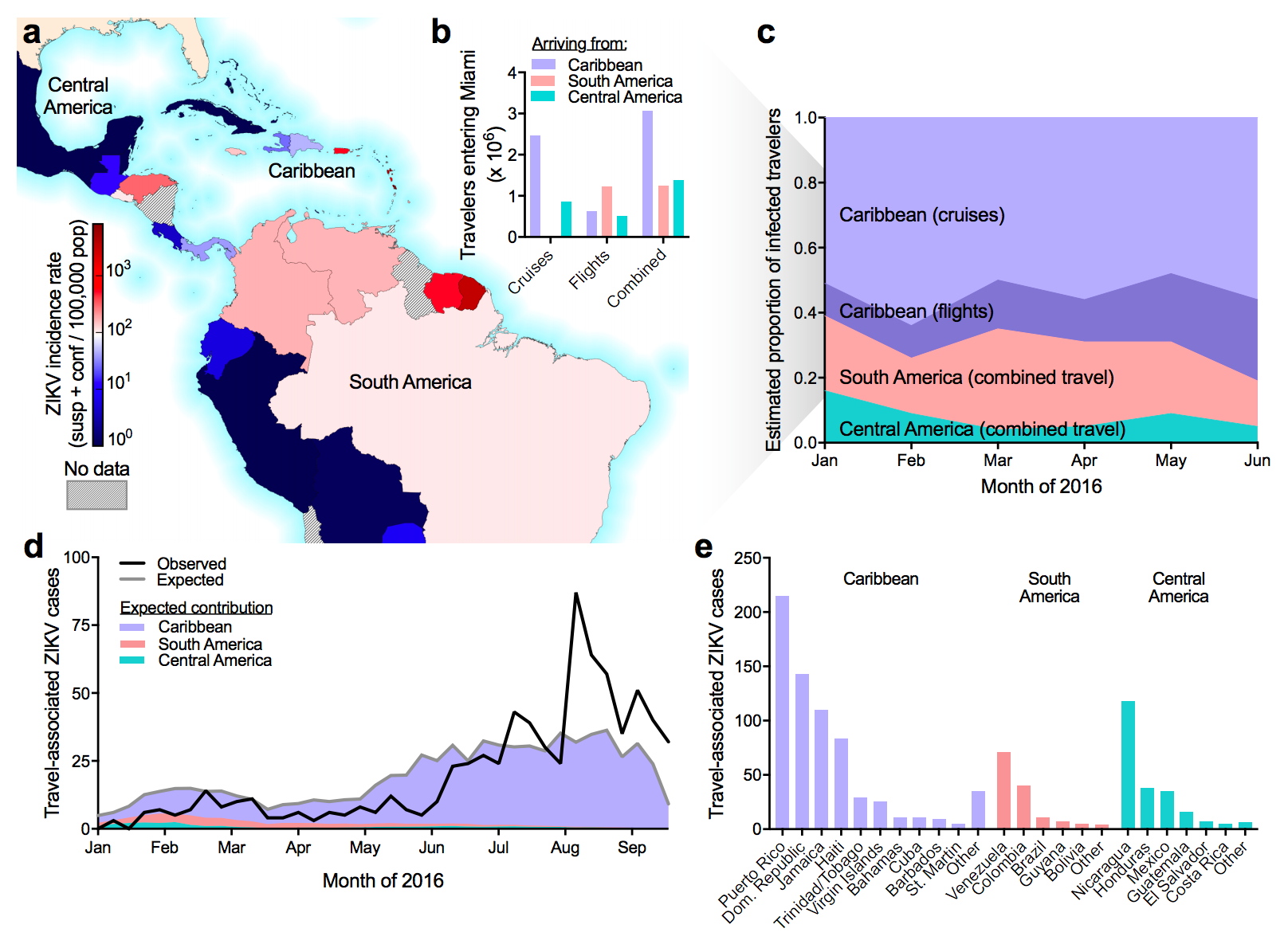
Flow of infected travelers greatest from Caribbean
Clustering suggests fewer, longer transmission chains and higher R0
Genomic epidemiology of Zika in the US Virgin Islands
with ![]() Alli Black,
Alli Black,
![]() Barney Potter,
Barney Potter,
![]() Gytis Dudas,
Esther Ellis, Brett Ellis,
Gytis Dudas,
Esther Ellis, Brett Ellis,
Kristian Andersen, Nathan Grubaugh, Leora Feldstein, and others
(and special thanks to Adam Geballe)
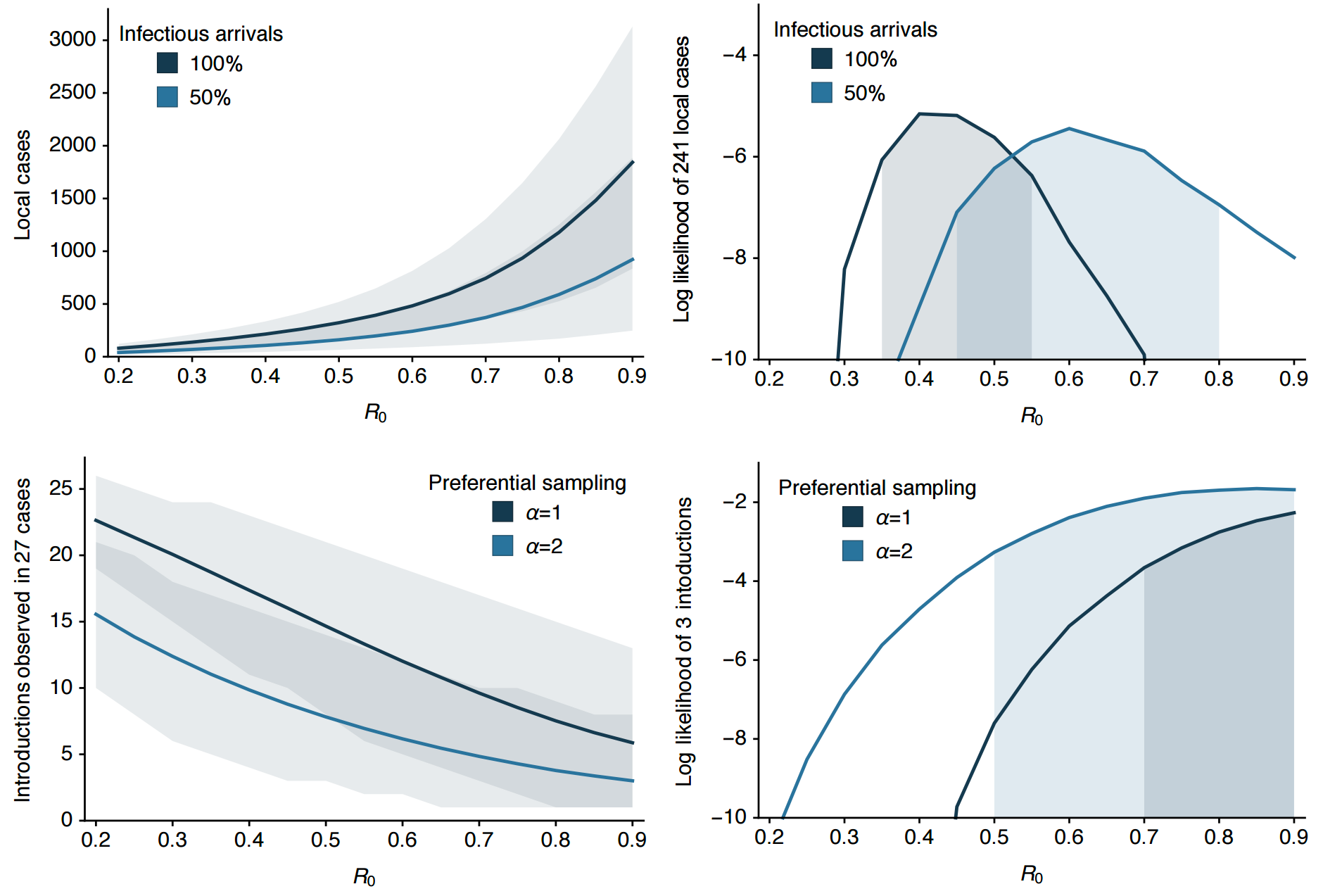
Preliminary analysis of 31 genomes shows multiple introductions to USVI
Important analyses, let's make them more rapid and more automated
Key challenges
- Timely analysis and sharing of results critical
- Dissemination must be scalable
- Integrate many data sources
- Results must be easily interpretable and queryable
nextstrain
Project to conduct real-time molecular epidemiology and evolutionary analysis of emerging epidemics
with
![]() Richard Neher,
Richard Neher,
![]() James Hadfield,
James Hadfield,
![]() Colin Megill,
Colin Megill,
