Real-time genomic surveillance of pathogen evolution and spread
Trevor Bedford (@trvrb)
4 Apr 2017
D-BSSE Seminar Series
ETH Zurich
Slides at bedford.io/talks/
Phylogenies
Phylogenies describe history
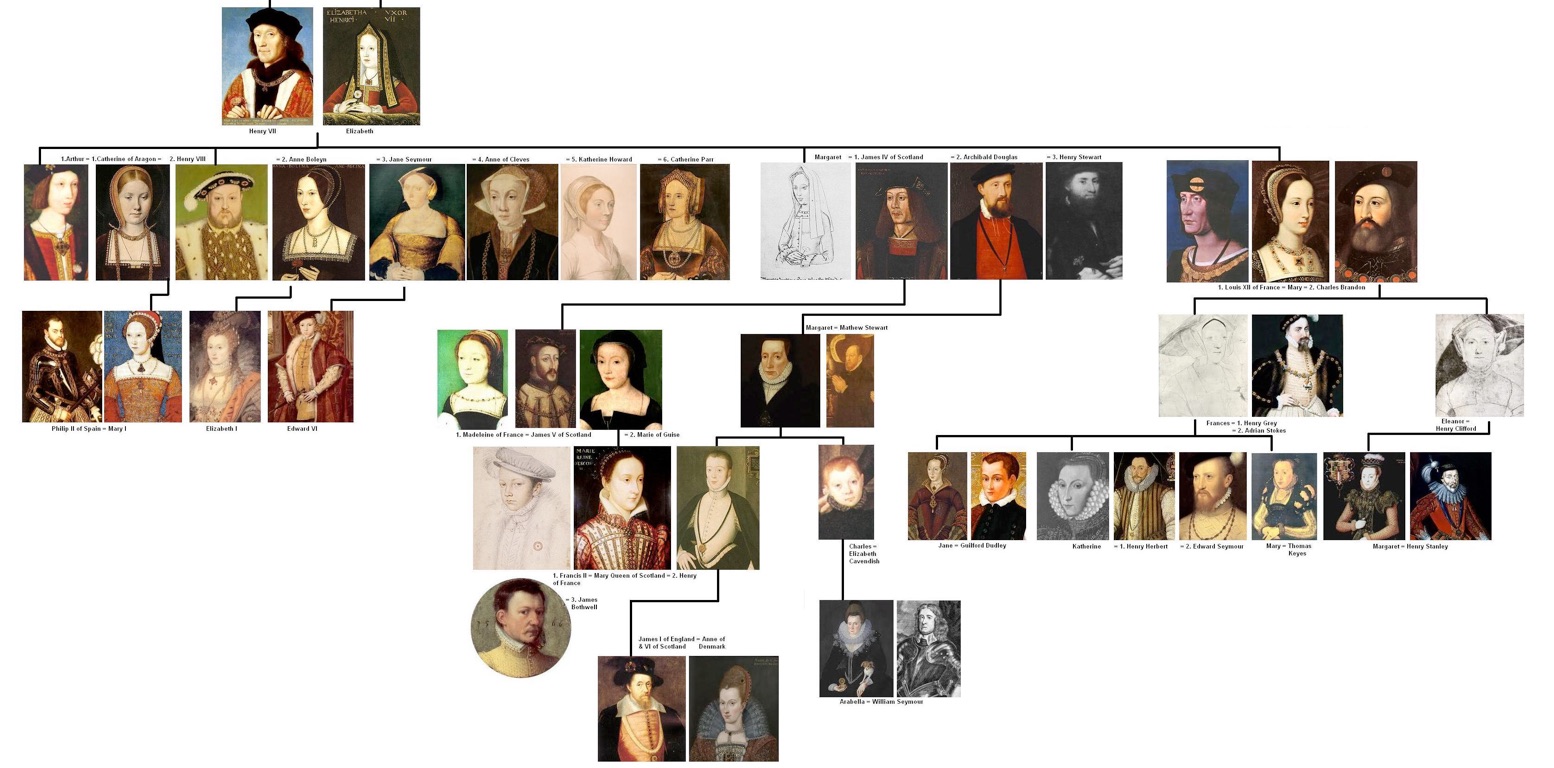
Phylogenies describe history
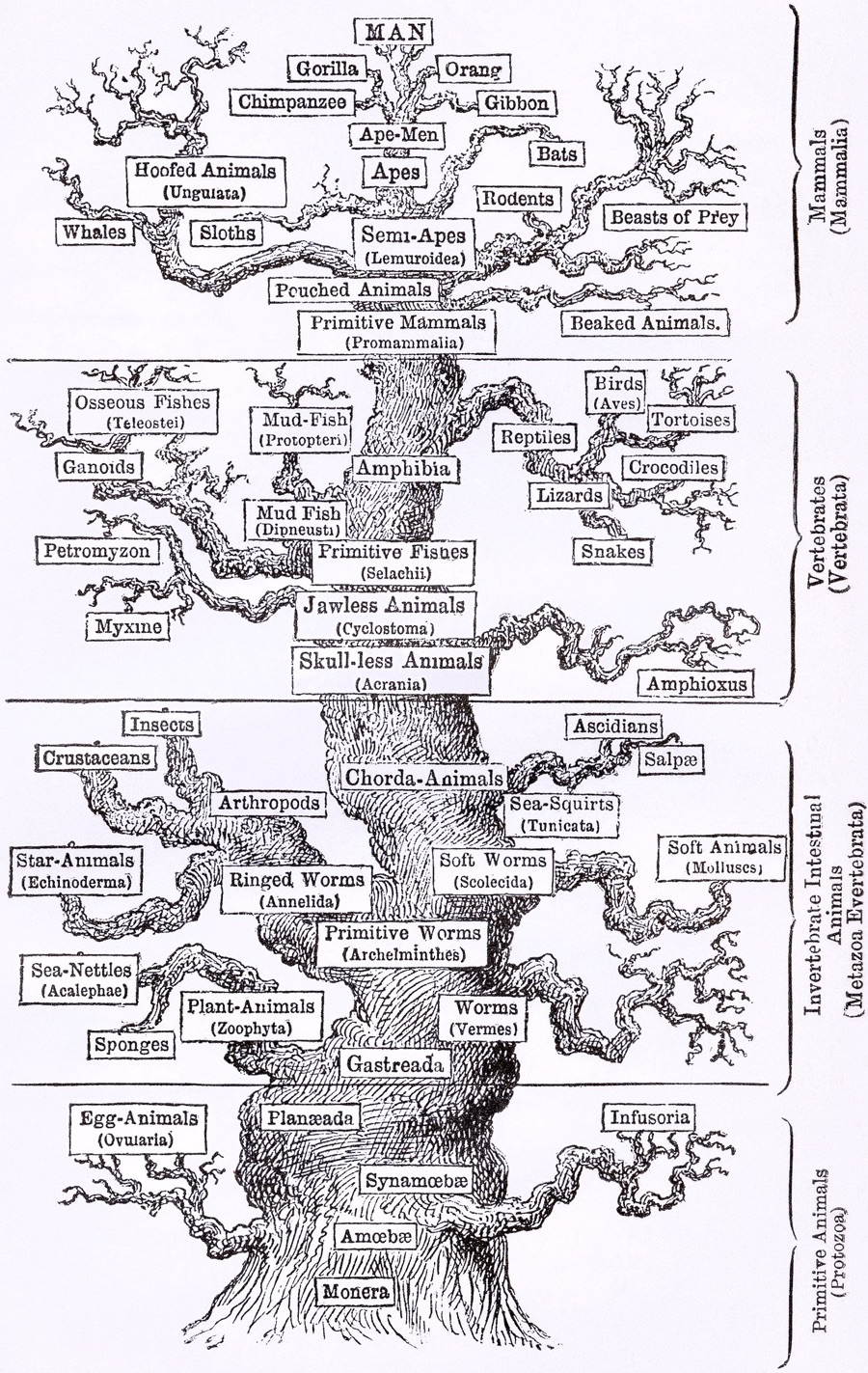
Phylogenies describe history
