Real-time tracking of virus evolution
Trevor Bedford (@trvrb)
29 Mar 2019
Population Biology, Ecology, and Evolution Seminar
Emory University
We work at the interface of virology, evolution and epidemiology
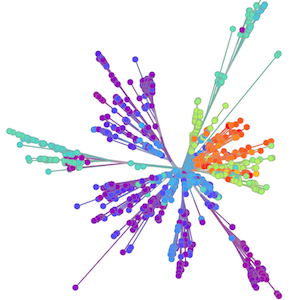
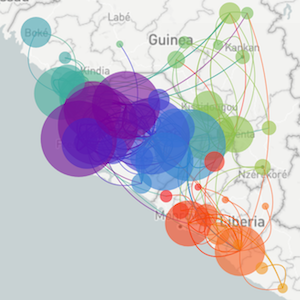

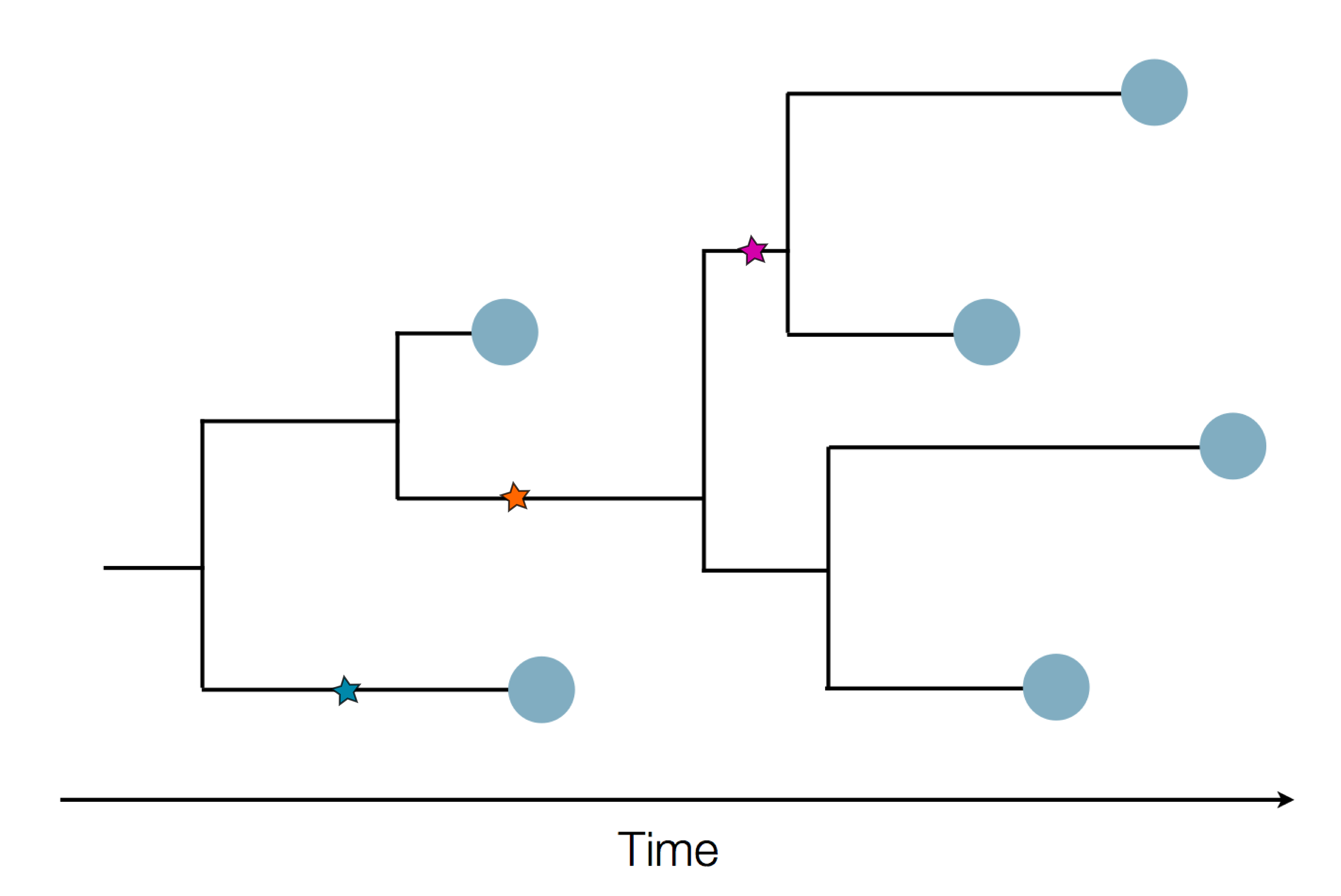
Sequencing to reconstruct pathogen spread
Epidemic process
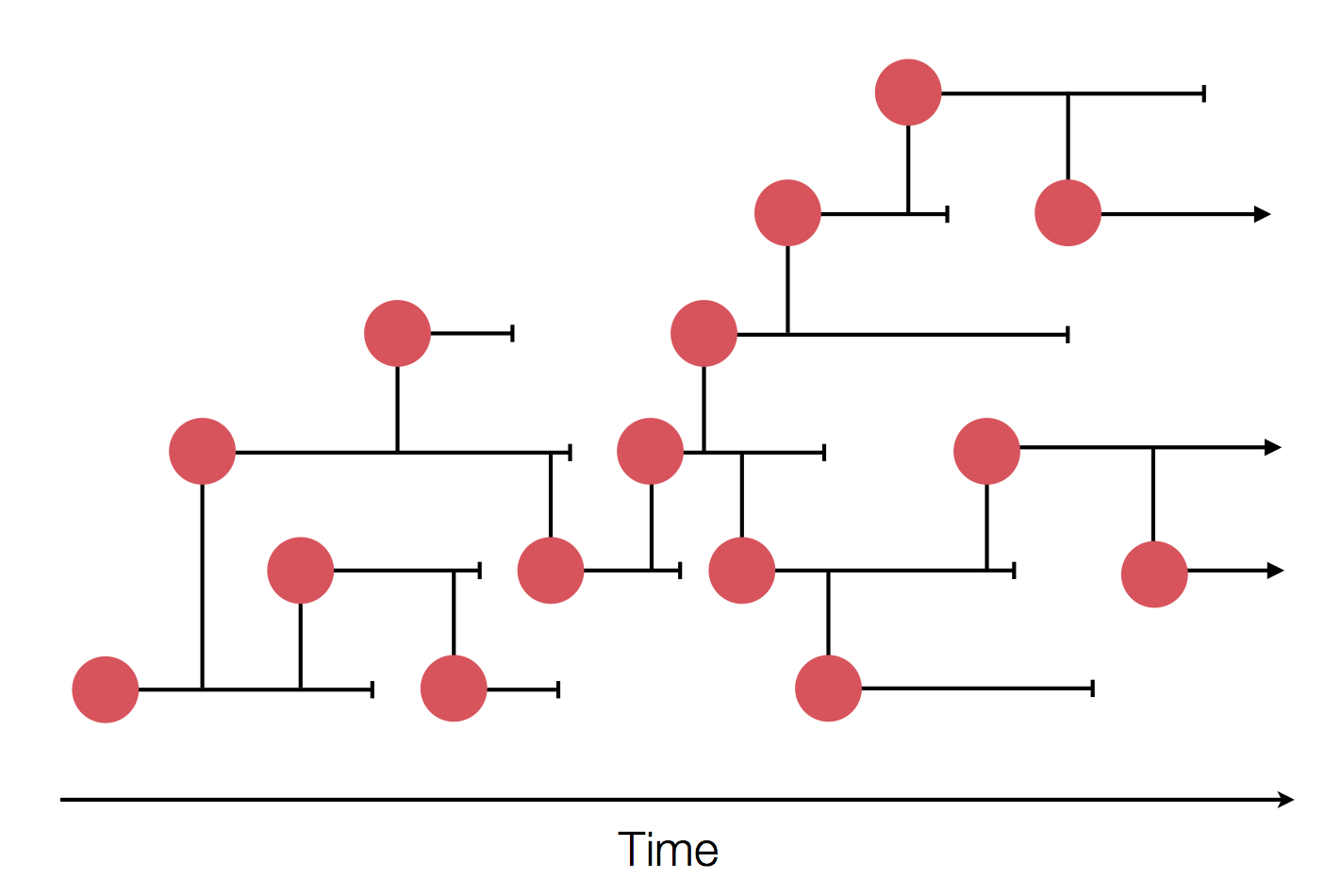
Sample some individuals
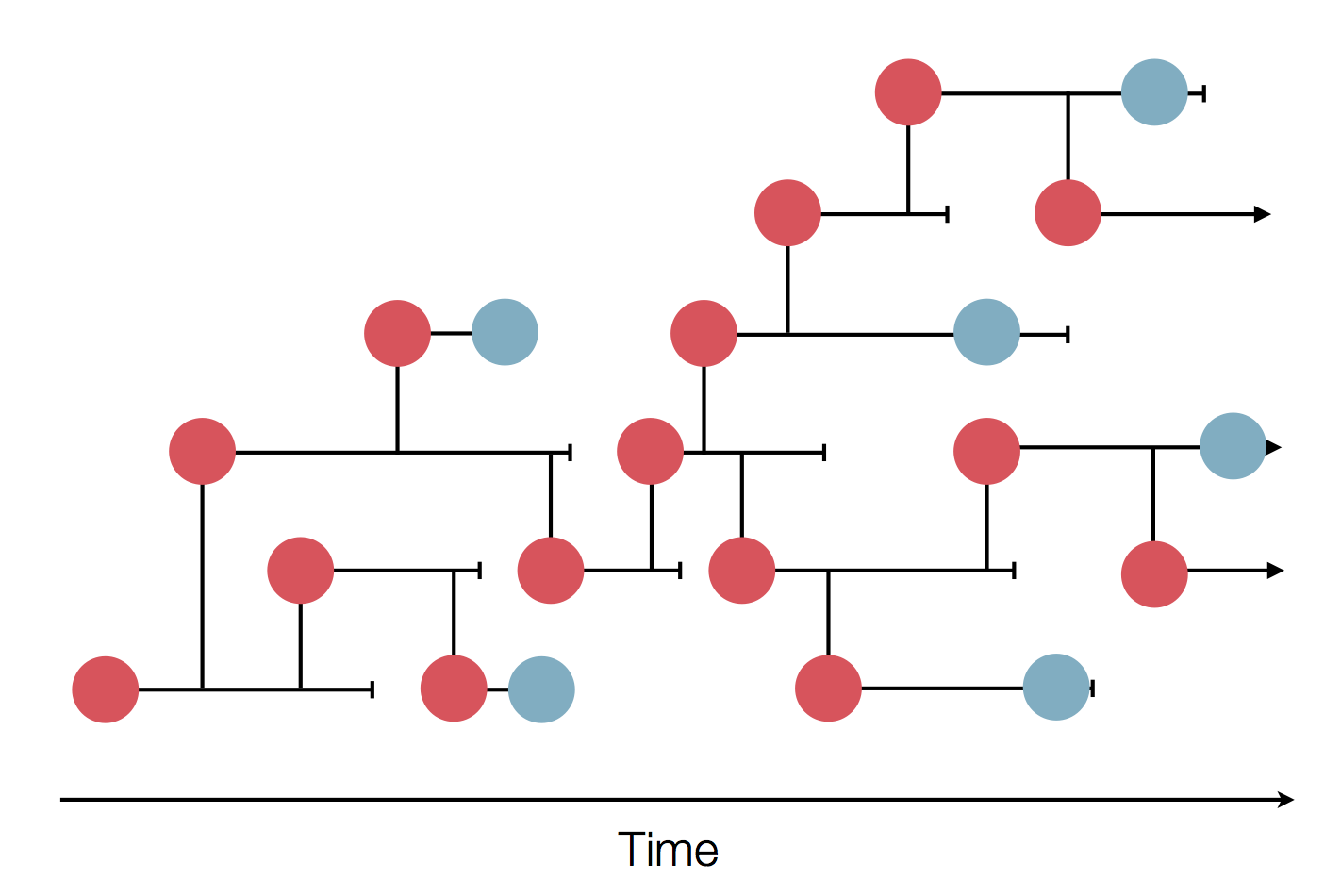
Sequence and determine phylogeny
Sequence and determine phylogeny
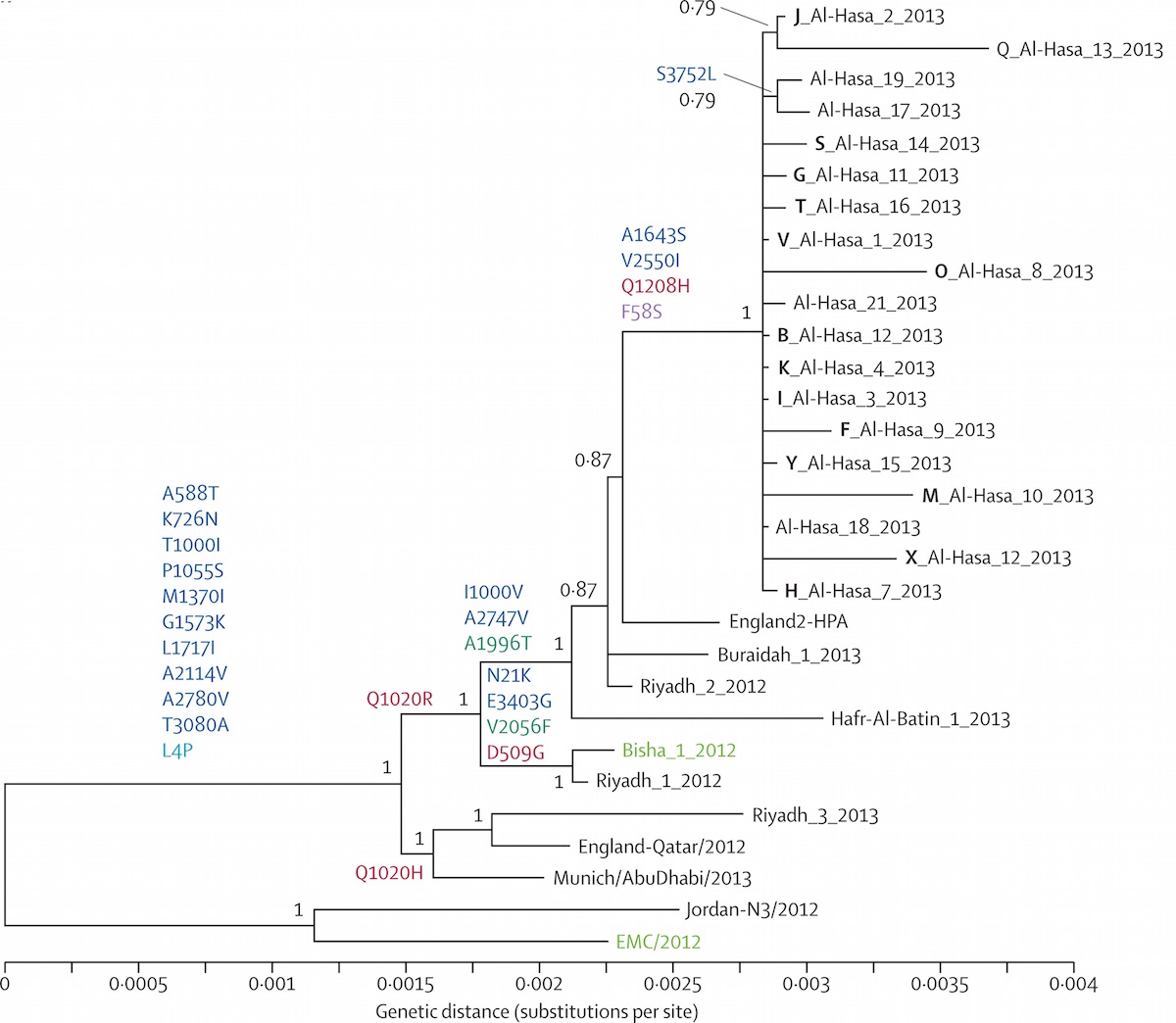
Localized Middle Eastern MERS-CoV phylogeny
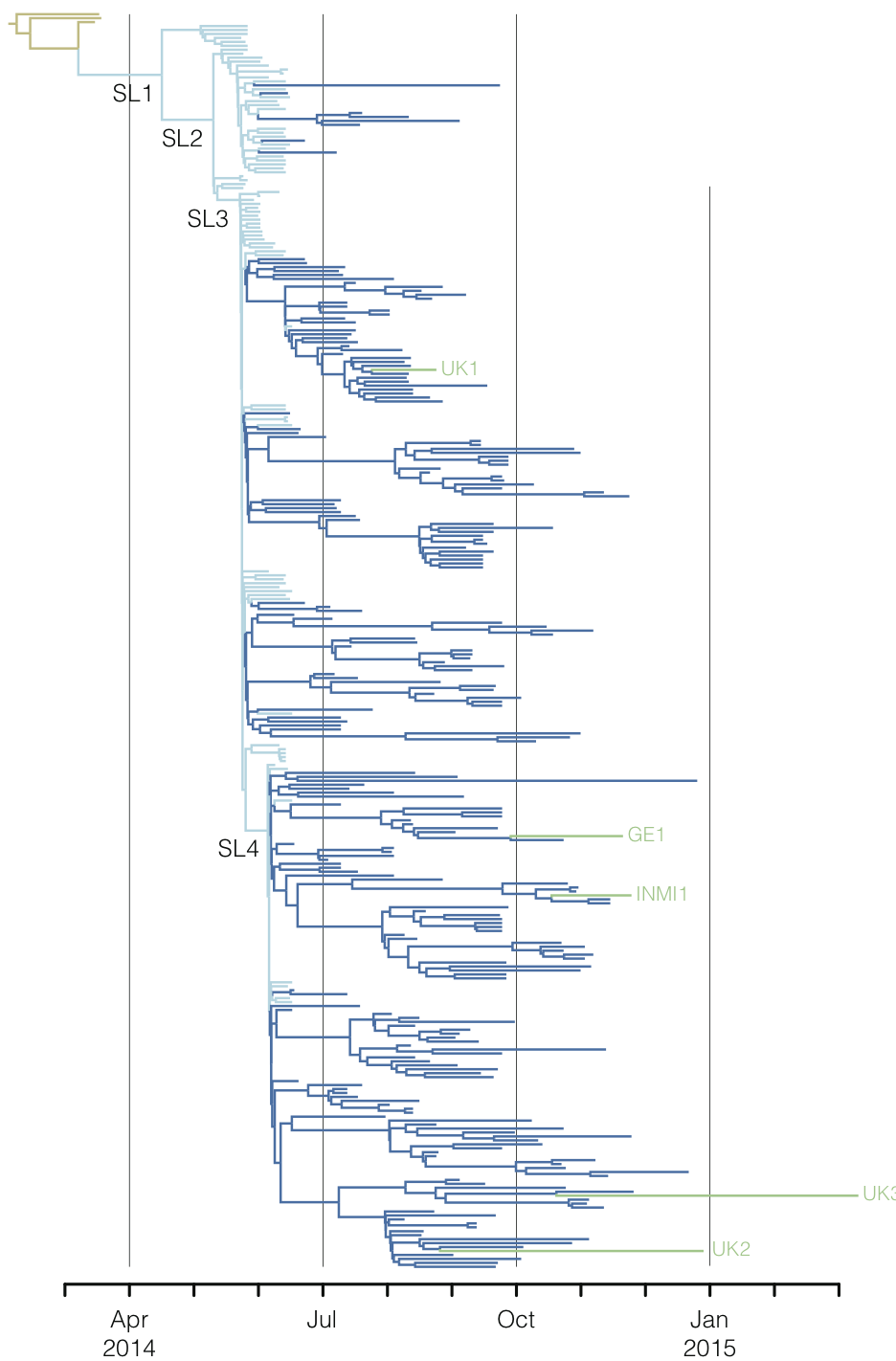
Regional West African Ebola phylogeny
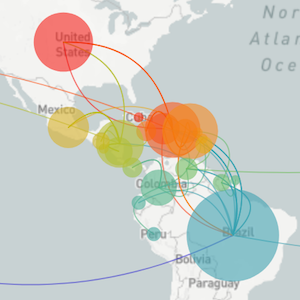
Global influenza phylogeny
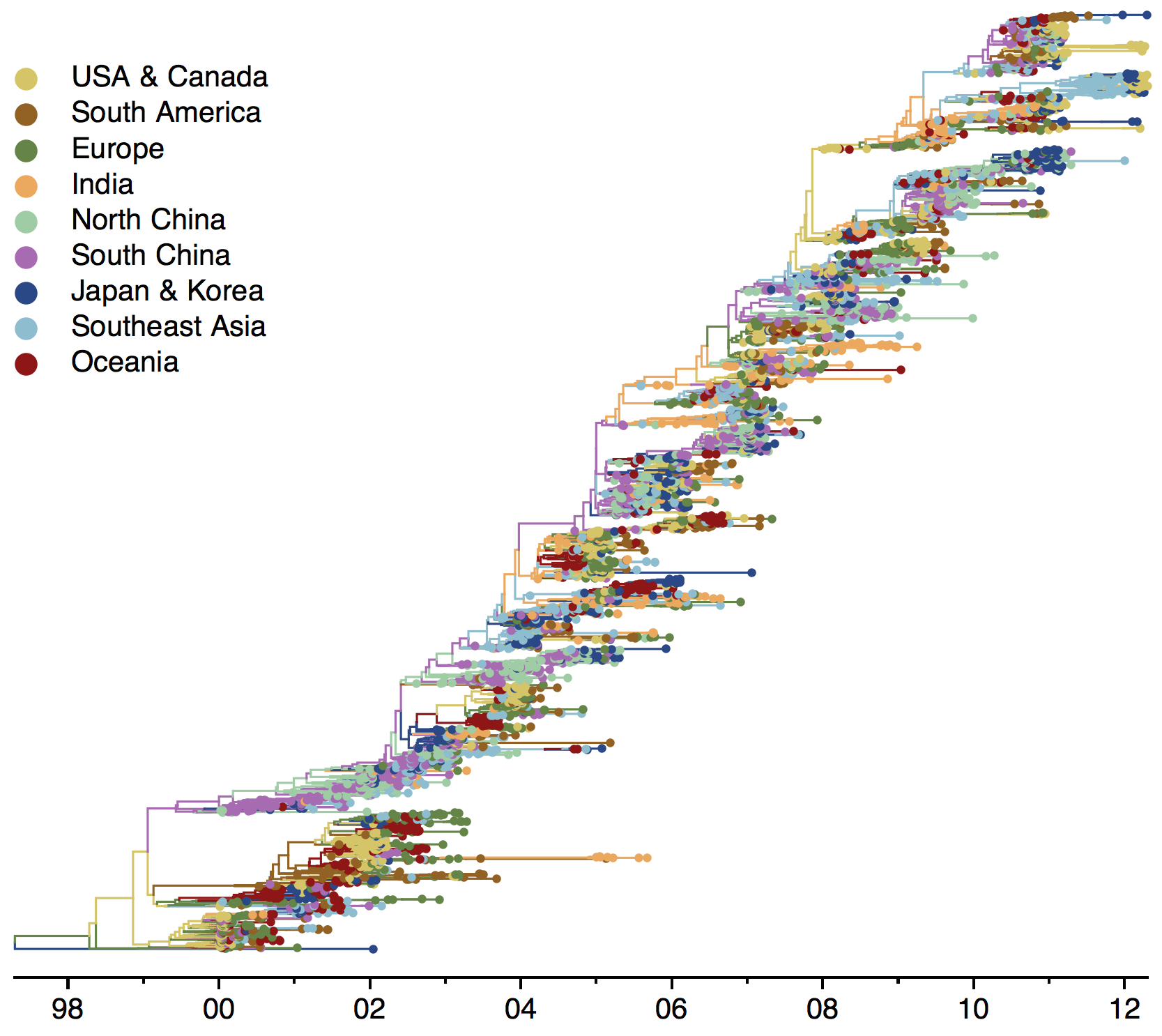
Phylogenetic tracking has the capacity to revolutionize epidemiology
Stuttering chains and animal-to-human spillover
MERS spillover in the Arabian Peninsula
Epidemic growth and human-to-human transmission