![]() Sidney Bell,
Sidney Bell,
![]() Charlton Callender,
Charlton Callender,
![]() Barney Potter,
Barney Potter,
and ![]() John Huddleston
John Huddleston
Nextstrain architecture
All code open source at github.com/nextstrain
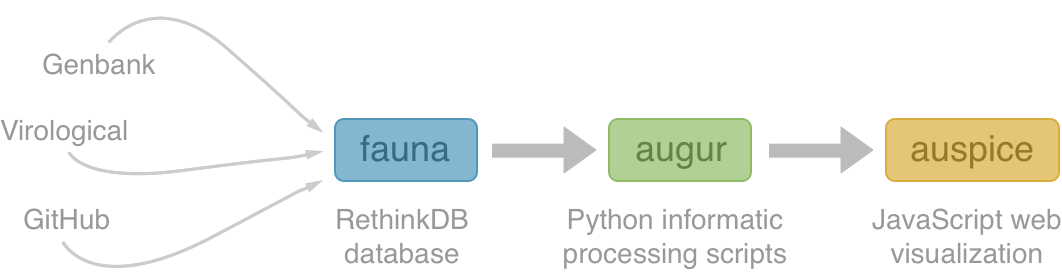
Fauna
Rethink database of virus and titer data
- Harmonizes data from different sources
- Integrates different types of data (serology, sequences, case details)
- Provides an interface for downstream analysis
Augur
Build scripts to align sequences, build trees and annotate
- Flexible build scripts to incorporate different viruses and analyses
- Constructs time-resolved phylogenies
- Annotates with geographic transitions and mutation events
Example augur pipeline for 1600 Ebola genomes
- Align with MAFFT (34 min)
- Build ML tree with RAxML (54 min)
- Temporally resolve tree and geographic ancestry with TreeTime (16 min)
- Total pipeline (1 hr 46 min)
Auspice
Web visualization of resulting trees
- Interactive data exploration and filtering
- Framework through React / D3