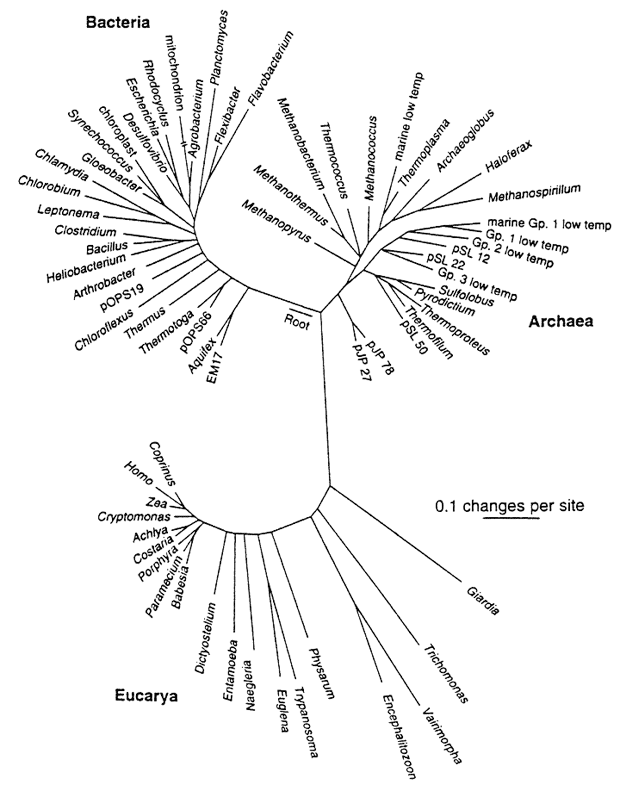
Phylogenies reveal process
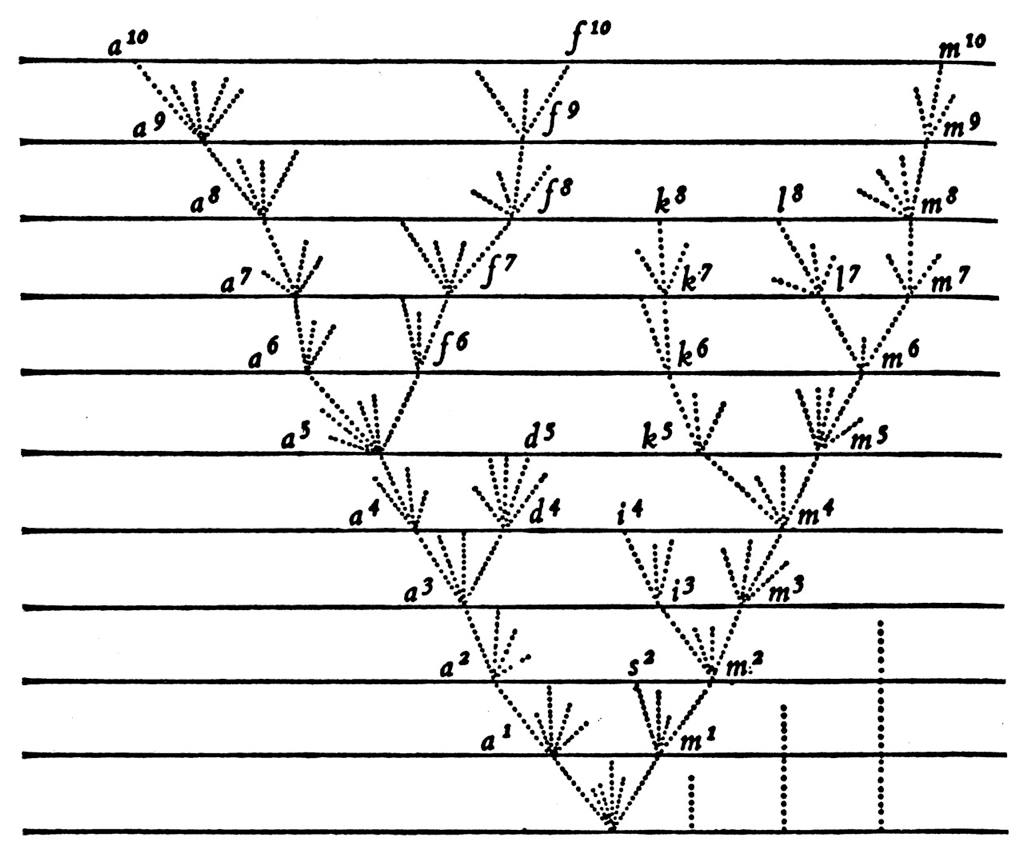
Epidemic process
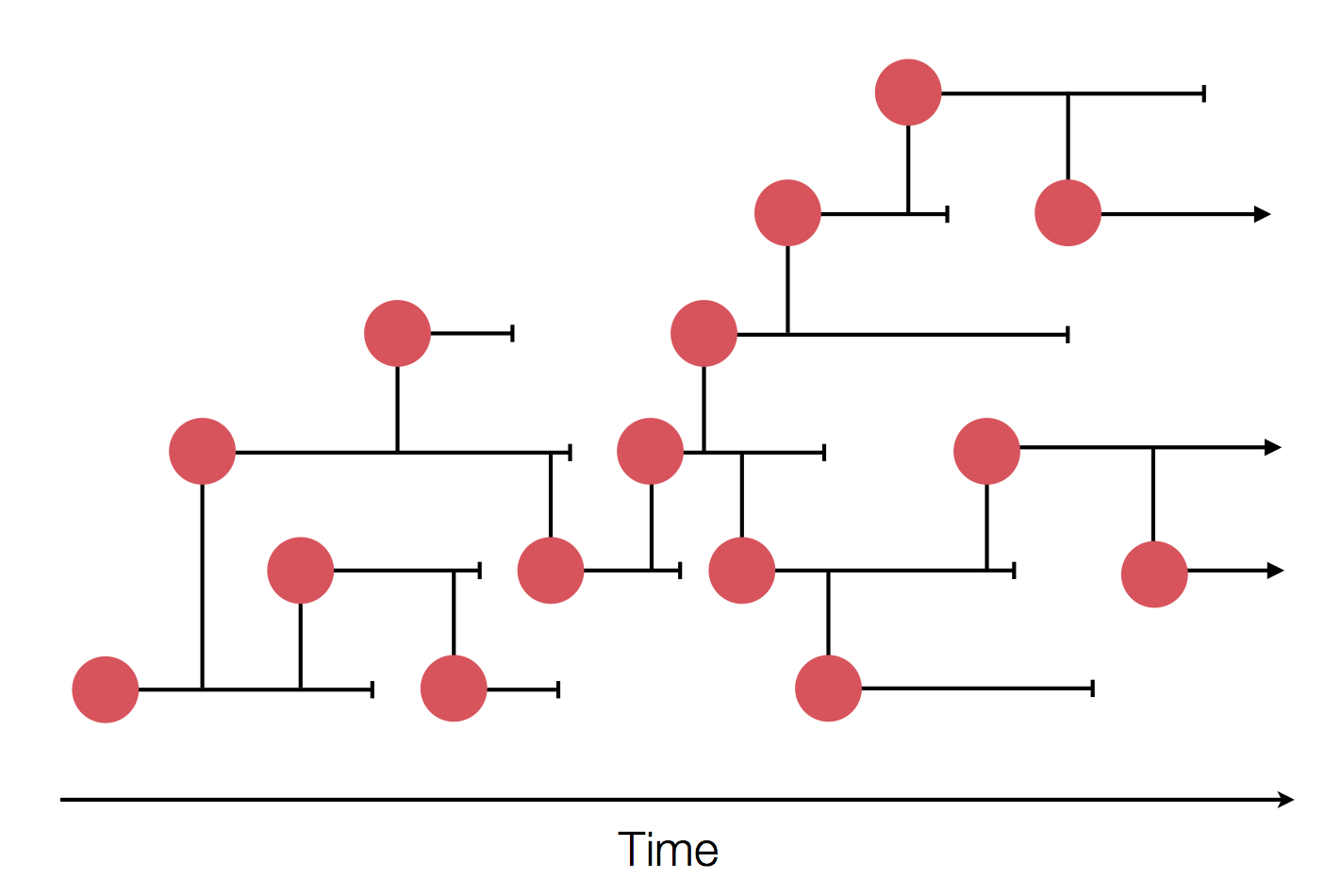
Sample some individuals