Ebola spread in West Africa
New methods for rapid phylogenetics and visualization
MERS-CoV
Middle East respiratory syndrome coronavirus (MERS-CoV)
- First identified in Saudi Arabia in 2012
- 2229 confirmed cases to date and 791 deaths
- Camels thought to be the intermediate host
- 30% of common colds due to endemic human coronaviruses
Ongoing incidence, but lack of epidemic growth
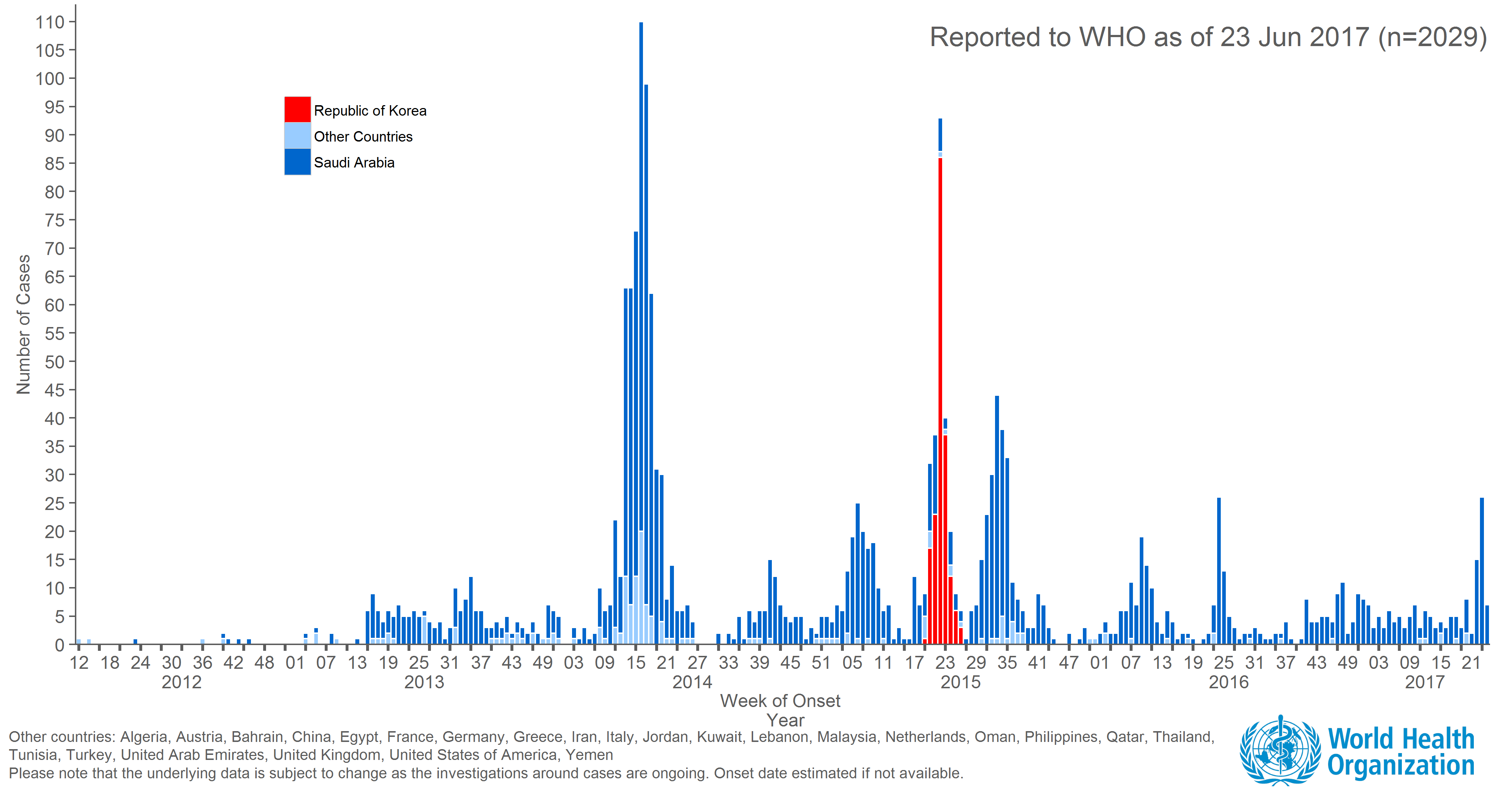
Cases localized to the Arabian Peninsula
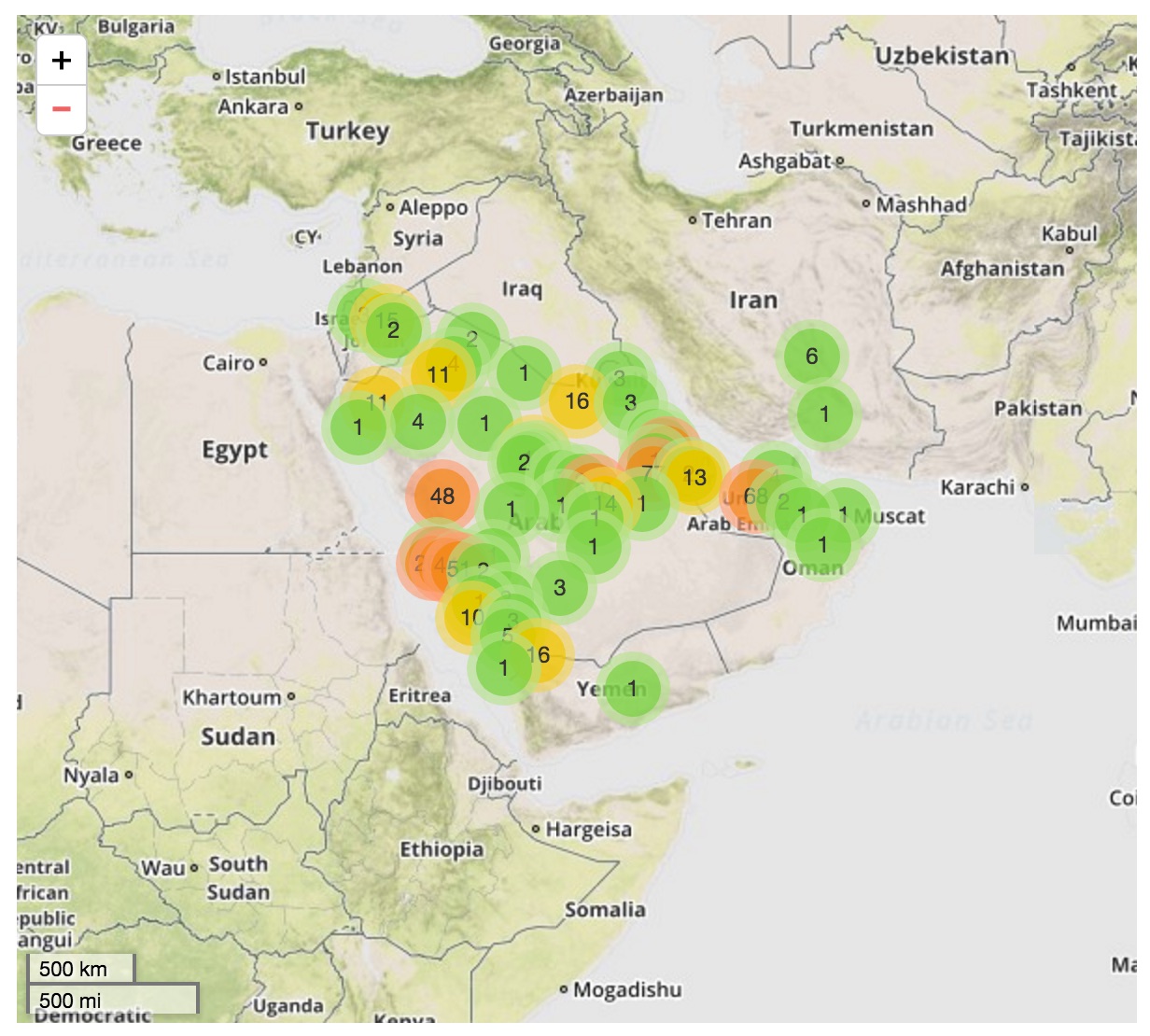
Hypotheses for MERS transmission
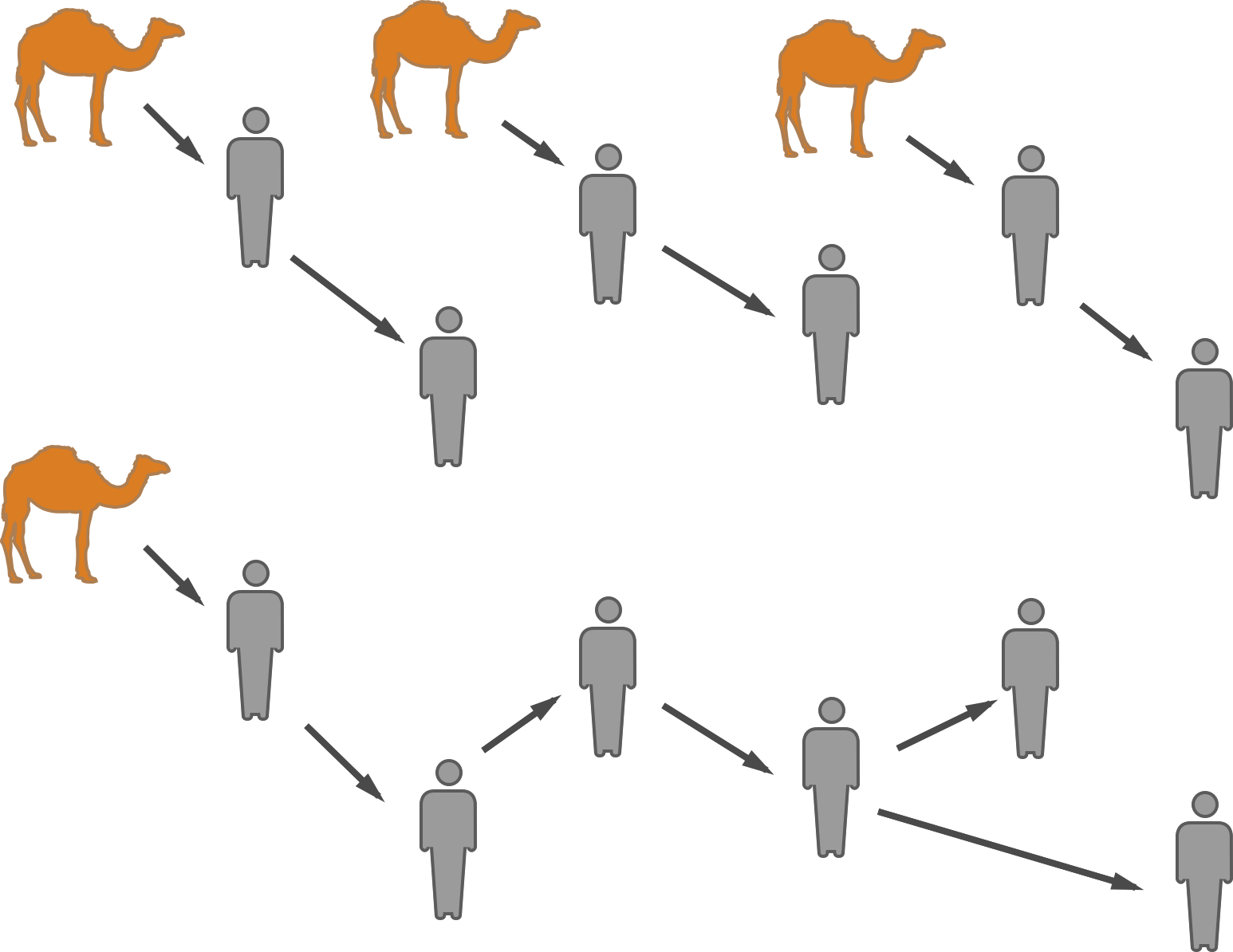
MERS-CoV spillover at the camel-human interface
with ![]() Gytis Dudas, Luiz Carvalho and Andrew Rambaut
Gytis Dudas, Luiz Carvalho and Andrew Rambaut
Genomic dataset
- 174 virus genomes from human infections
- 100 virus genomes from camel infections
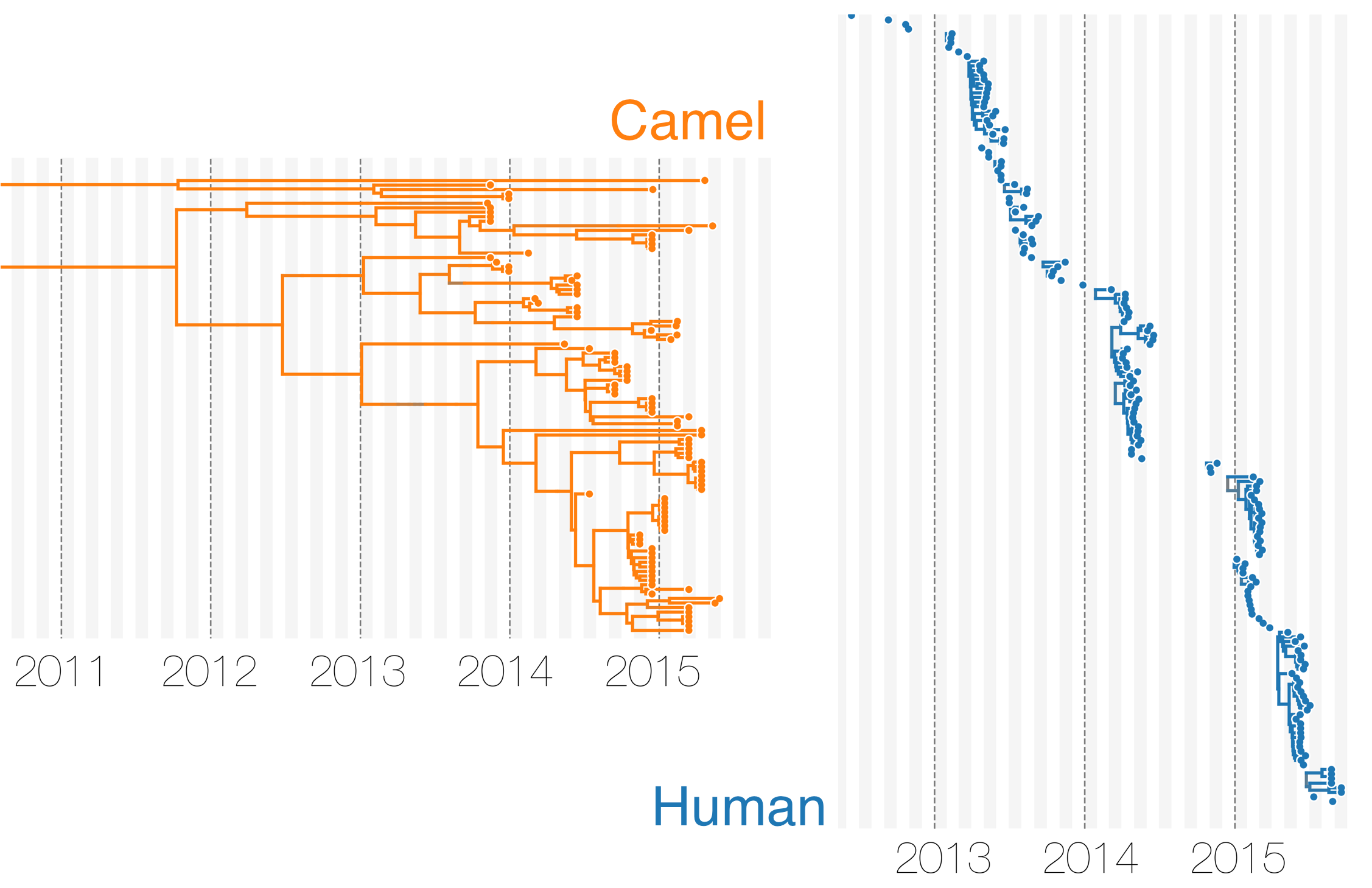
MERS tree with host state
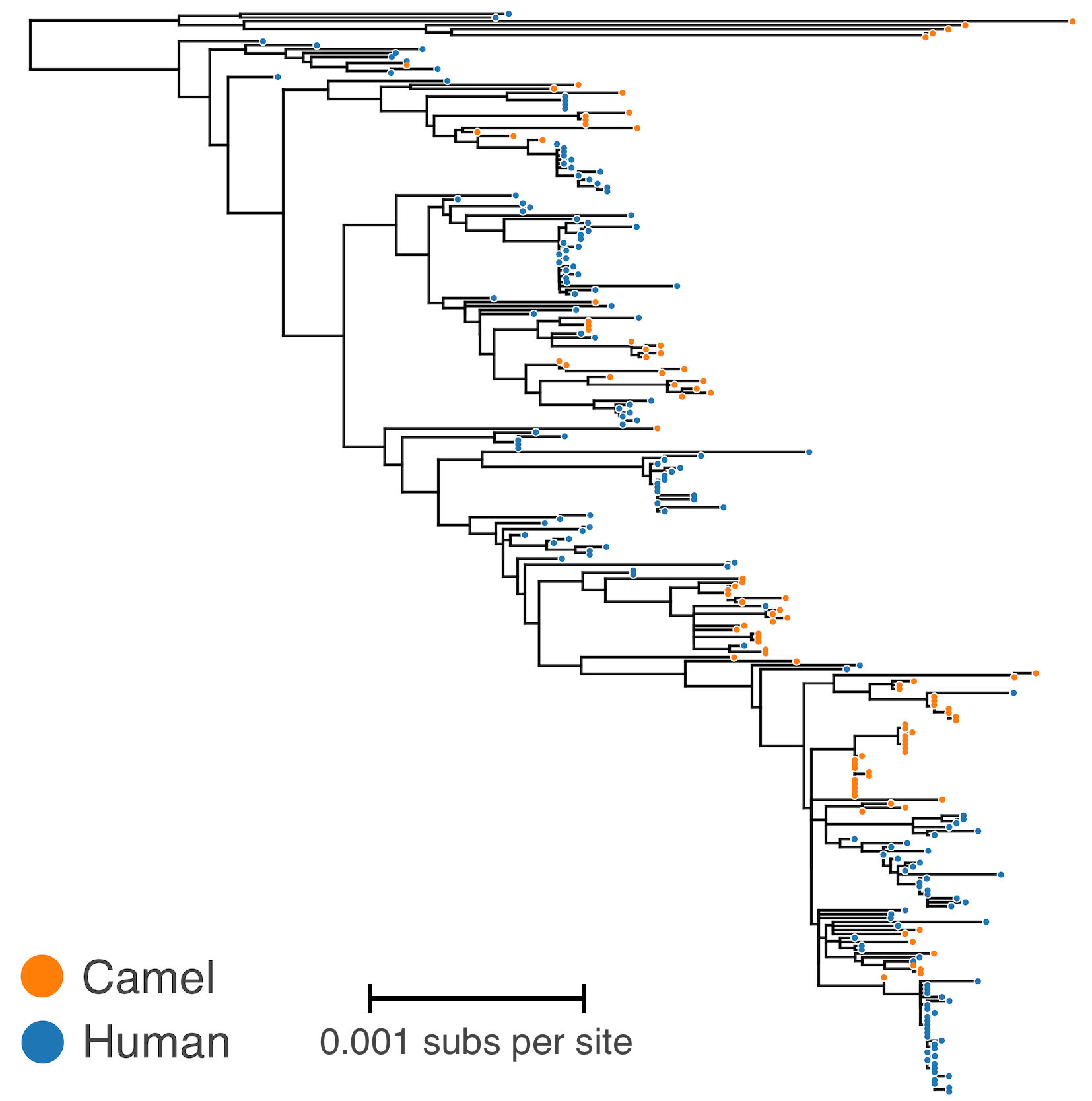
Phylodynamic reconstruction of host state
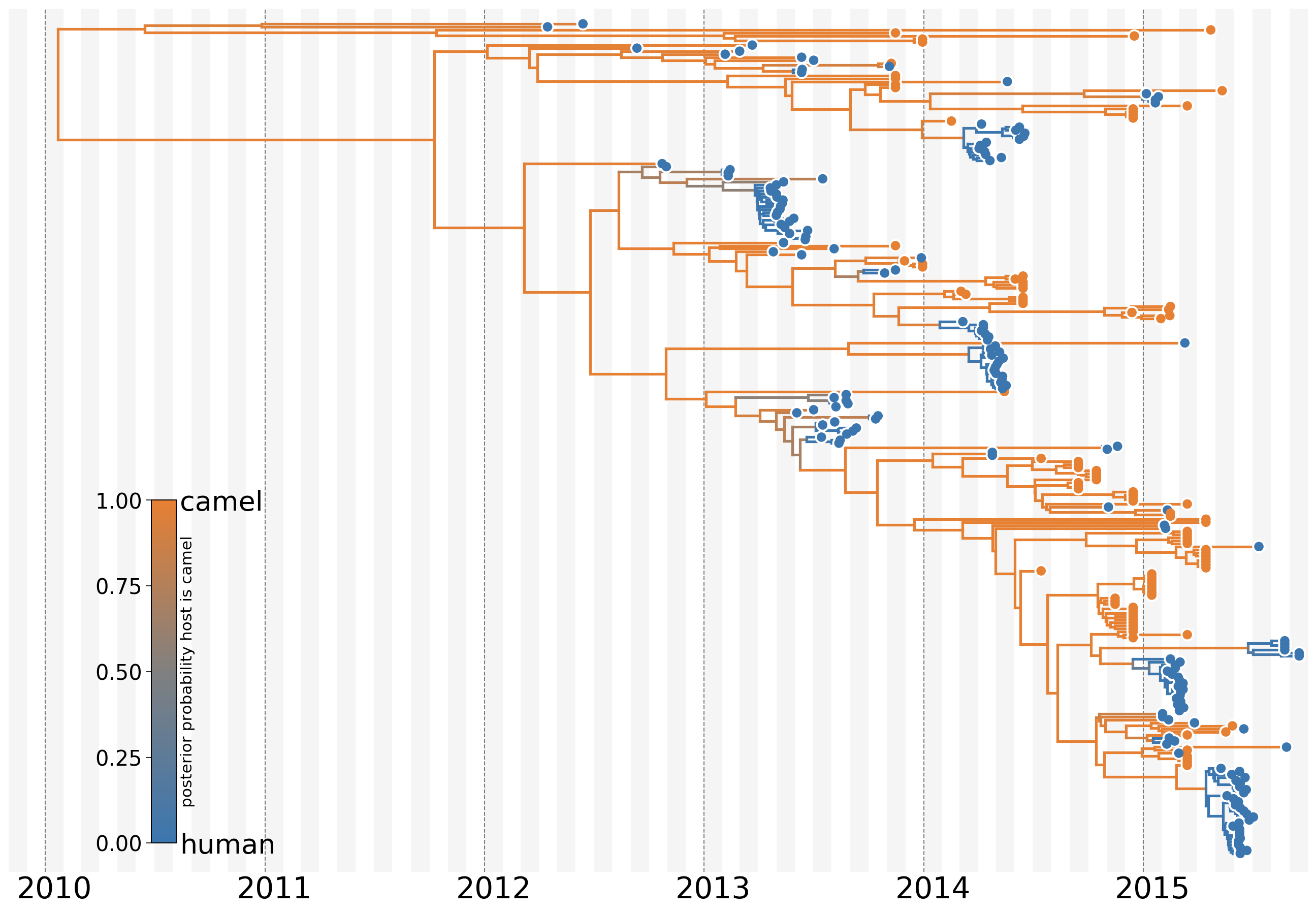