- Connects phylogeny, geography and genotypes
nextstrain.org
Rapid on-the-ground sequencing by Ian Goodfellow, Matt Cotten and colleagues
Build out pipelines for different pathogens,
improve databasing and lower
bioinformatics bar
Acknowledgements
Bedford Lab:
![]() Alli Black,
Alli Black,
![]() Sidney Bell,
Sidney Bell,
![]() Gytis Dudas,
Gytis Dudas,
![]() John Huddleston,
John Huddleston,
![]() Barney Potter,
Barney Potter,
![]() James Hadfield,
James Hadfield,
![]() Louise Moncla
Louise Moncla
Influenza: WHO Global Influenza Surveillance Network, GISAID, Richard Neher, Colin Russell, Andrew Rambaut, Marc Suchard, Philippe Lemey, Steven Riley
Ebola: Gytis Dudas, Andrew Rambaut, Luiz Carvalho, Philippe Lemey, Marc Suchard, Andrew Tatem
Zika: Nick Loman, Nuno Faria, Oli Pybus, Josh Quick, Kristian Andersen, Nathan Grubaugh, Jason Ladner, Gustavo Palacios, Sharon Isern, Gytis Dudas, Alli Black, Barney Potter, Esther Ellis
Nextstrain: Richard Neher, James Hadfield, Colin Megill, Sidney Bell, Charlton Callender, Barney Potter, John Huddleston