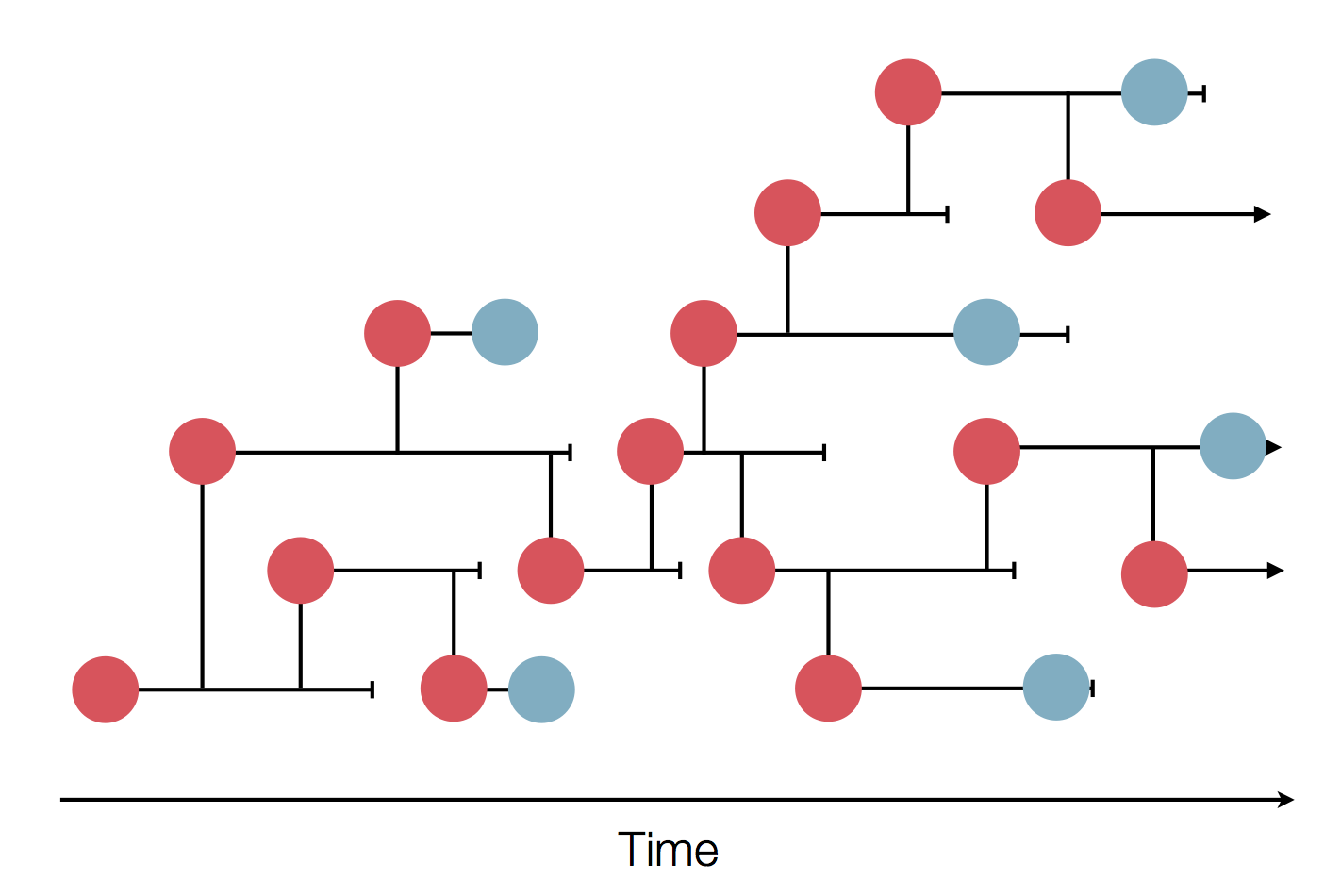
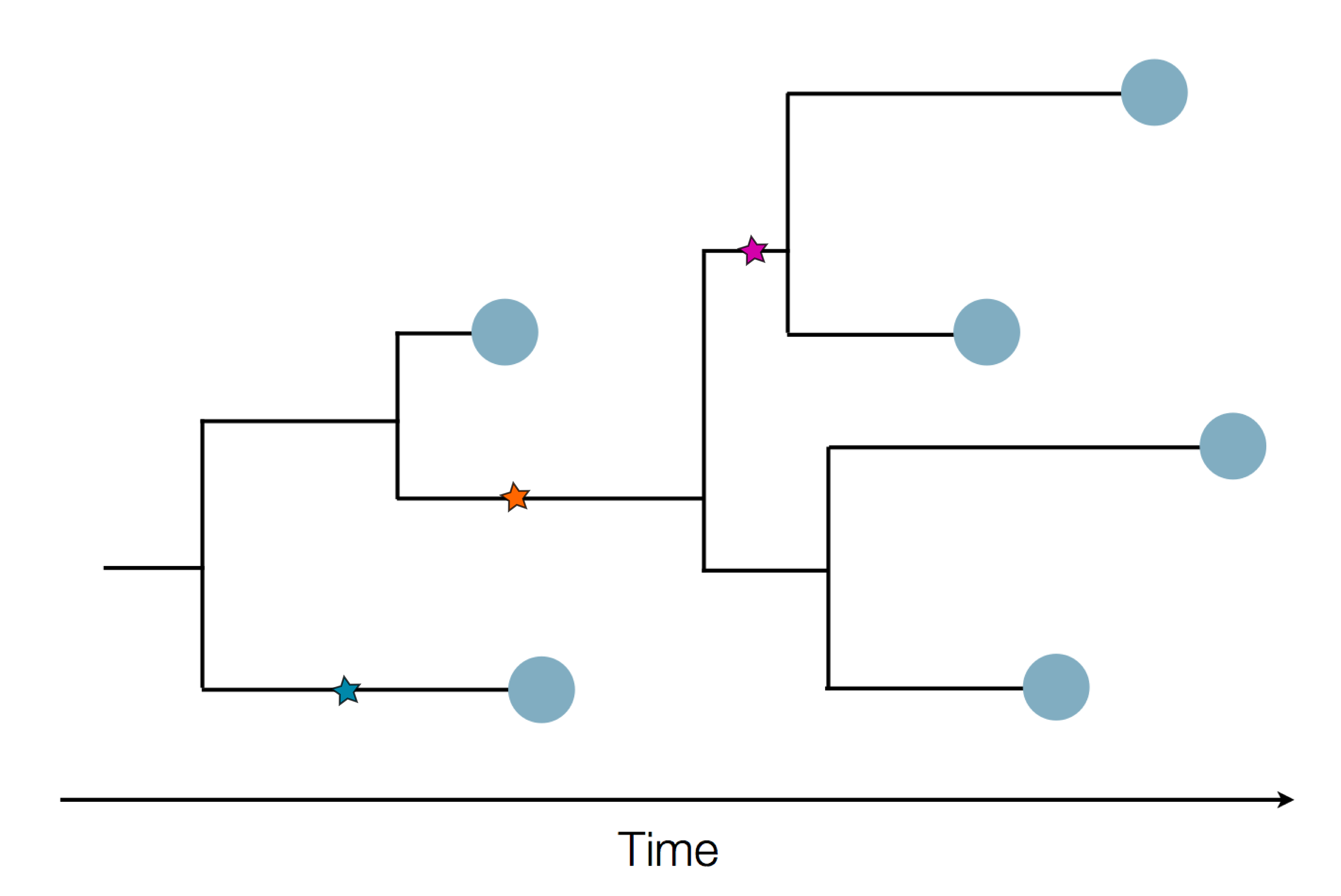
Sequence and determine phylogeny
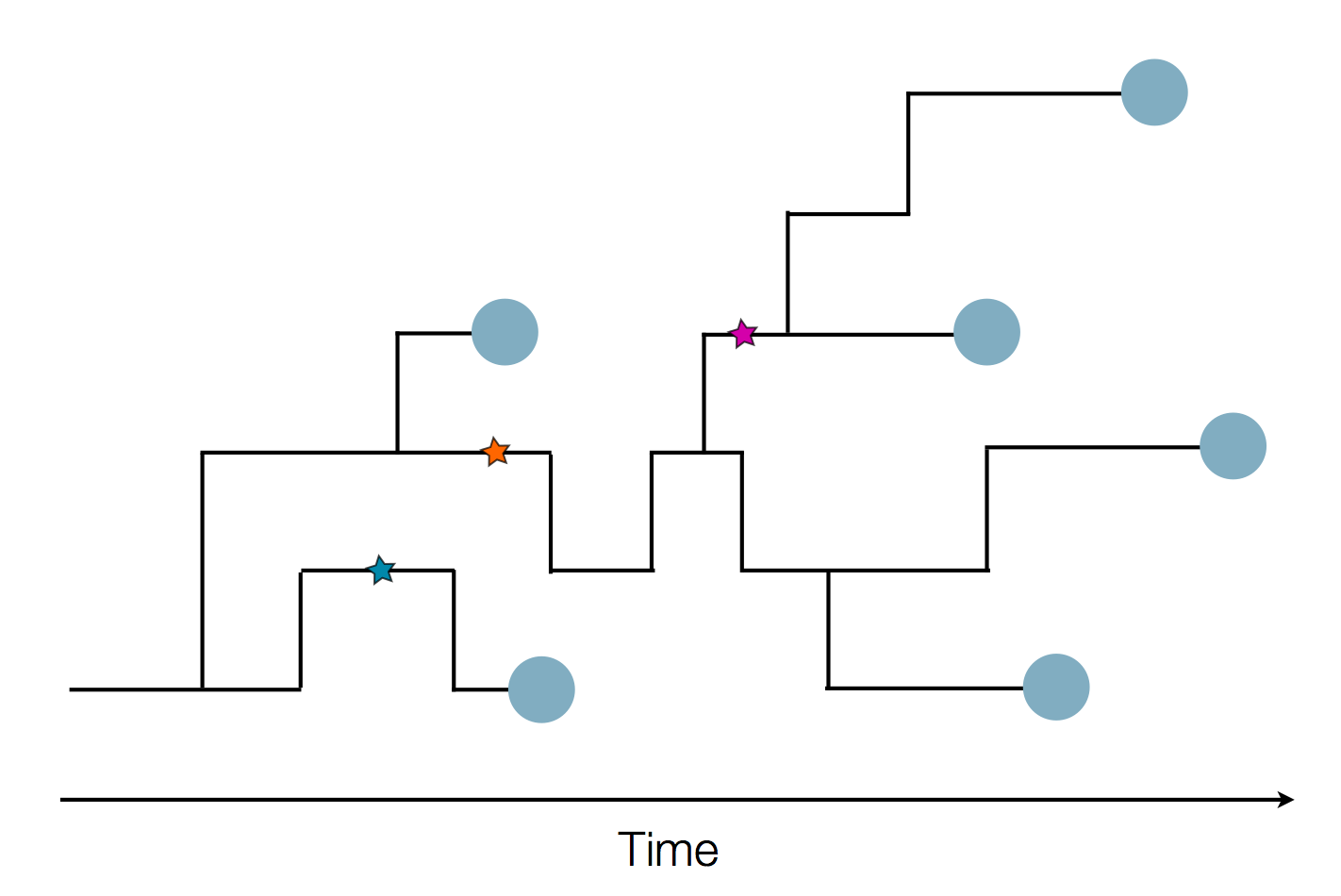