Humans are transient hosts
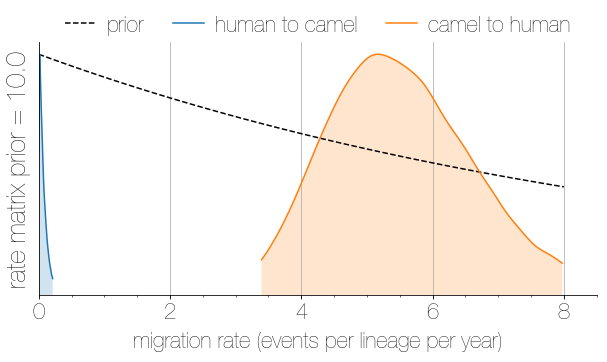
Asymmetric migration rates
- 56 (48–63) camel-to-human transmission events resulting in 174 sequenced human infections
- 3 (0-12) human-to-camel transmission events
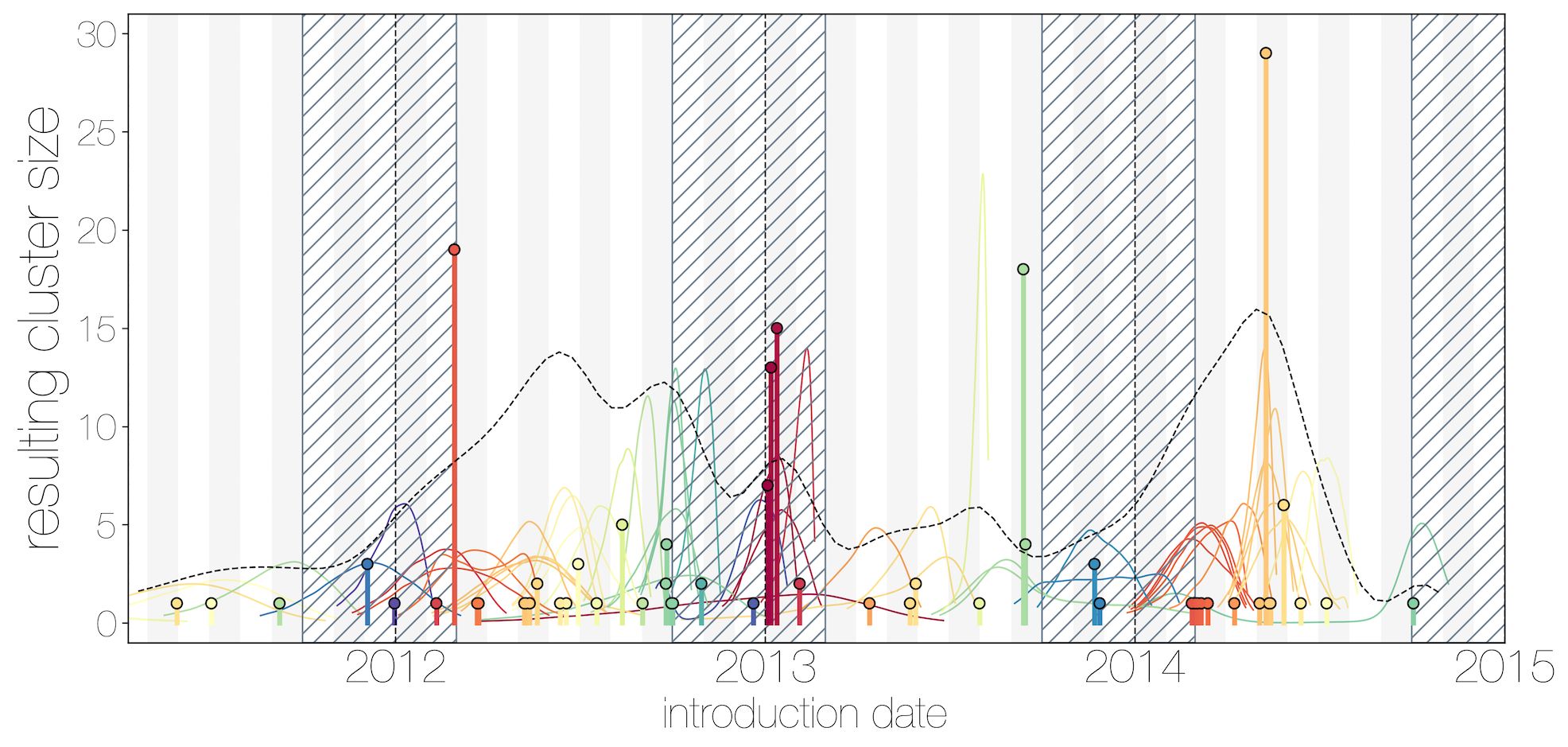
Introduction events tend to occur between April and July
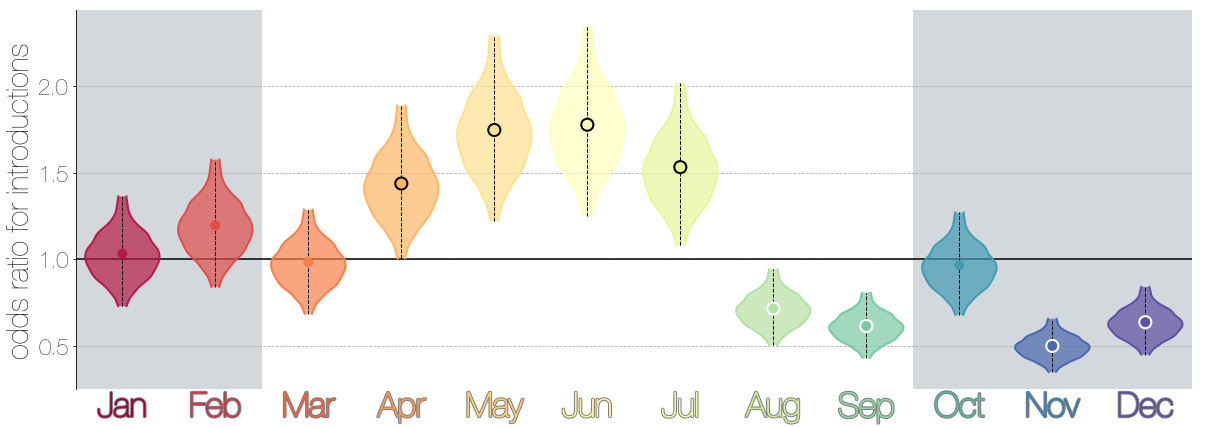
Dromedary camel calving occurs between Nov and Feb
Monte Carlo simulation
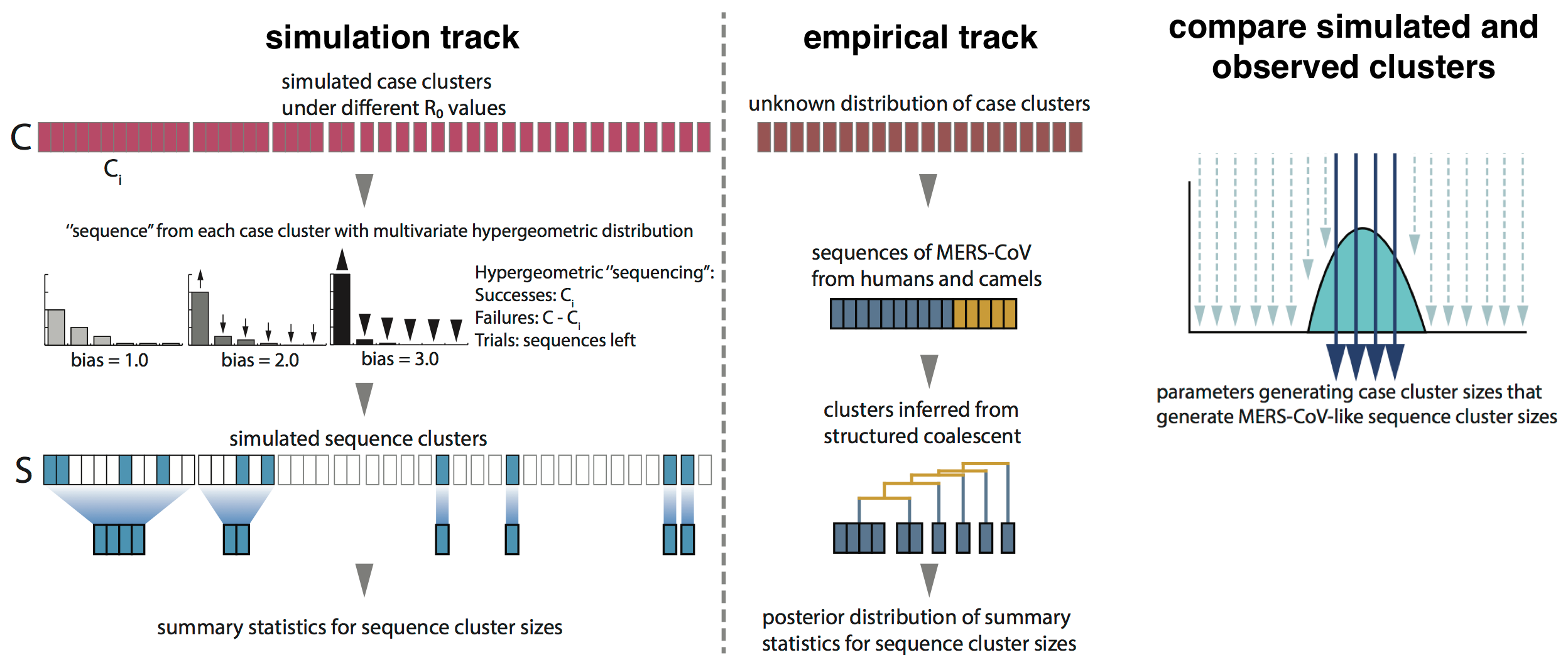
Phylogenetic clustering suggests $R_0$ below 1.0 and ~2000 human cases driven by ~600 introduction events
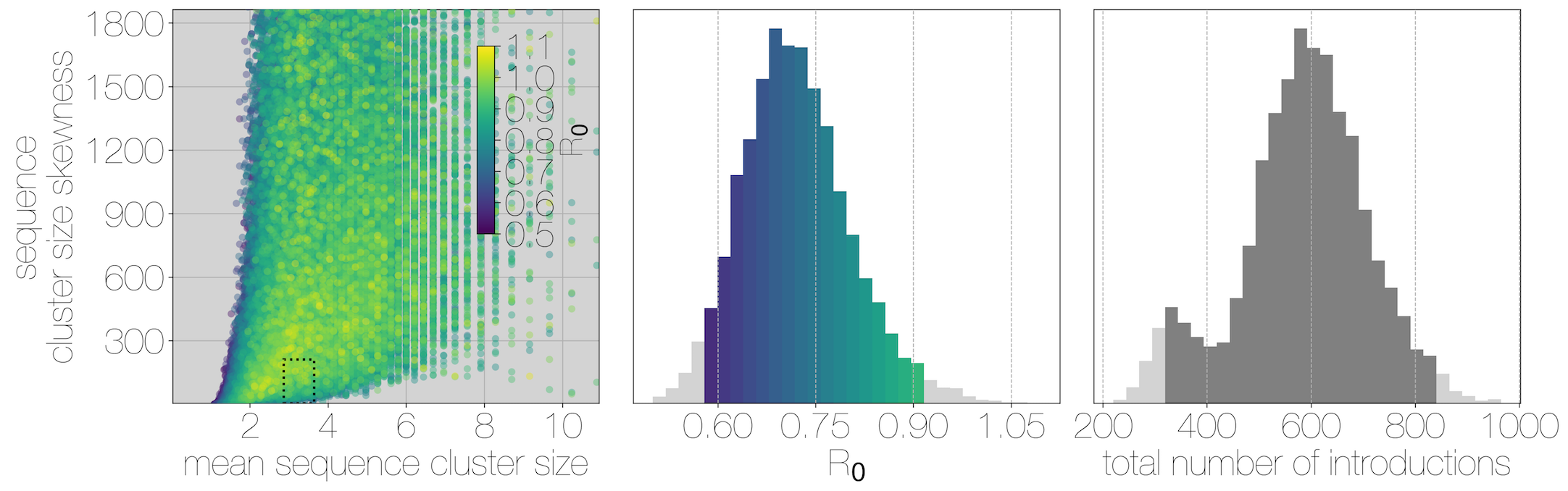
Critically, no evidence of increasing cluster sizes through time
Many other viruses that exhibit stuttering chains of human infection
- Nipah virus (fruit bats / pigs, Southeast Asia)
- Lassa virus (rodents, West Africa)
- Avian influenza (birds, mainland China)
Sylvatic introductions of yellow fever virus show similar dynamics
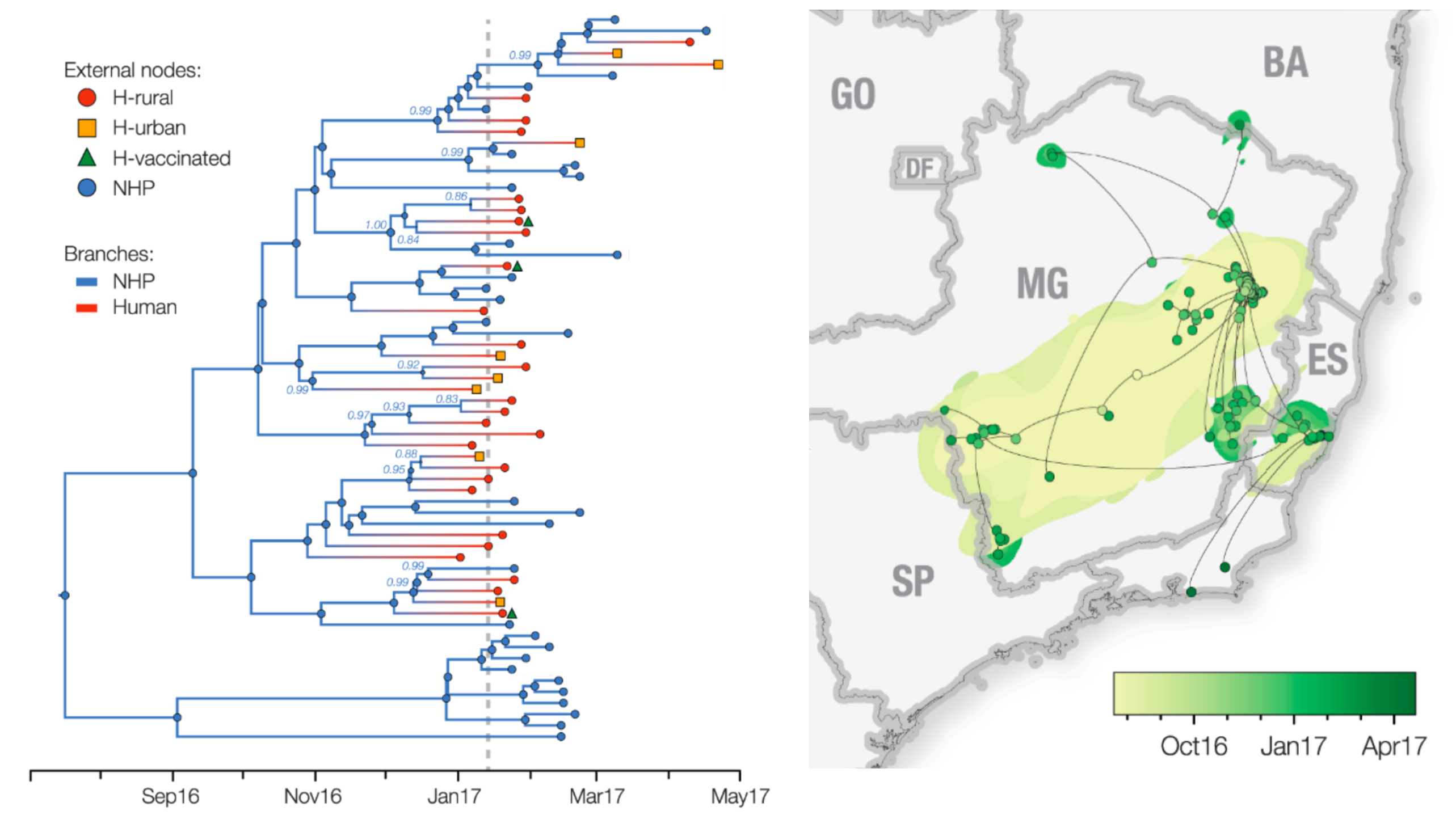
Ebola