Sequence and determine phylogeny
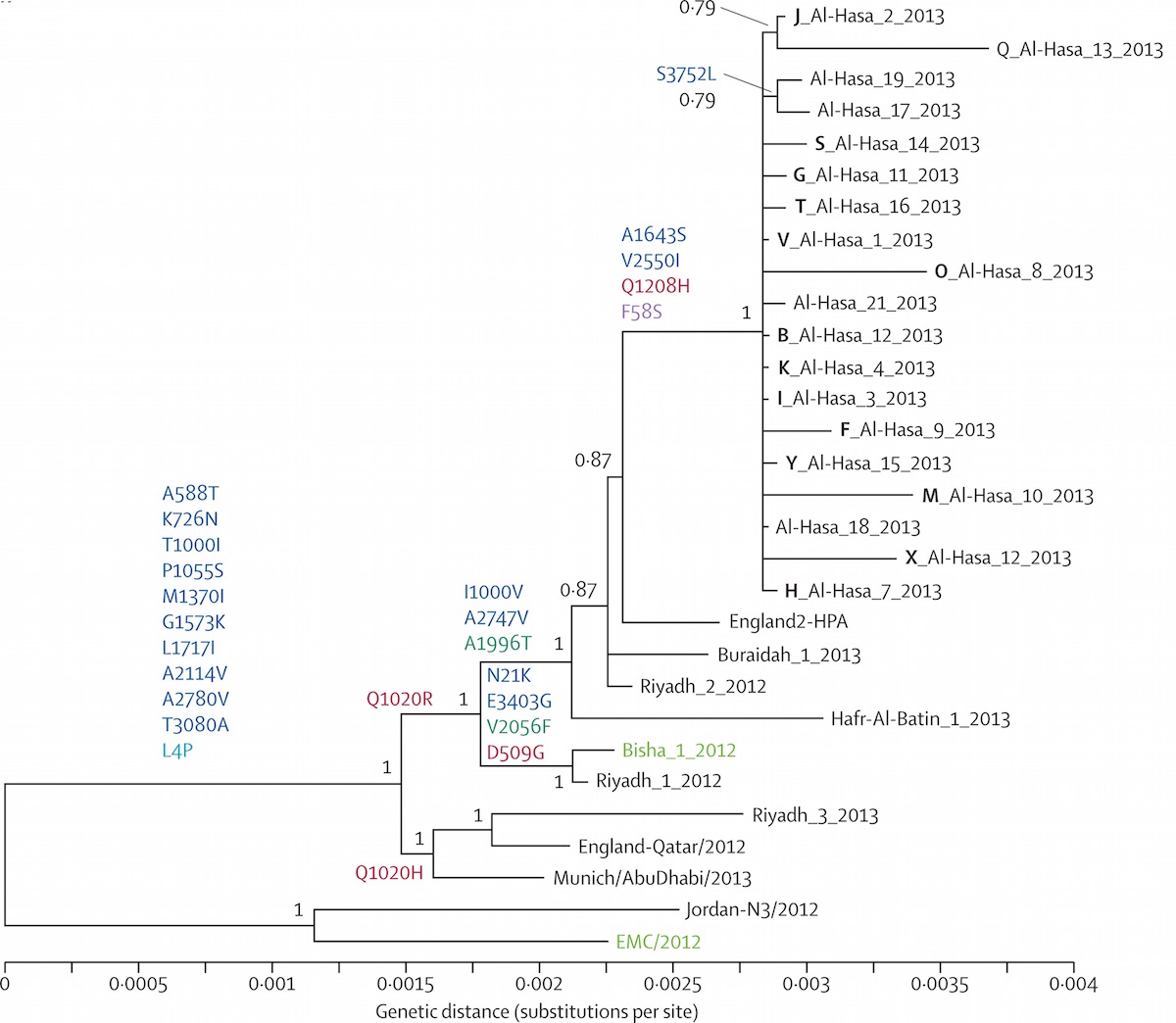
Localized Middle Eastern MERS-CoV phylogeny
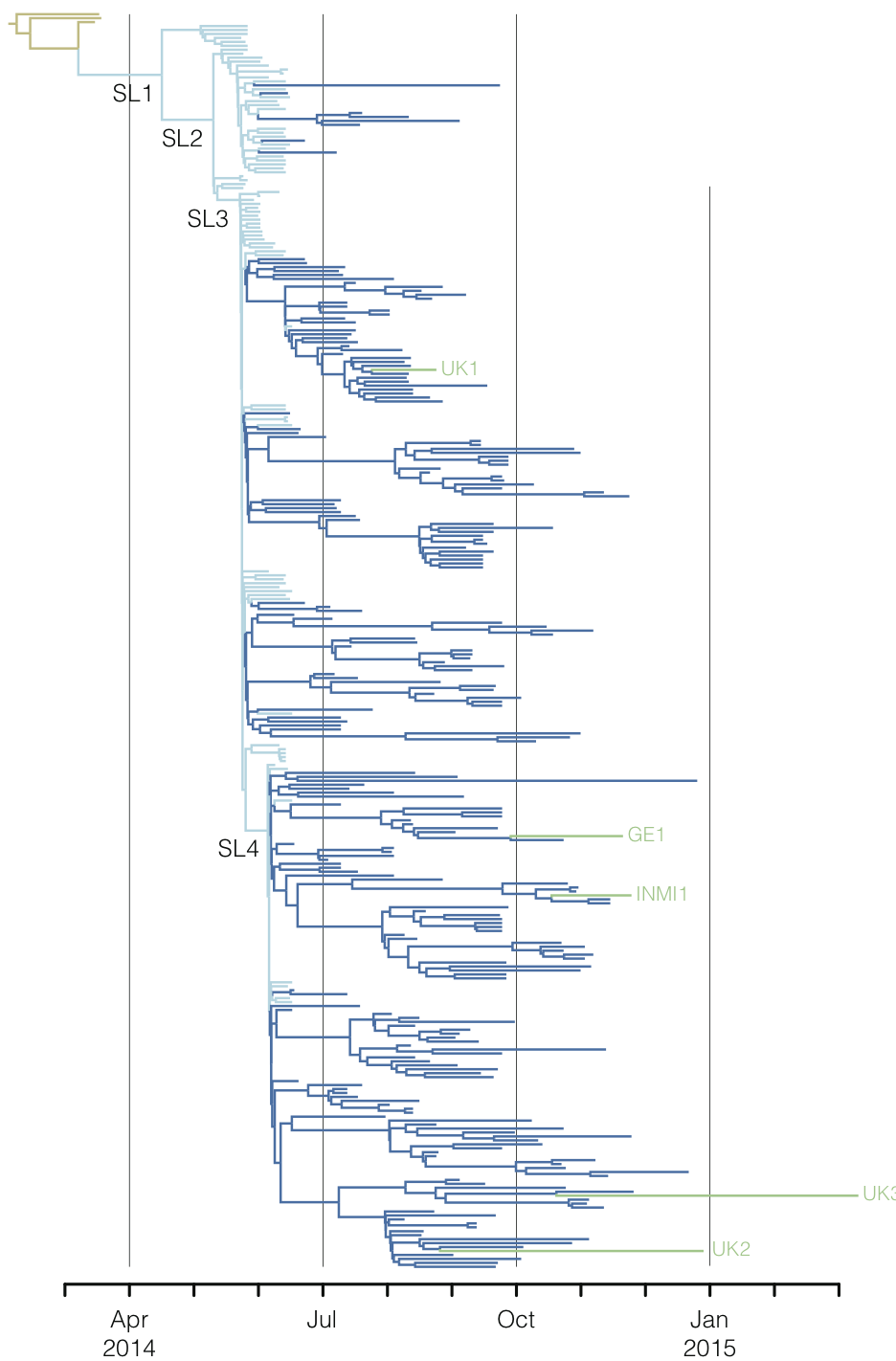
Regional West African Ebola phylogeny