Ebola epidemic of 2014-2016 was unprecedented in scope
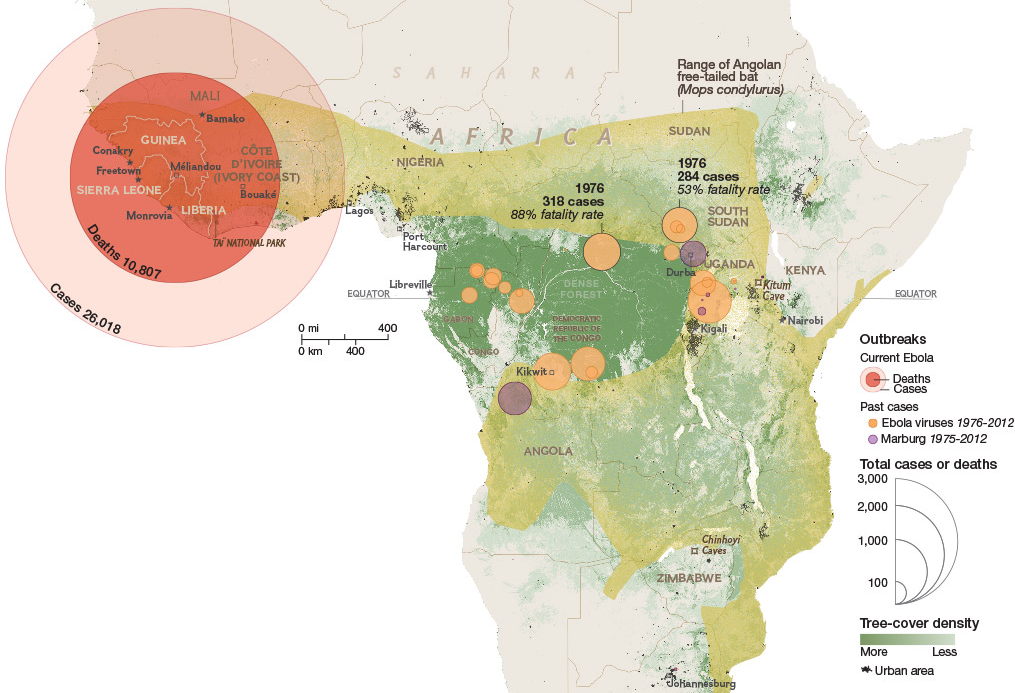
Ebola epidemic in West Africa
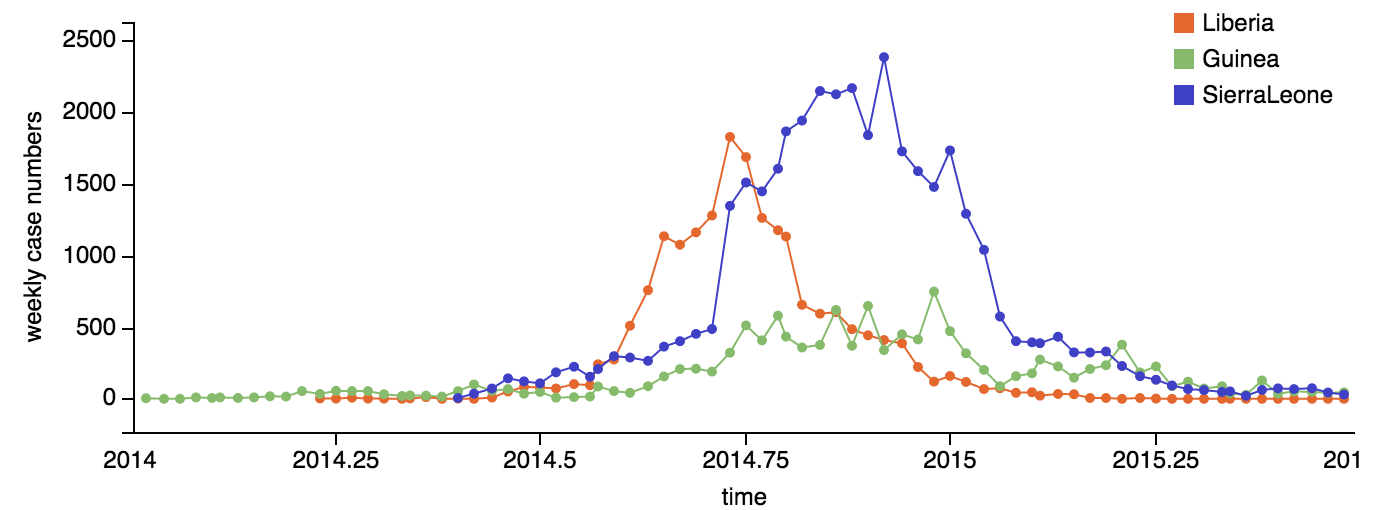
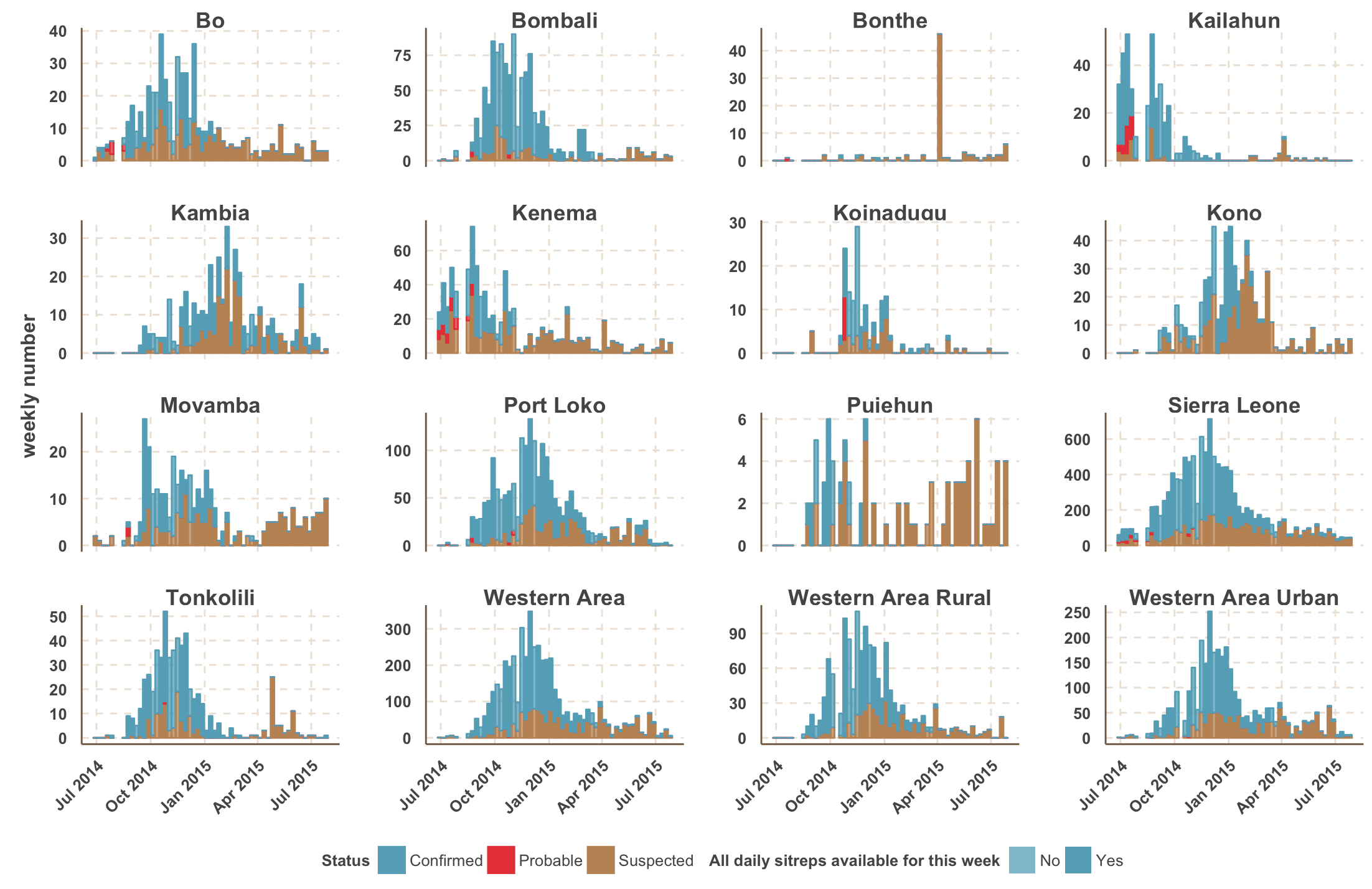
Ebola epidemic within Sierra Leone
Virus genomes reveal factors that spread and sustained the Ebola epidemic
with ![]() Gytis Dudas, Andrew Rambaut, Luiz Carvalho, Marc Suchard, Philippe Lemey,
Gytis Dudas, Andrew Rambaut, Luiz Carvalho, Marc Suchard, Philippe Lemey,
and many others
Sequencing of 1610 Ebola virus genomes collected during the 2013-2016 West African epidemic
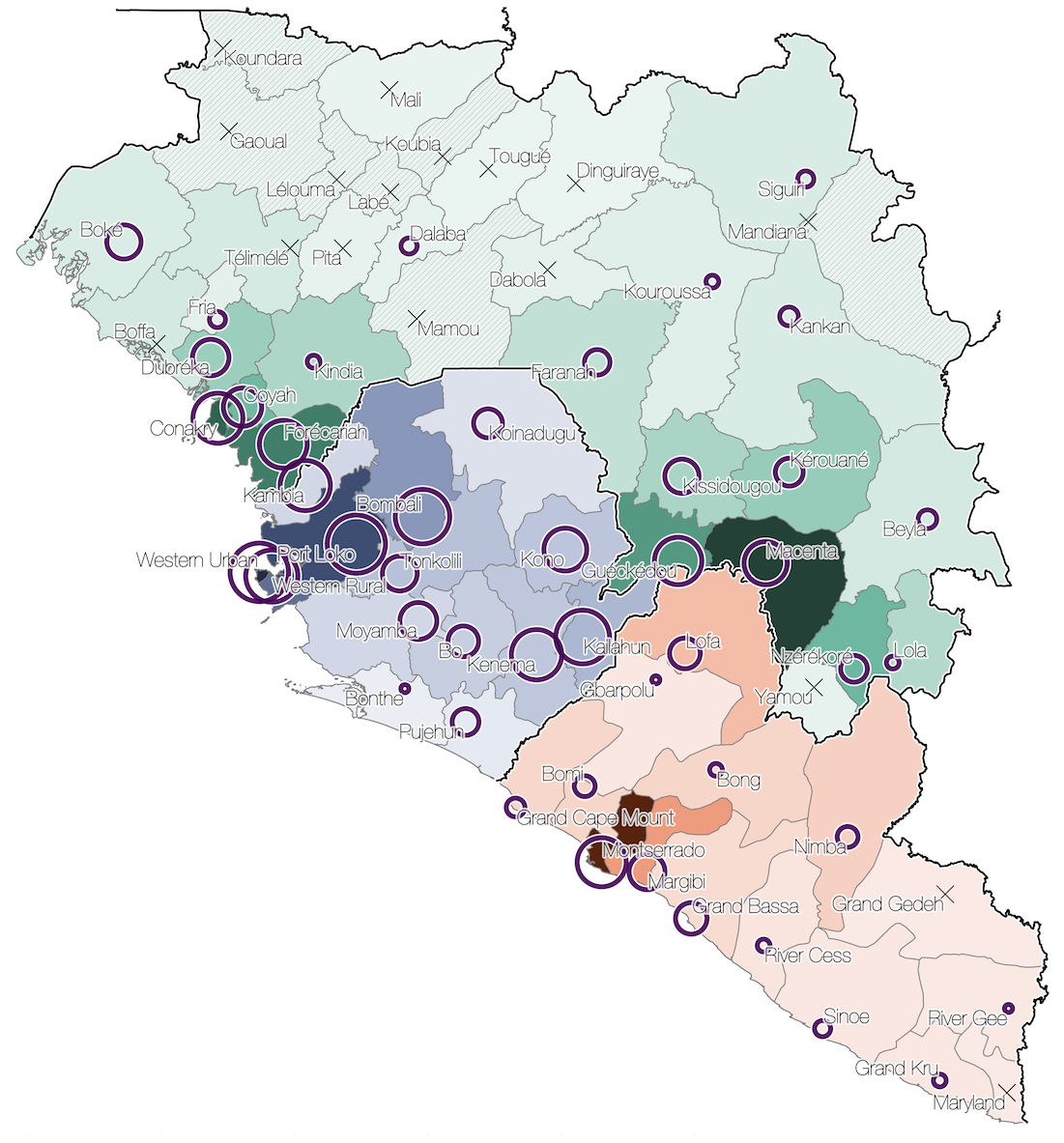
Sequenced genomes were representative of spatiotemporal diversity
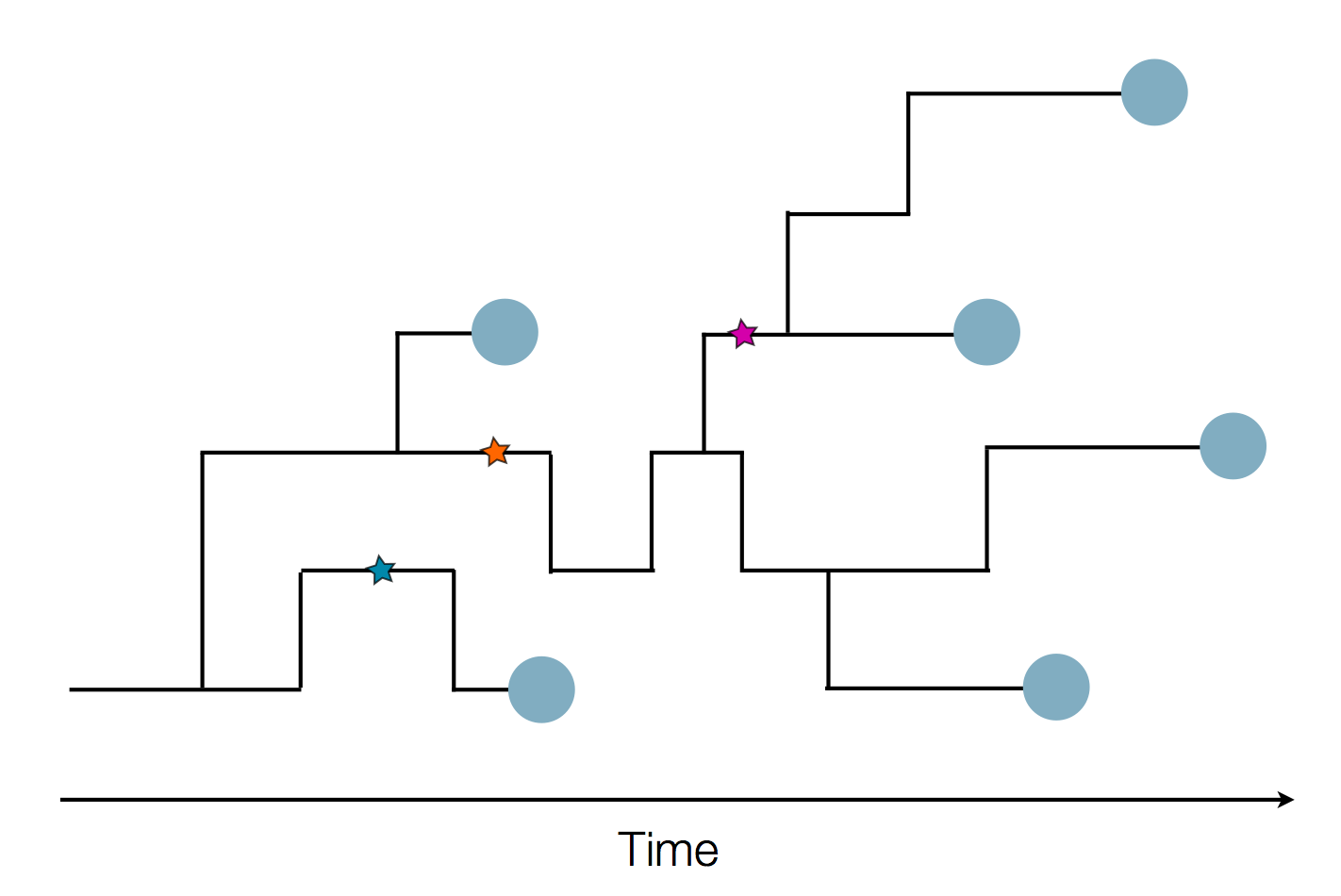
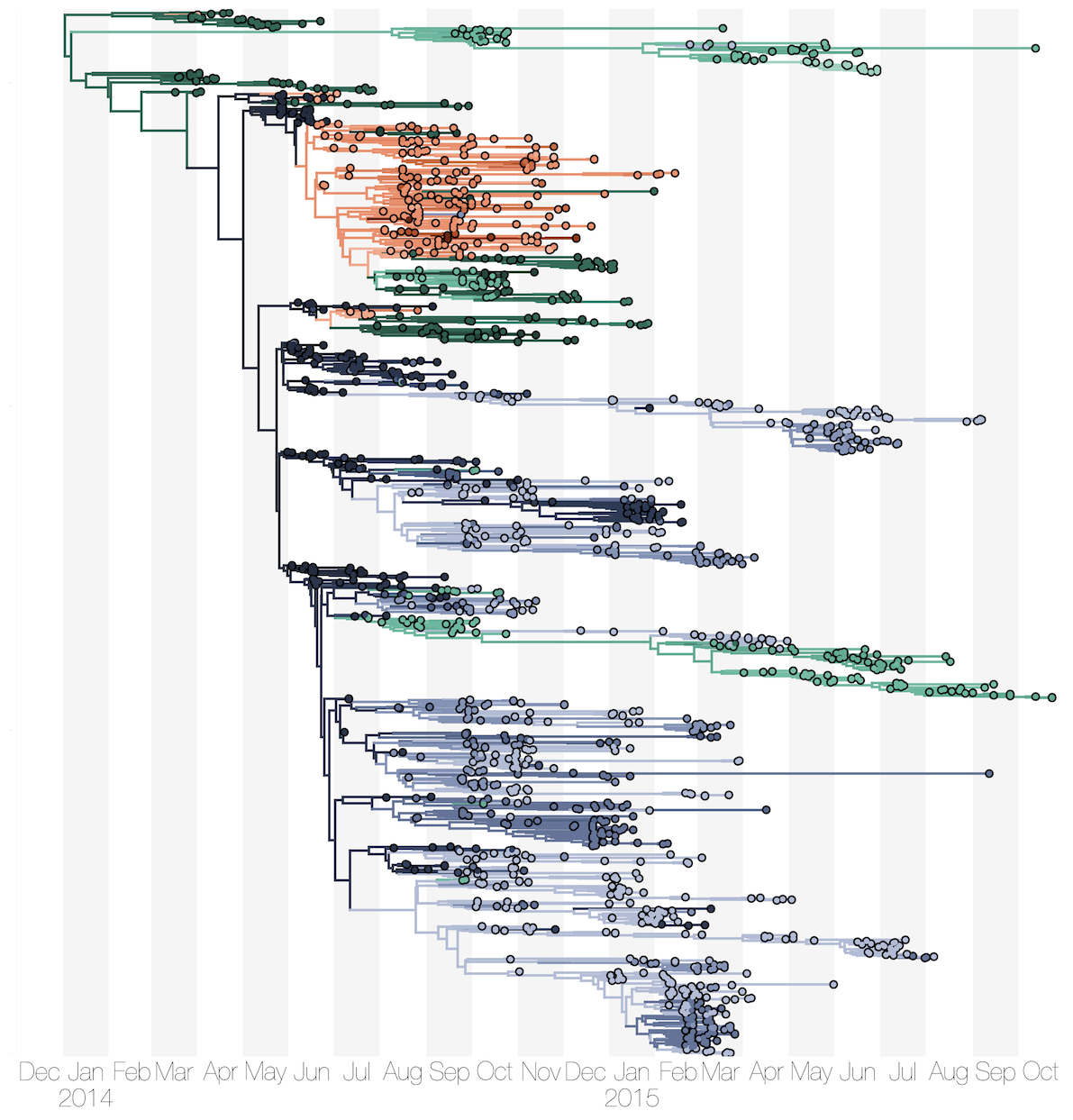
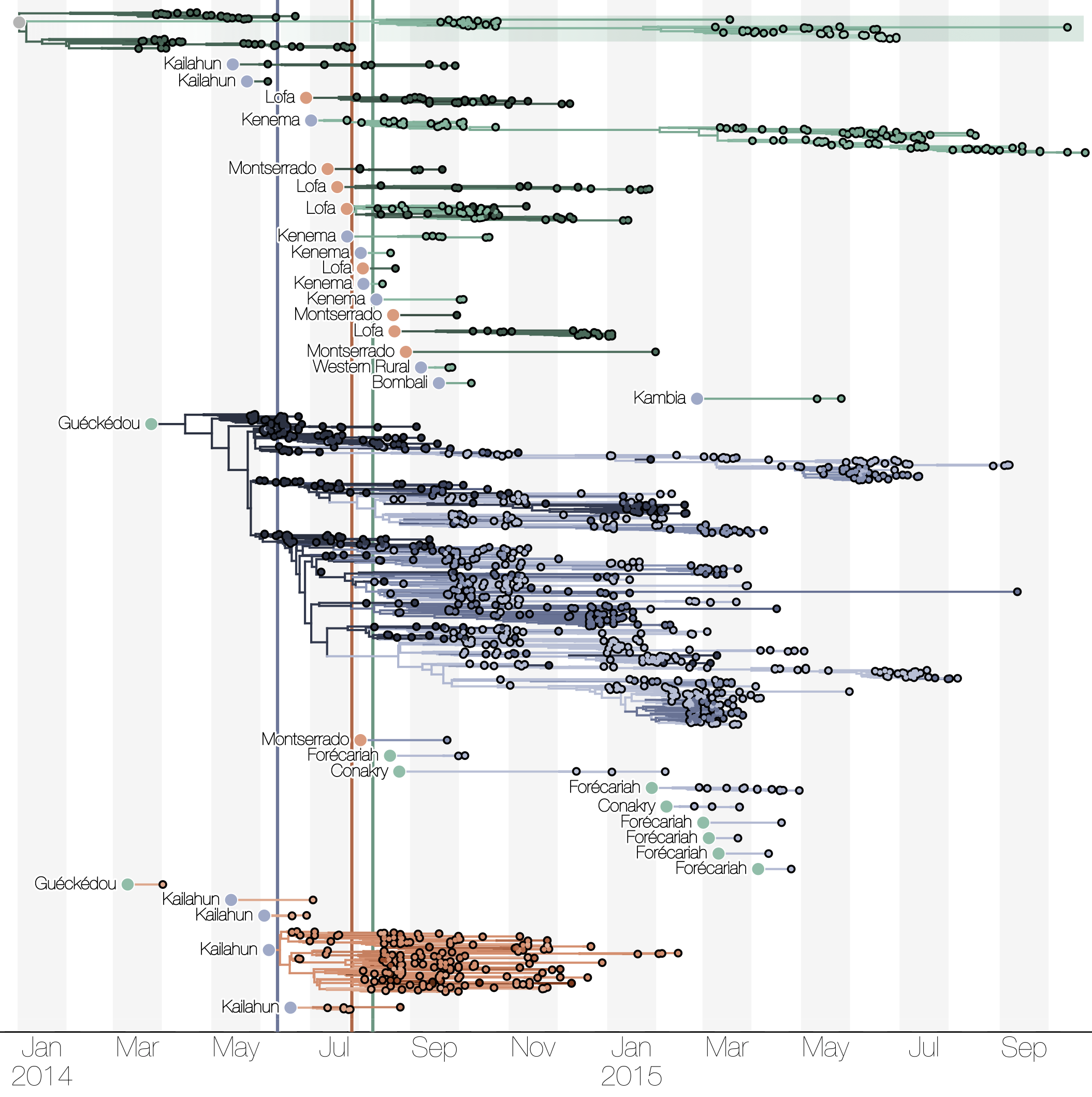
Phylogenetic reconstruction of epidemic
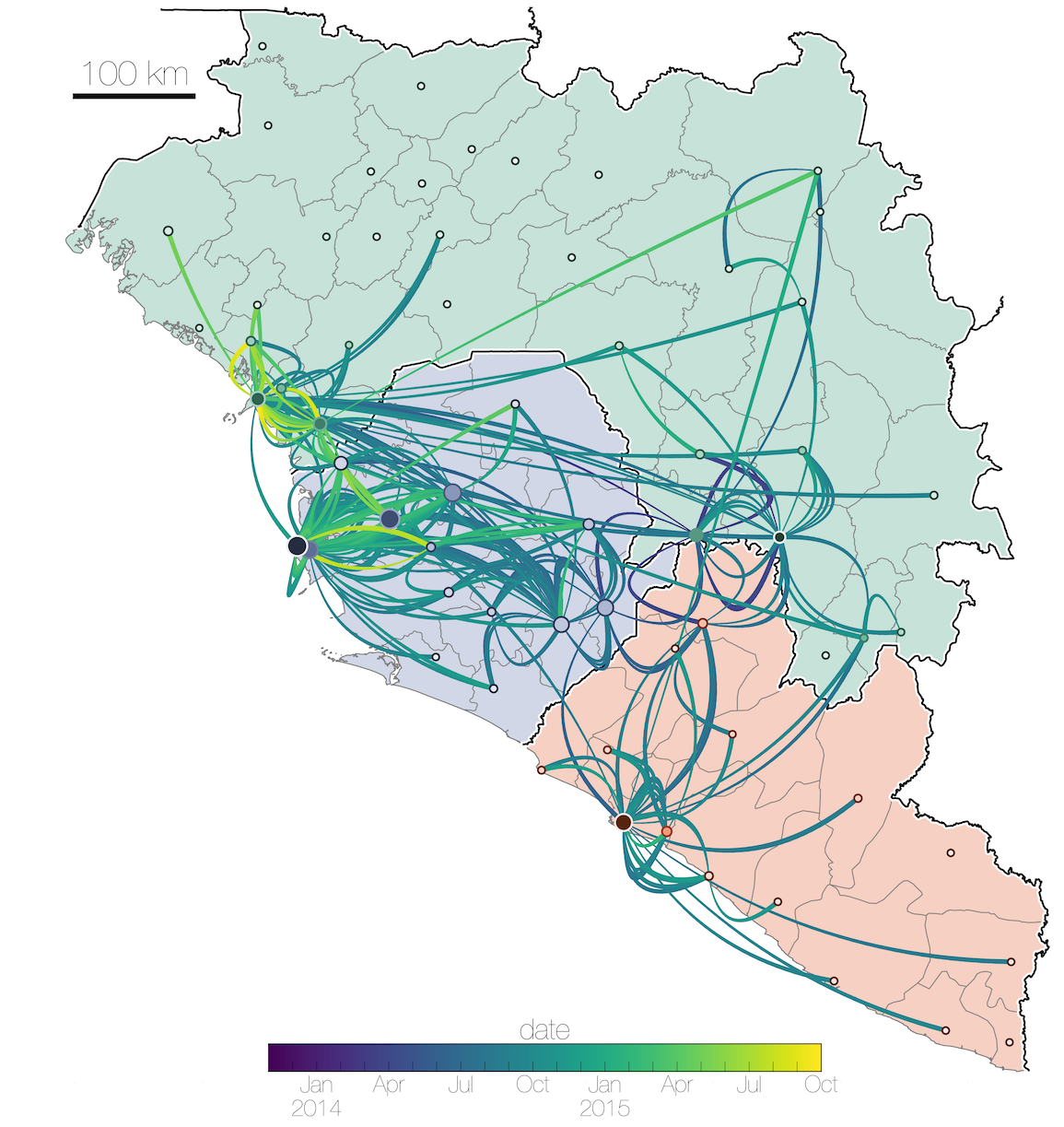
Tracking migration events
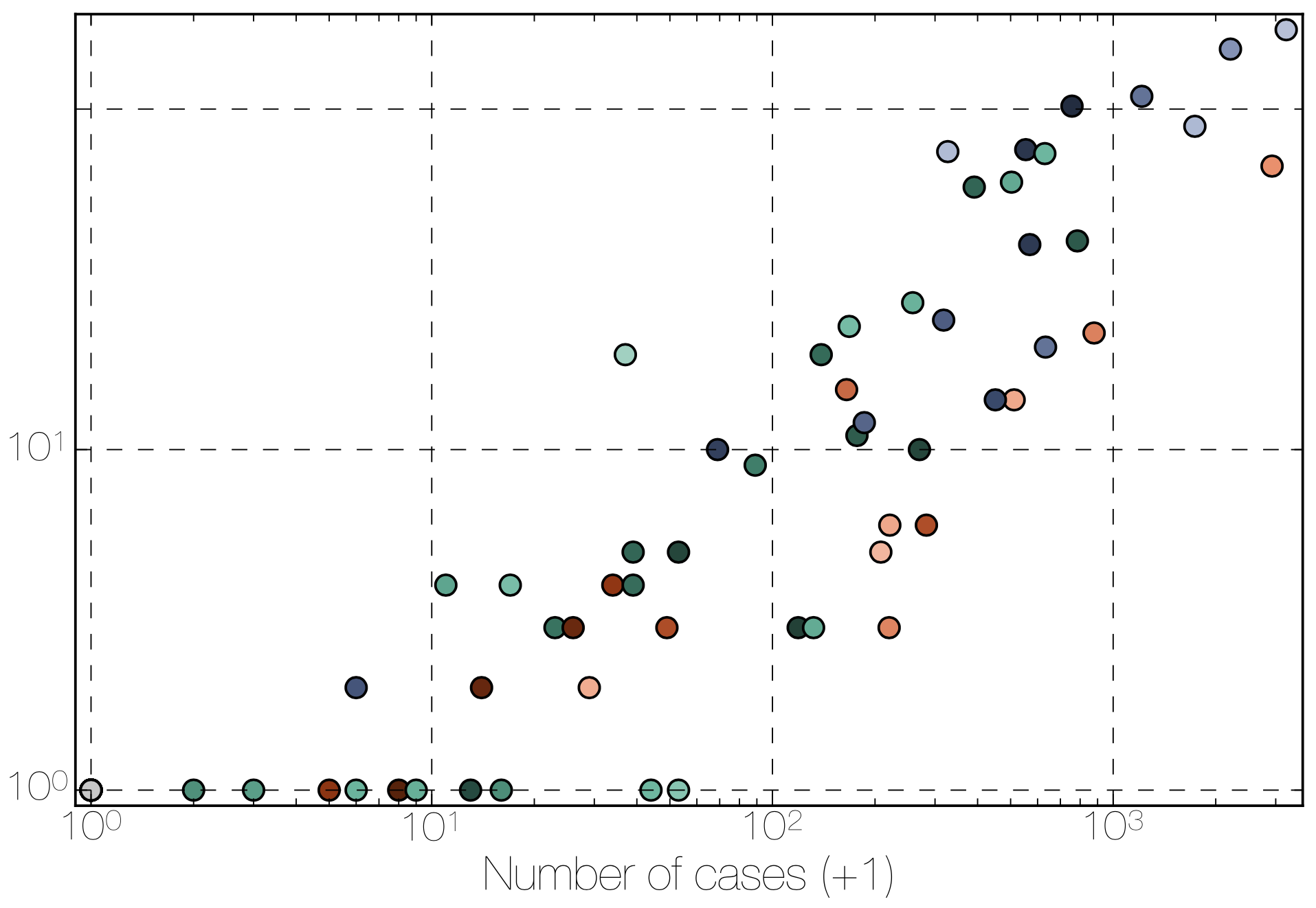
Factors influencing migration rates
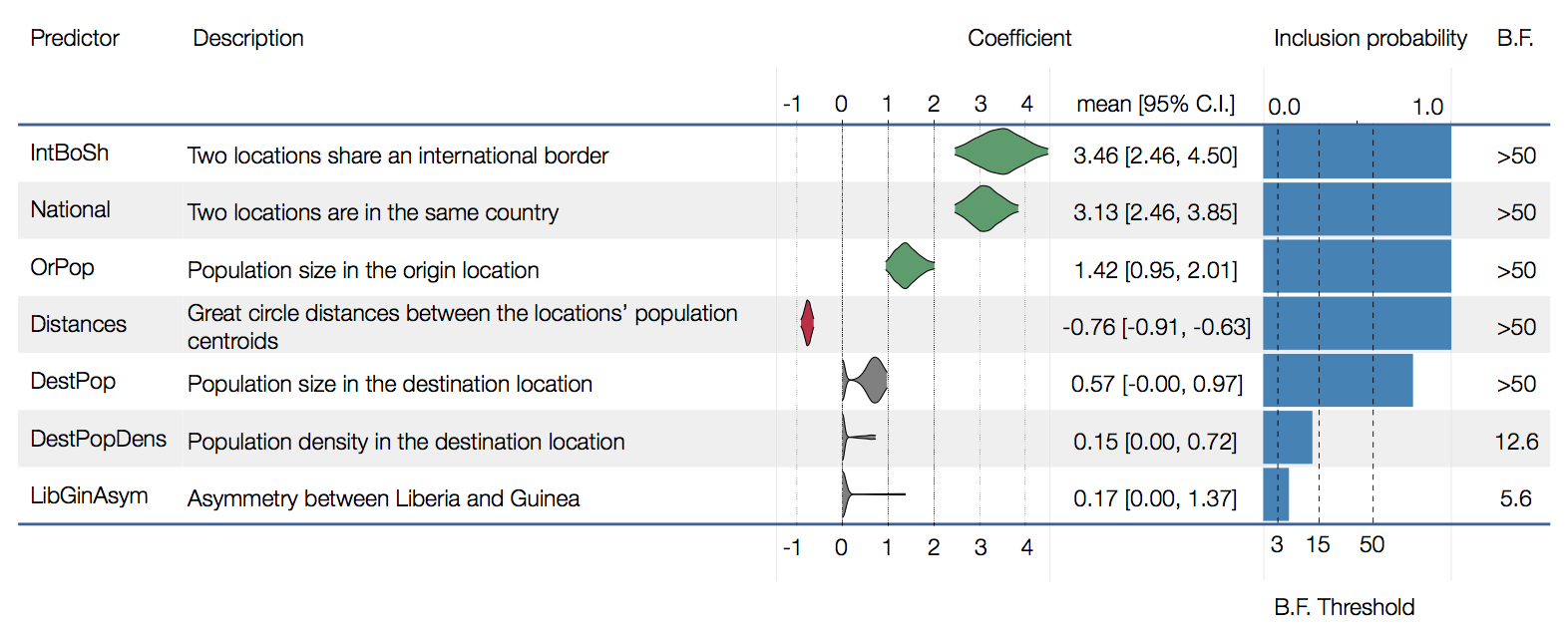
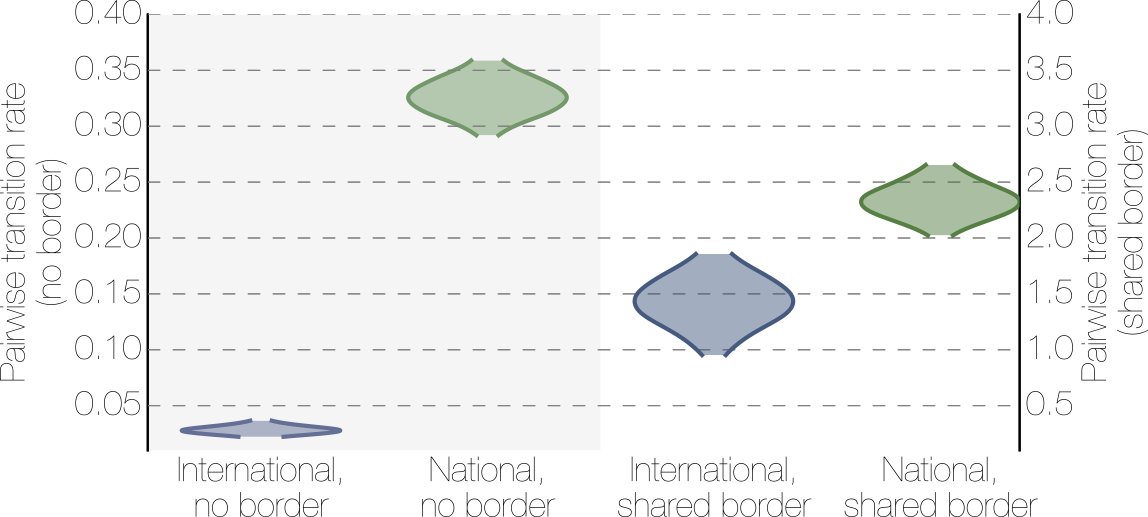
Effect of borders on migration rates
Spatial structure at the country level
Substantial mixing at the regional level
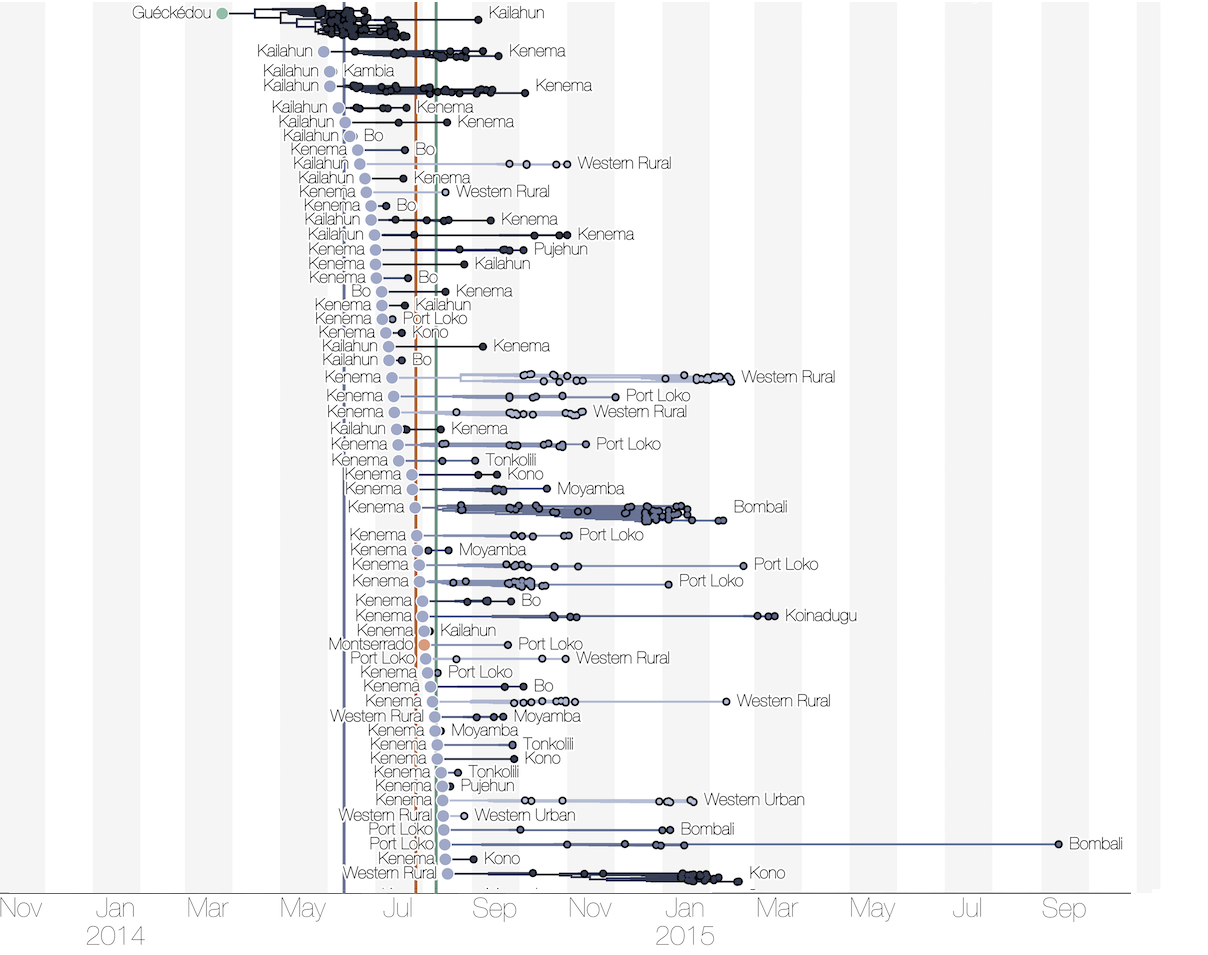
Each introduction results in a minor outbreak
Regional outbreaks due to multiple introductions
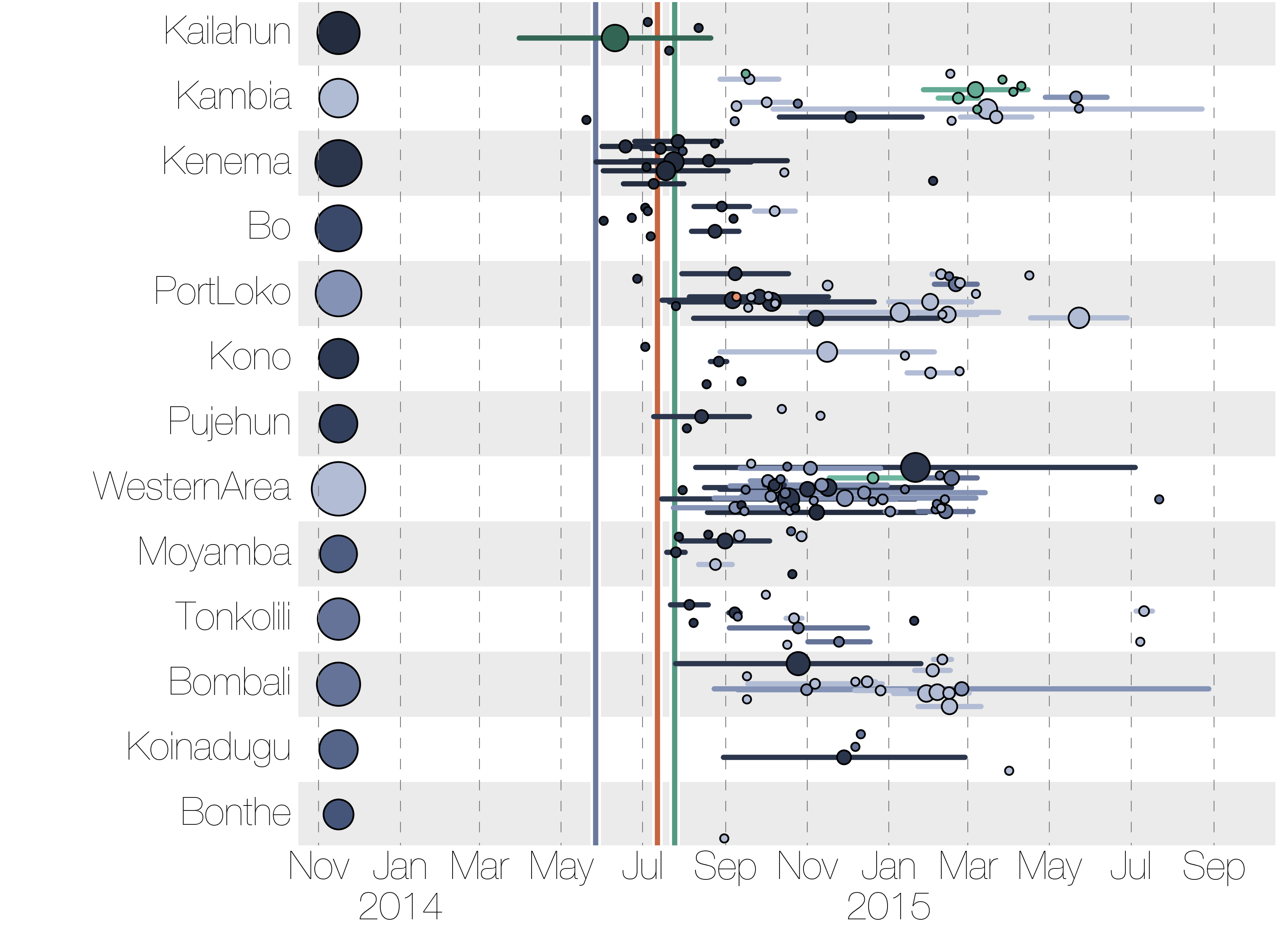
Regional outbreaks due to multiple introductions
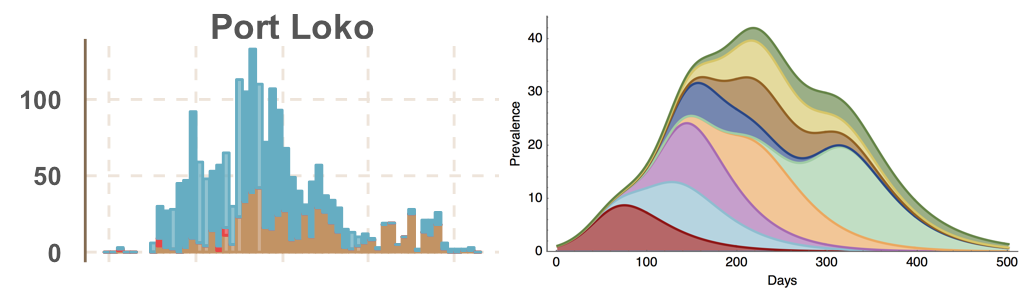
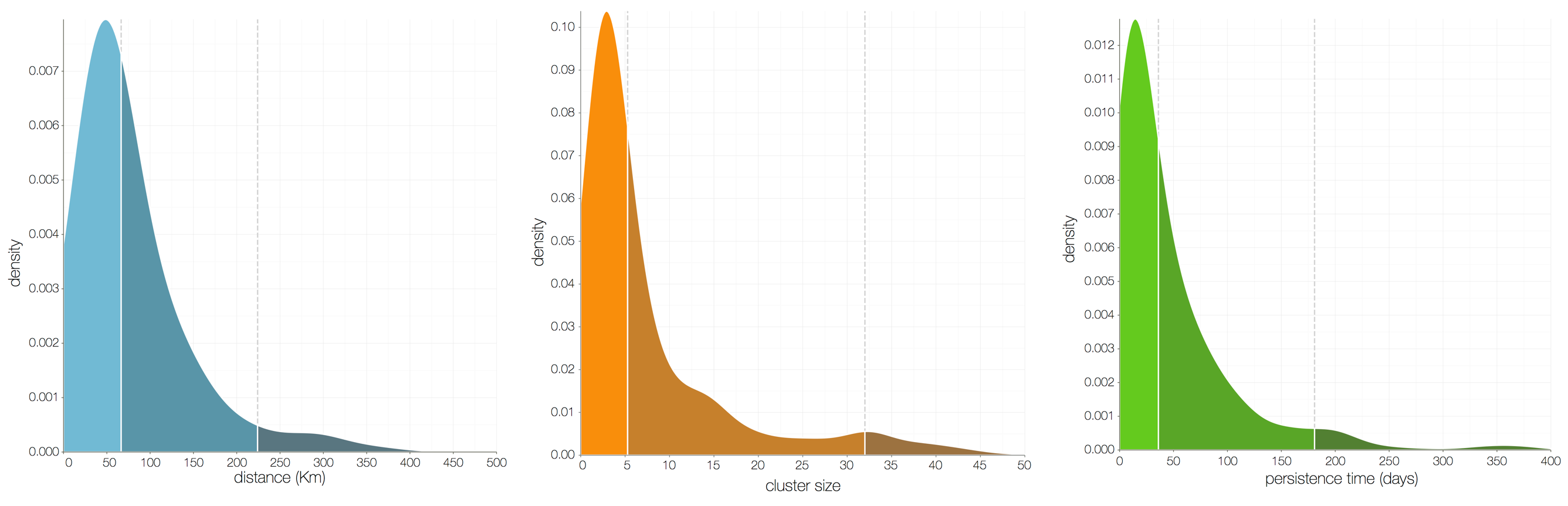
Ebola spread in West Africa followed a gravity model with moderate slowing by international borders, in which spread is driven by short-lived migratory clusters
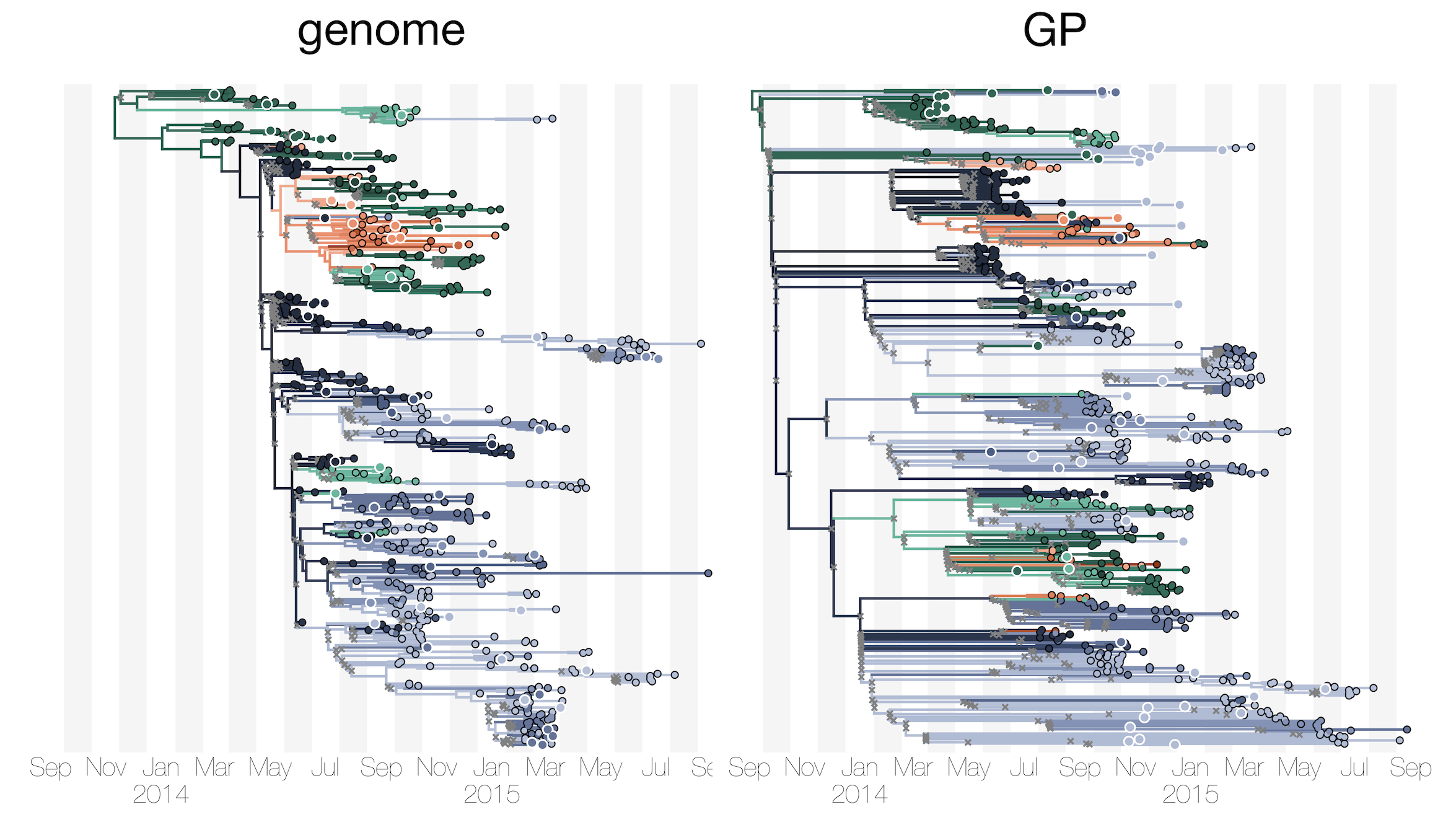
The ability of single genes vs full genomes to resolve time and space in outbreak analysis
with ![]() Gytis Dudas
Gytis Dudas
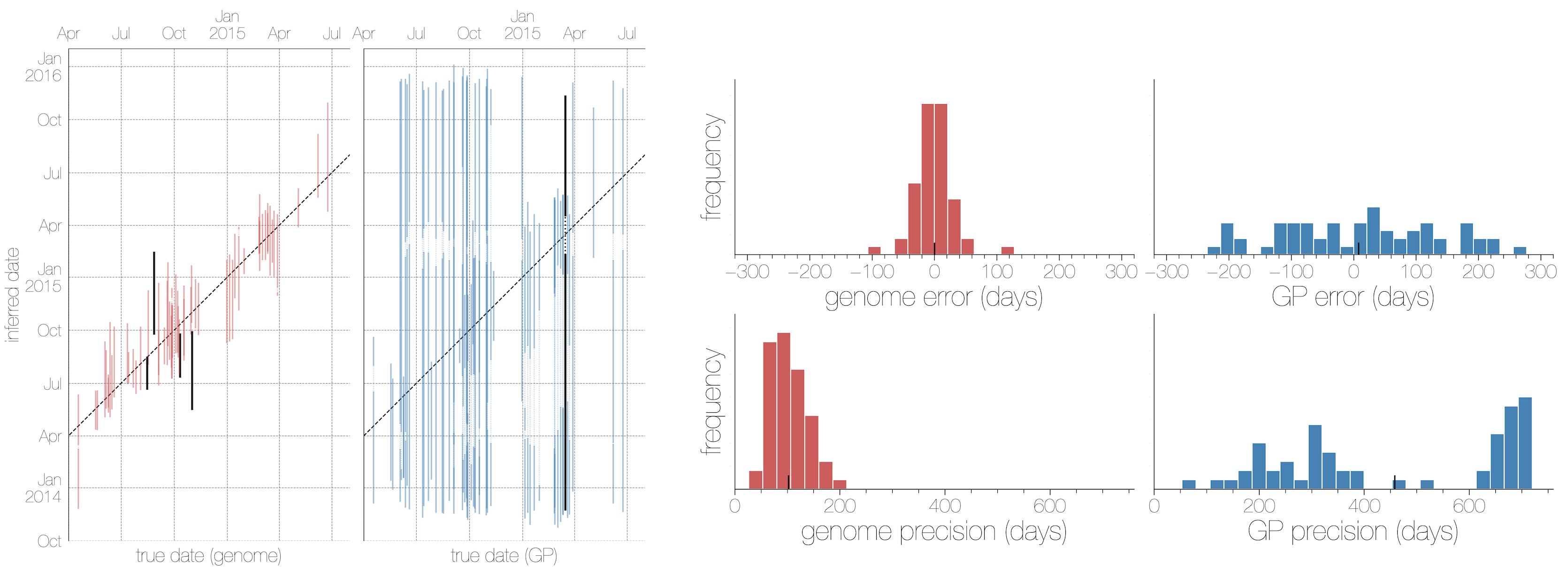
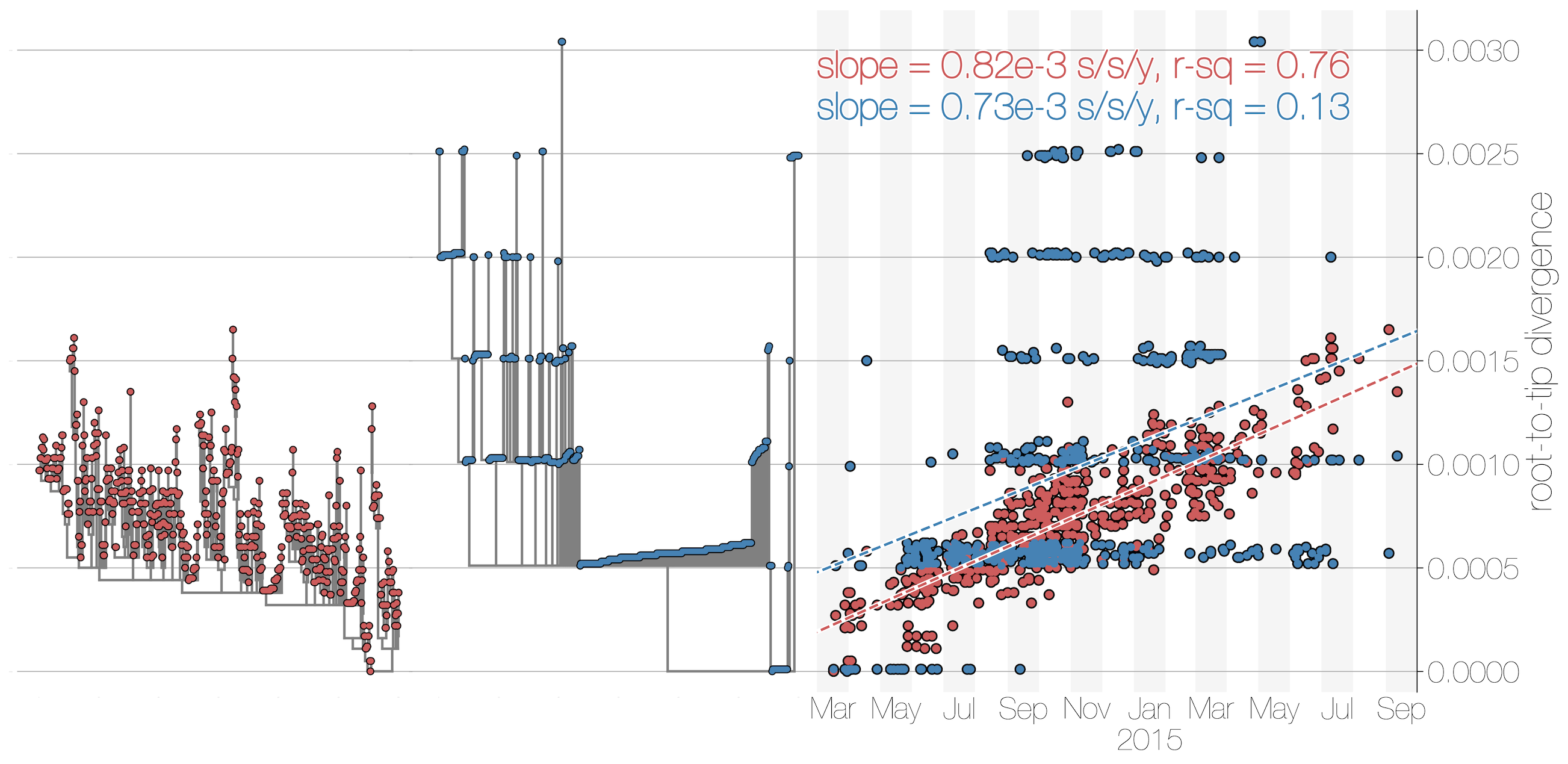
Accuracy vs precision
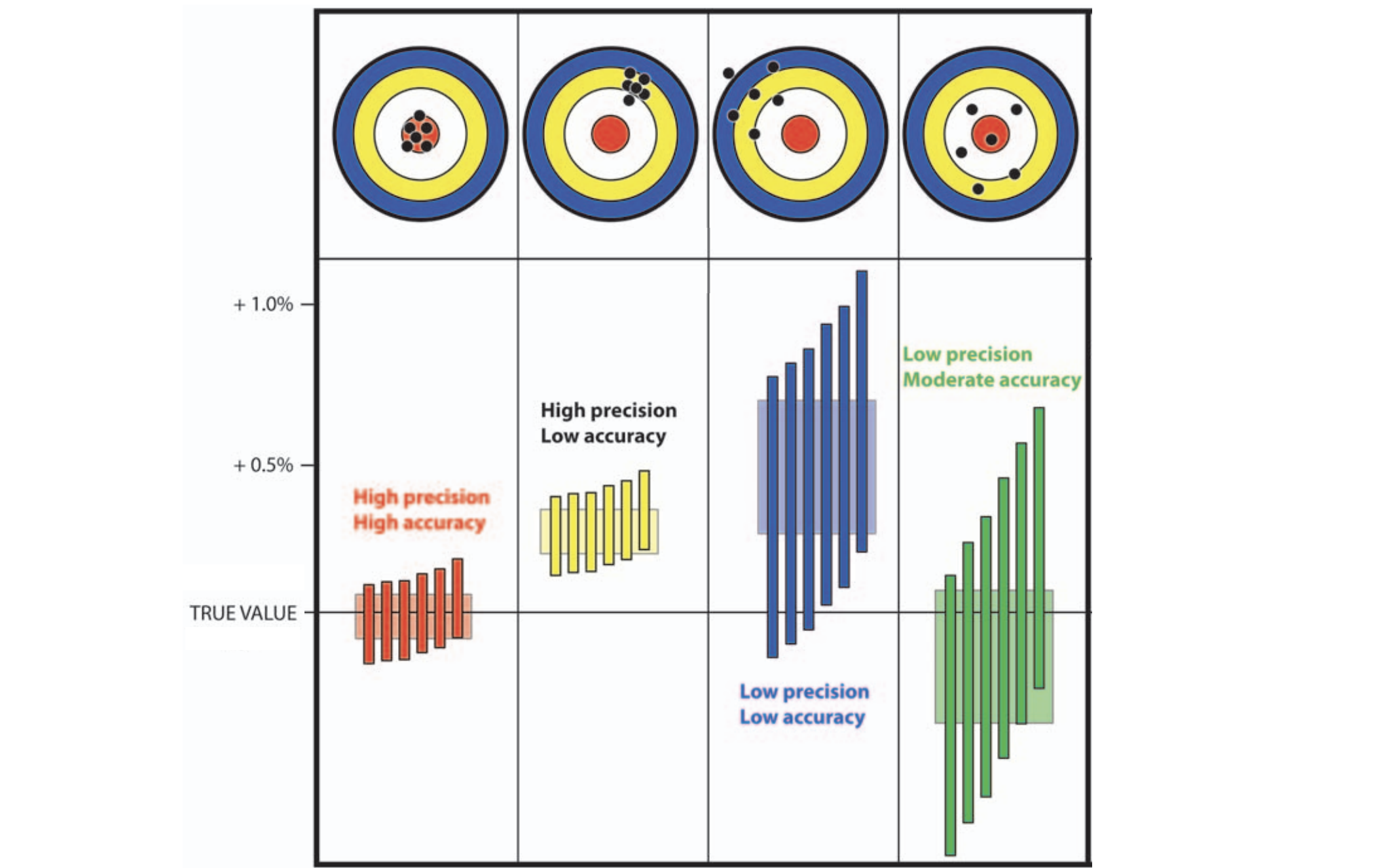
Approach accuracy and precision through a machine learning approach to model testing. Leave out 60/600 tips and predict time and location of these tips