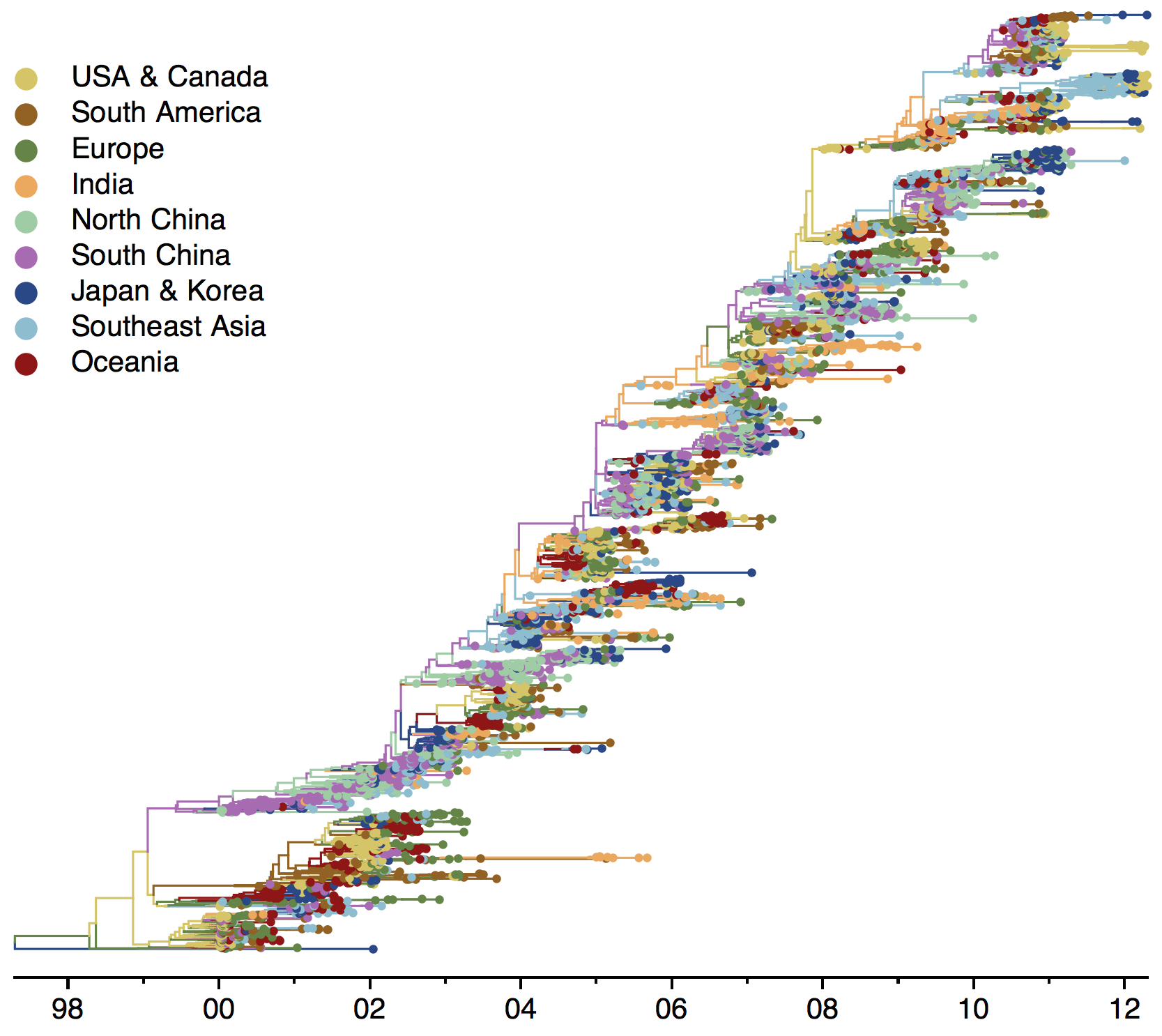
Global influenza phylogeny
Applications of evolutionary analysis for influenza vaccine strain selection and charting spread of Ebola and Zika