Maximum likelihood divergence trees
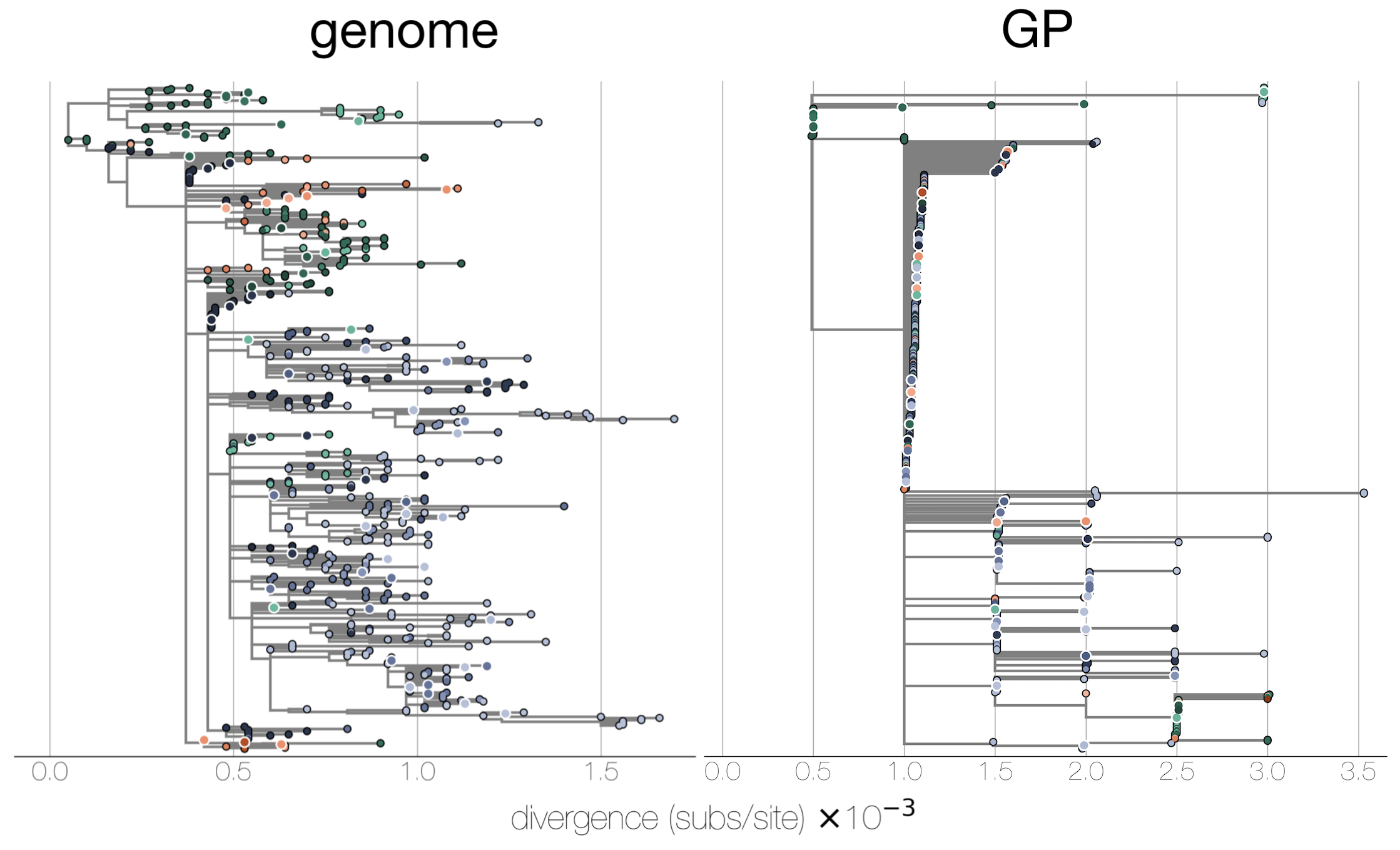
BEAST time trees
Date reconstruction
Evolutionary rate reconstruction
Location reconstruction
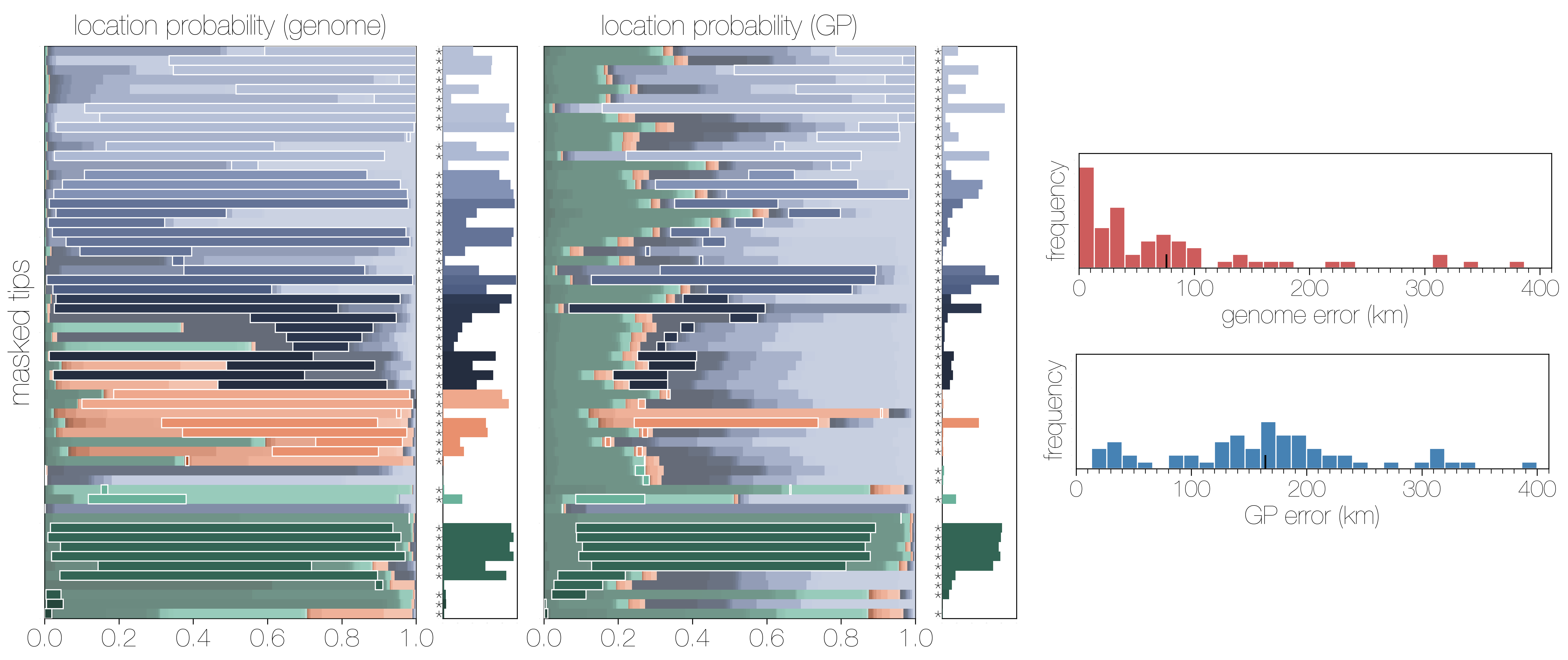
Estimates are generally well calibrated
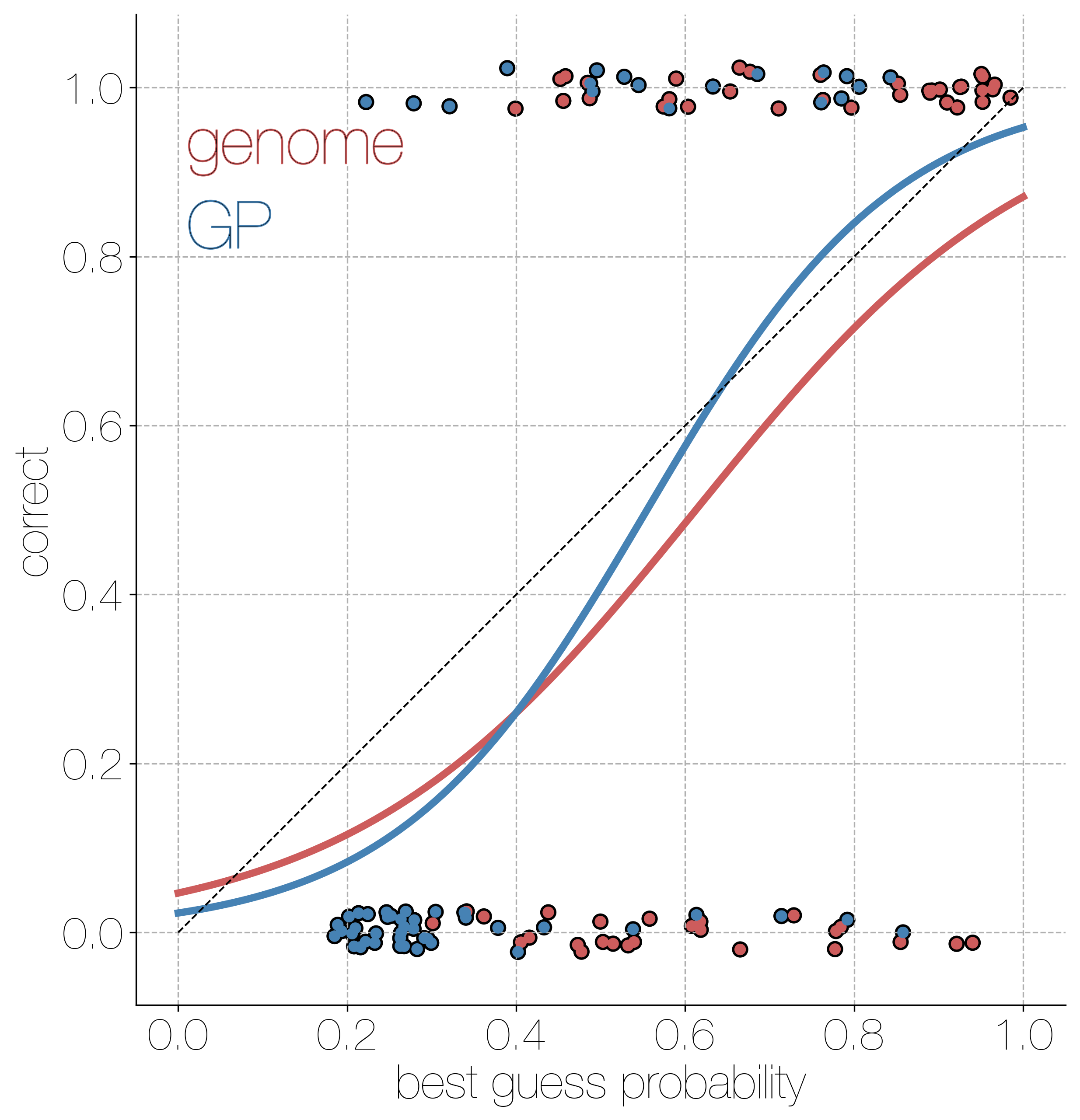
Actionable inferences
Genomic analyses were mostly done in a retrospective manner
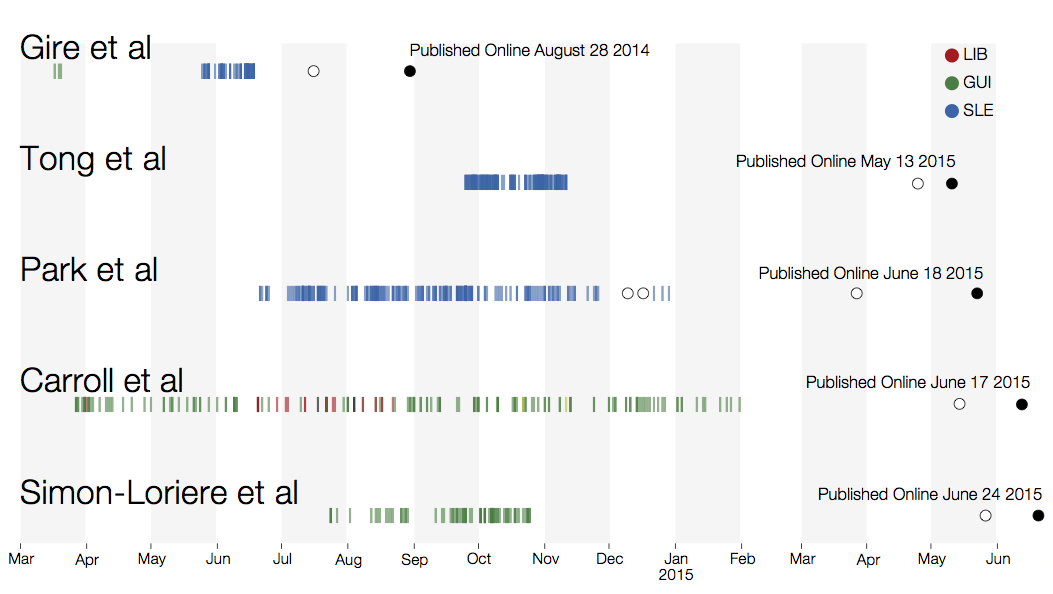
Key challenges to making genomic epidemiology actionable
- Timely analysis and sharing of results critical
- Dissemination must be scalable
- Integrate many data sources
- Results must be easily interpretable and queryable
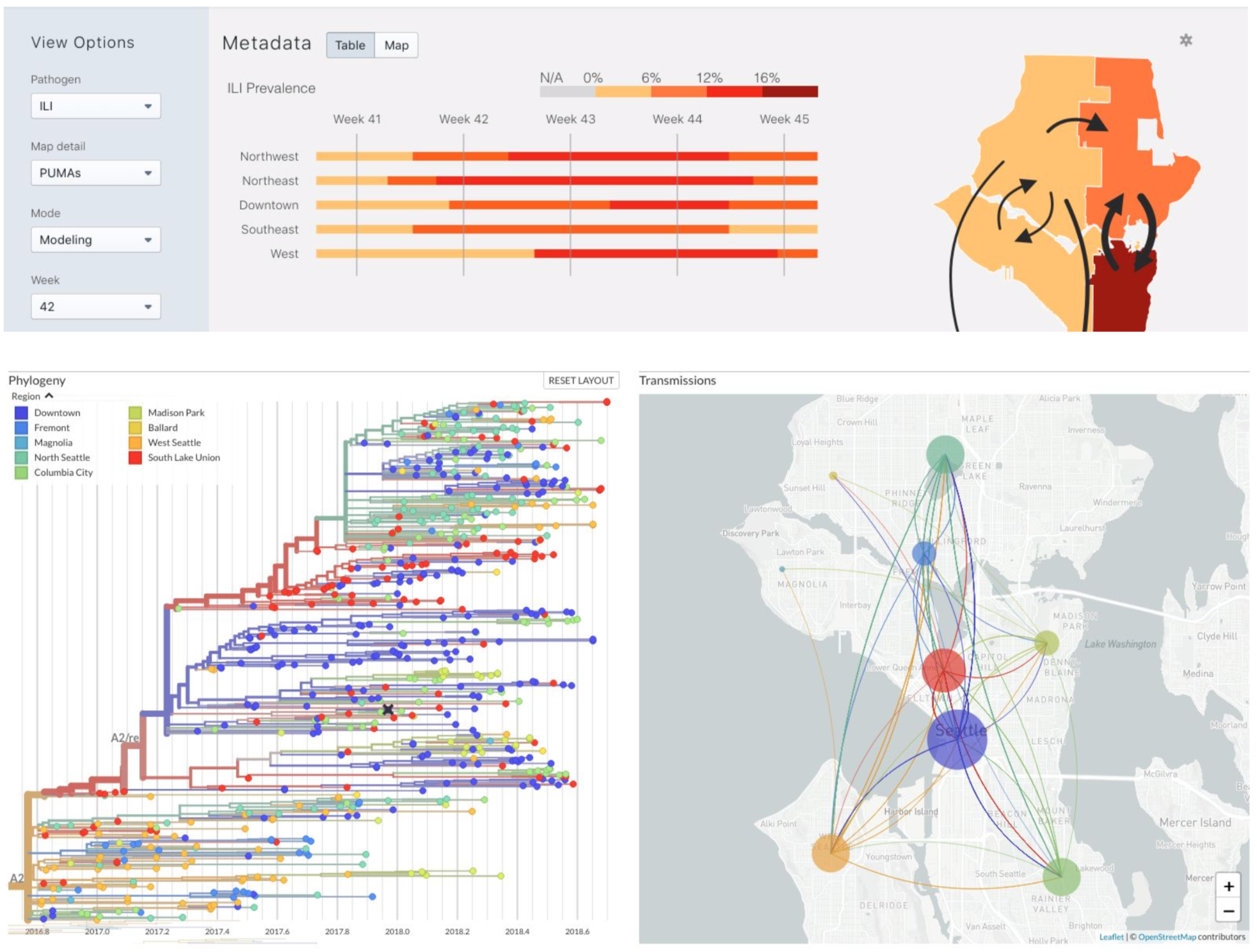
Nextstrain
Project to conduct real-time molecular epidemiology and evolutionary analysis of emerging epidemics
with
![]() Richard Neher,
Richard Neher,
![]() James Hadfield,
James Hadfield,
![]() Emma Hodcroft,
Emma Hodcroft,
![]() Tom Sibley,
Tom Sibley,
![]() John Huddleston,
John Huddleston,
![]() Colin Megill,