Influenza
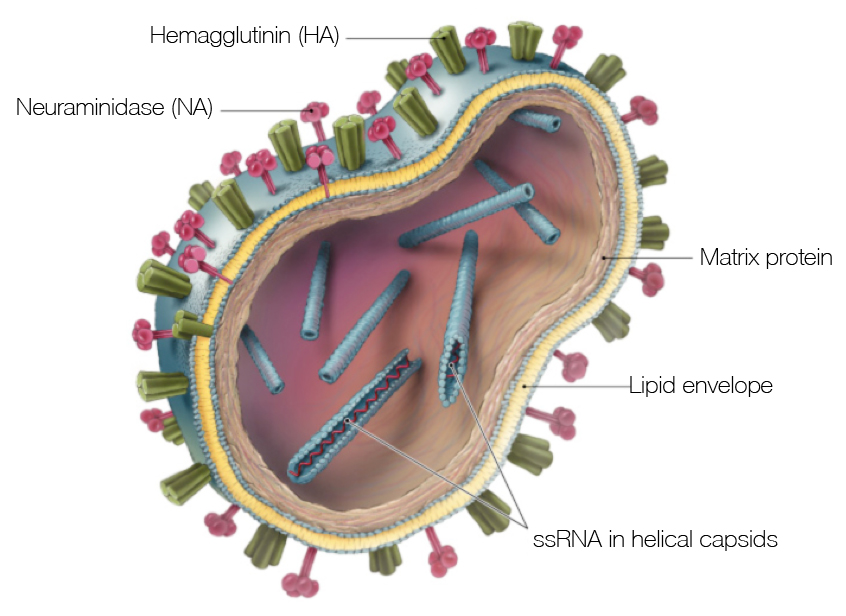
Influenza virion
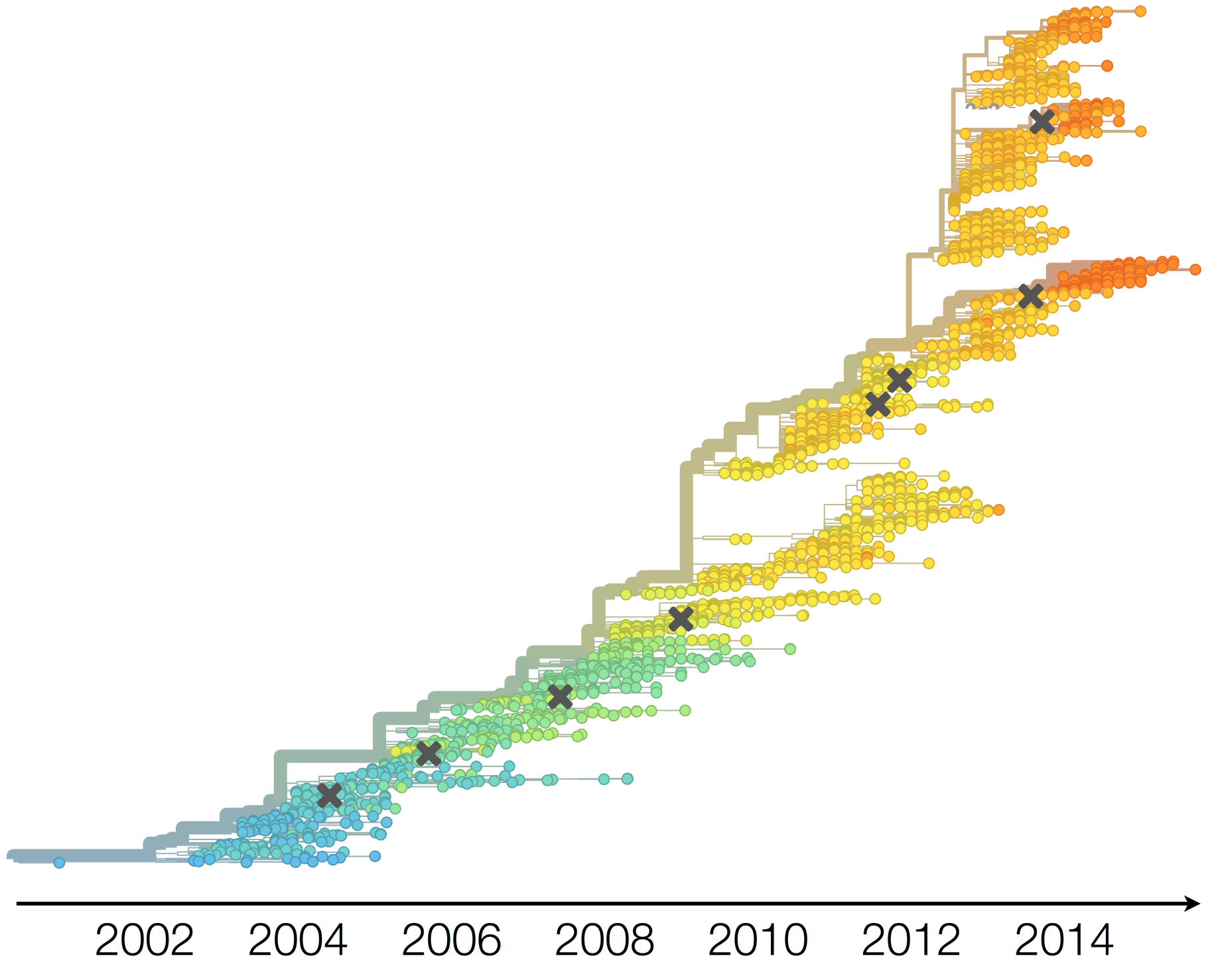
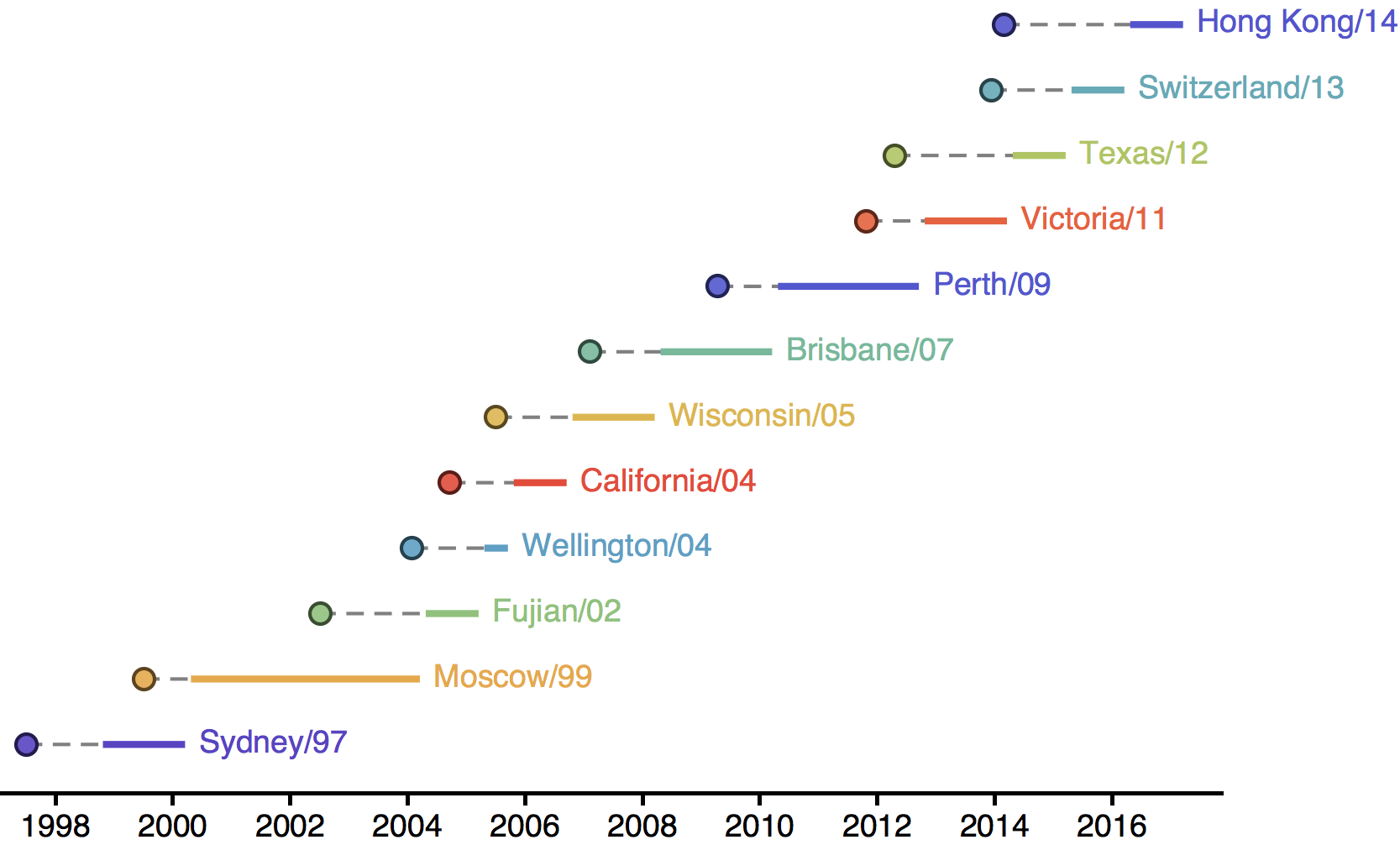