Colin Megill,
![]() Sidney Bell,
Sidney Bell,
![]() Barney Potter,
Barney Potter,
![]() Charlton Callender
Charlton Callender
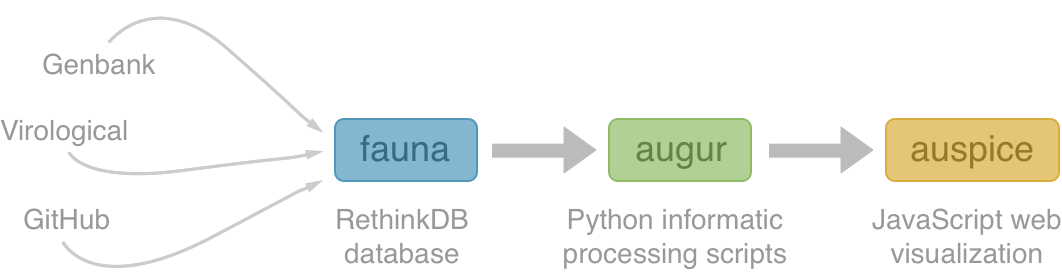
Nextstrain architecture
All code open source at github.com/nextstrain
Two central aims: (1) rapid and flexible phylodynamic analysis and
(2) interactive visualization
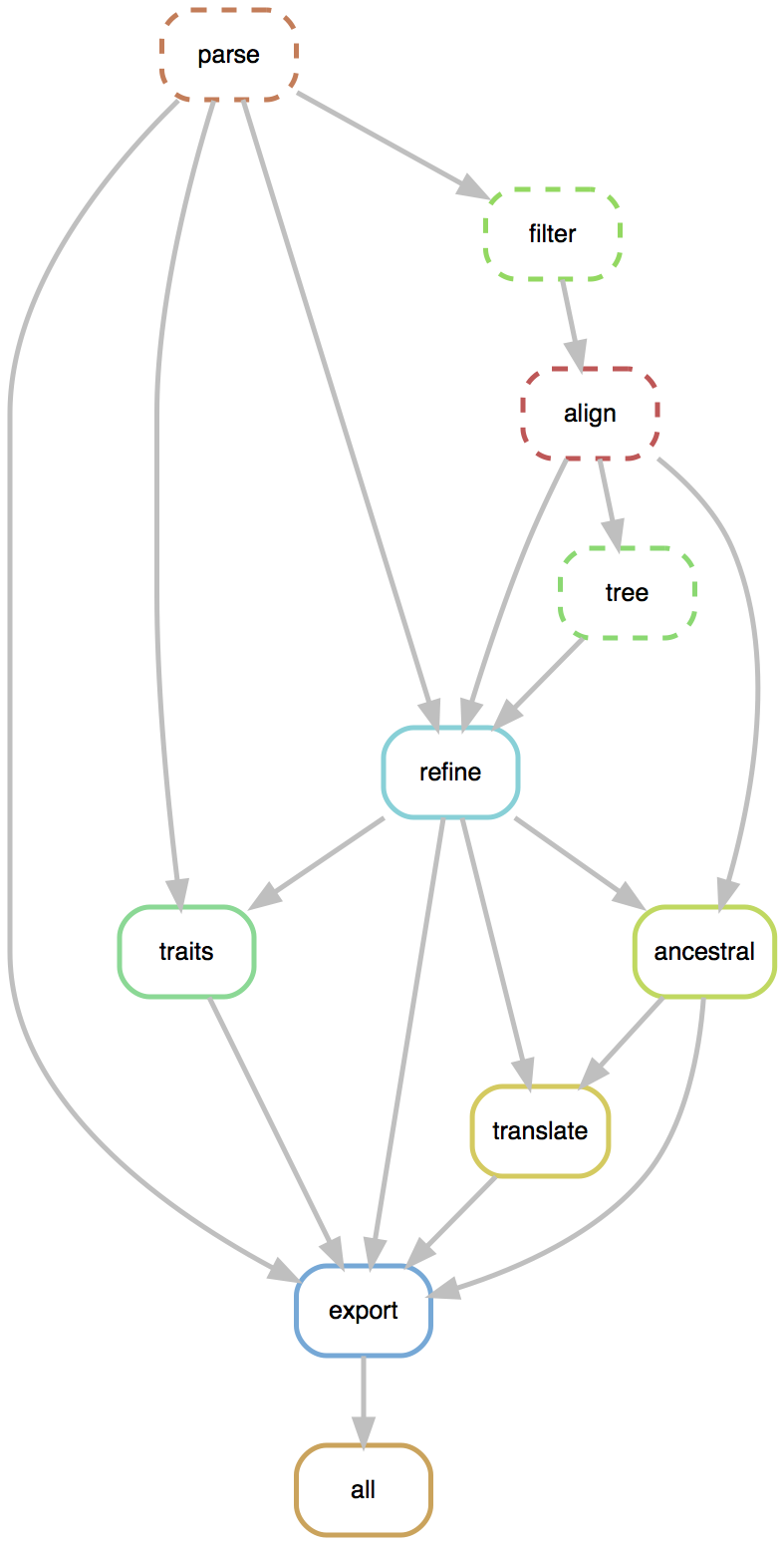
Rapid build pipeline for 1600 Ebola genomes
- Align with MAFFT (34 min)
- Build ML tree with RAxML (54 min)
- Temporally resolve tree and geographic ancestry with TreeTime (16 min)
- Total pipeline (1 hr 46 min)
Flexible pipelines constructed through command line modules
- Modules called via
augur filter,augur tree,augur traits, etc... - Designed to be composable across pathogen builds
- Defined pipeline, making steps obvious
- Provides dependency graph for fast recomputation
- Pathogen-specific repos give users an obvious foundation to build from
Nextstrain is two things
- a bioinformatics toolkit and visualization app, which can be used for a broad range of datasets
- a collection of real-time pathogen analyses kept up-to-date on the website nextstrain.org
nextstrain.org
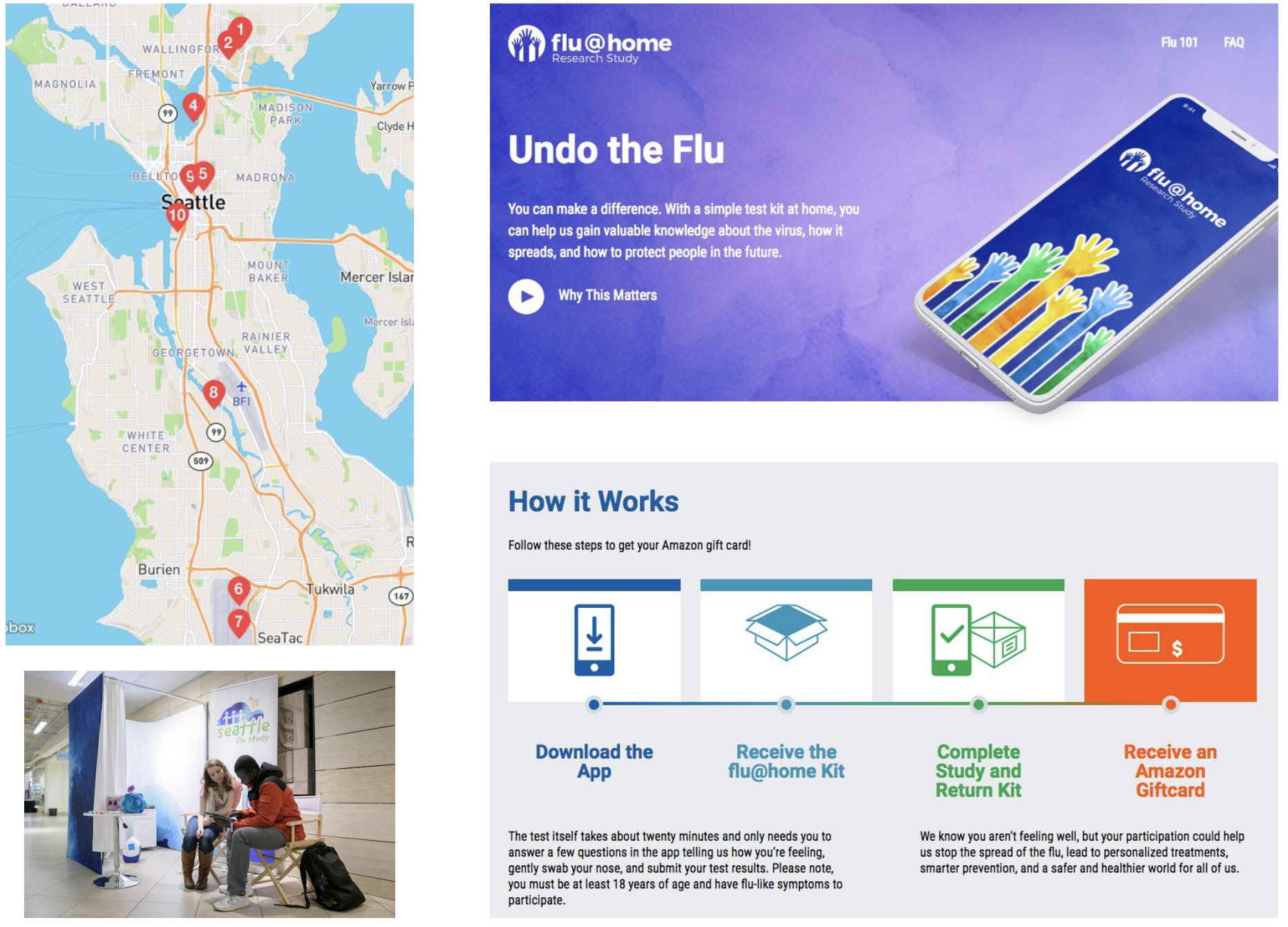
Rapid on-the-ground sequencing in Makeni, Sierra Leone
Newly released features
- Bacteria build pipelines using VCF rather than FASTA
- "Community" builds to promote frictionless sharing of results
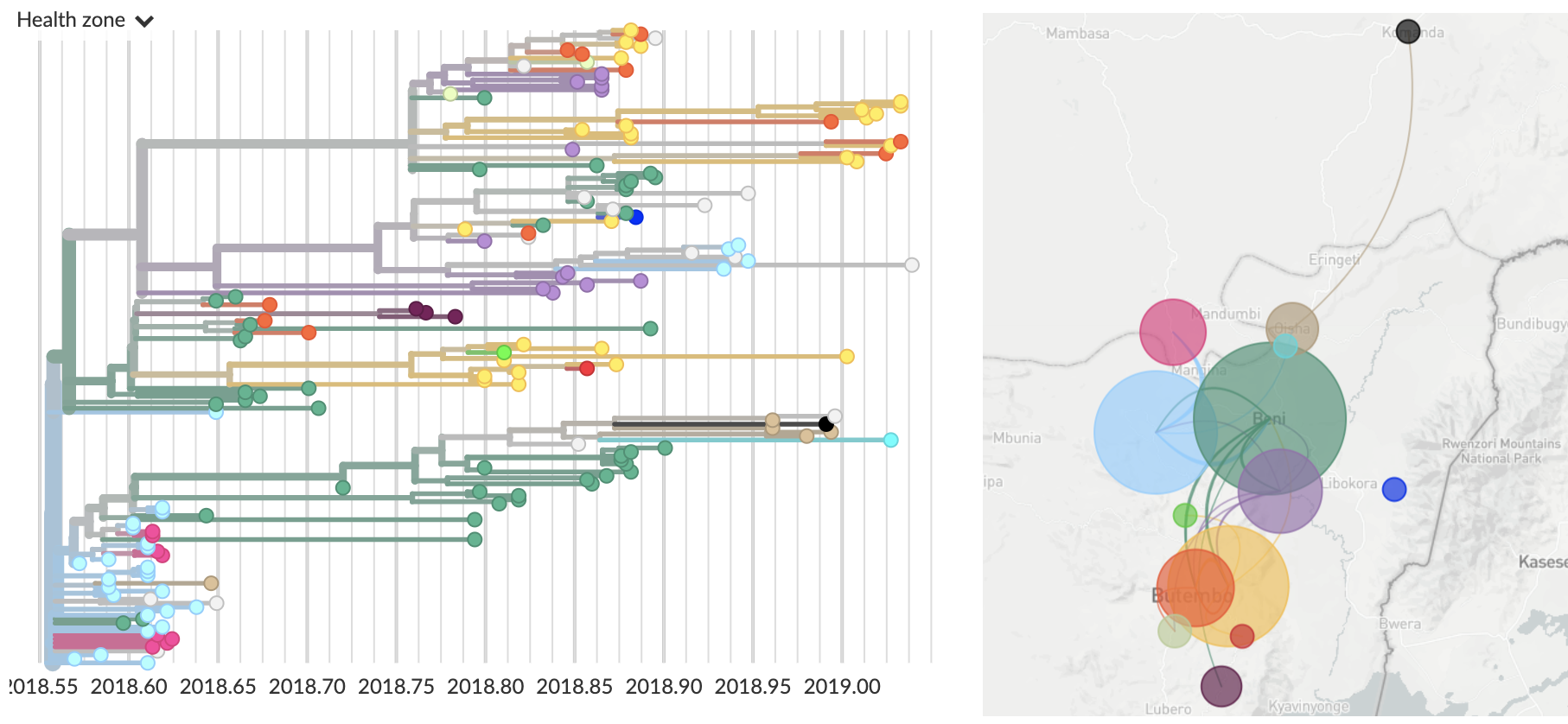
Genomic epidemiology of the ongoing DRC Ebola epidemic
Acknowledgements
Bedford Lab:
 Alli Black,
Alli Black,
 John Huddleston,
John Huddleston,
 Barney Potter,
Barney Potter,
 James Hadfield,
James Hadfield,
 Louise Moncla,
Louise Moncla,
 Tom Sibley,
Tom Sibley,
 Maya Lewinsohn,
Maya Lewinsohn,
 Katie Kistler
Katie Kistler
MERS: Gytis Dudas, Andrew Rambaut, Luiz Carvalho
Ebola: Gytis Dudas, Andrew Rambaut, Luiz Carvalho, Philippe Lemey, Marc Suchard, Andrew Tatem
Nextstrain: Richard Neher, James Hadfield, Emma Hodcroft, Tom Sibley, John Huddleston, Sidney Bell, Barney Potter, Colin Megill, Charlton Callender