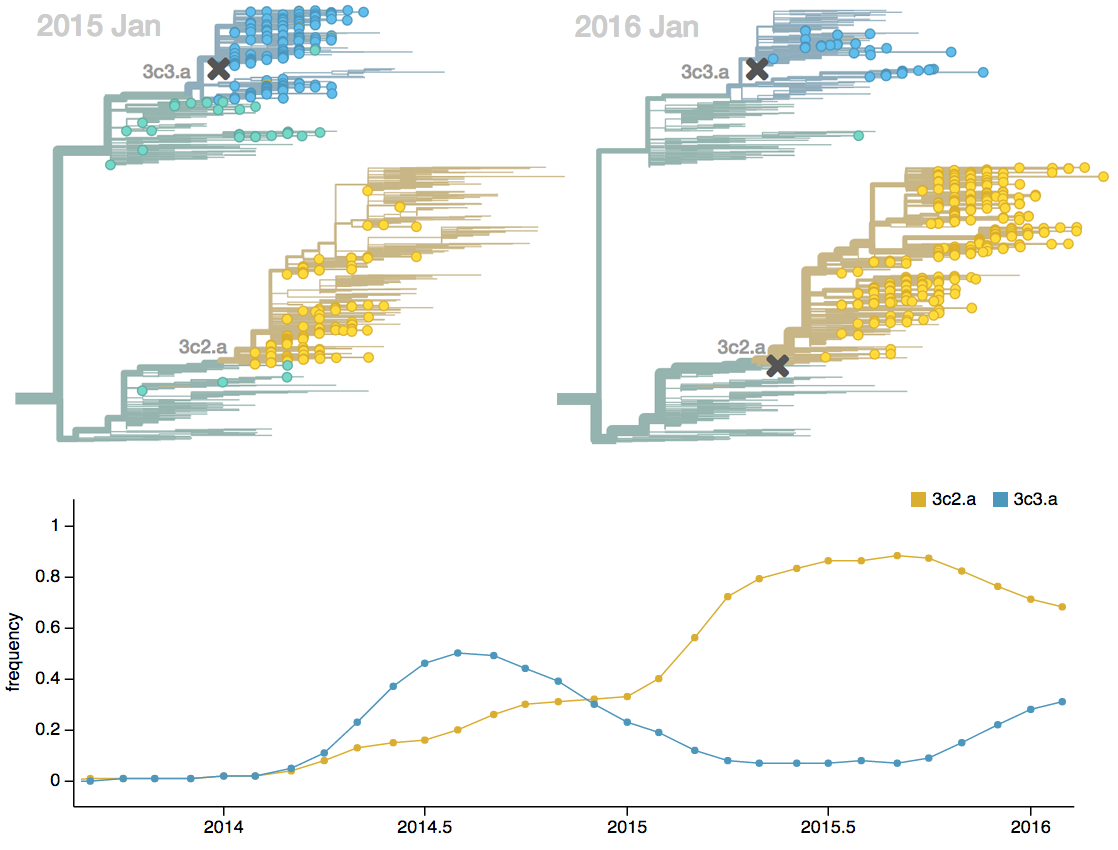
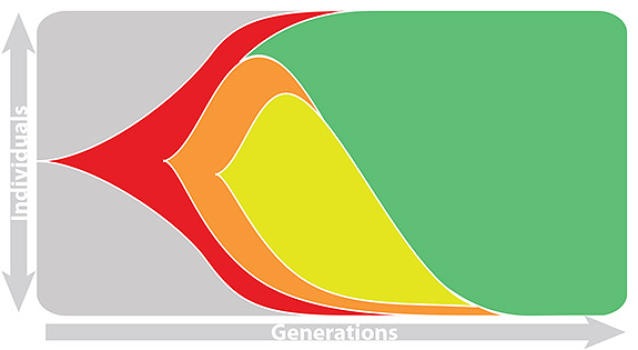
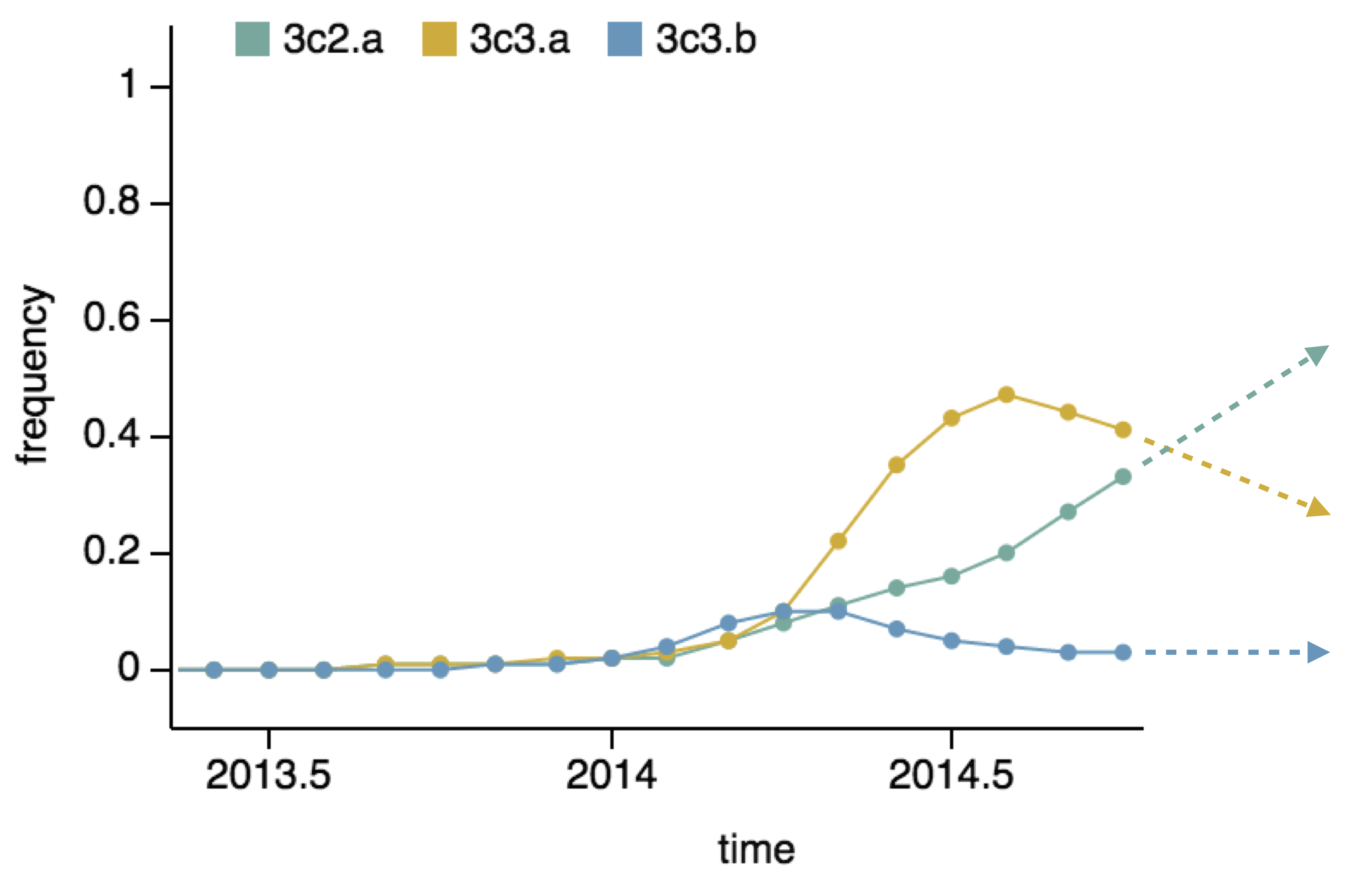
Population turnover (in H3N2) is extremely rapid
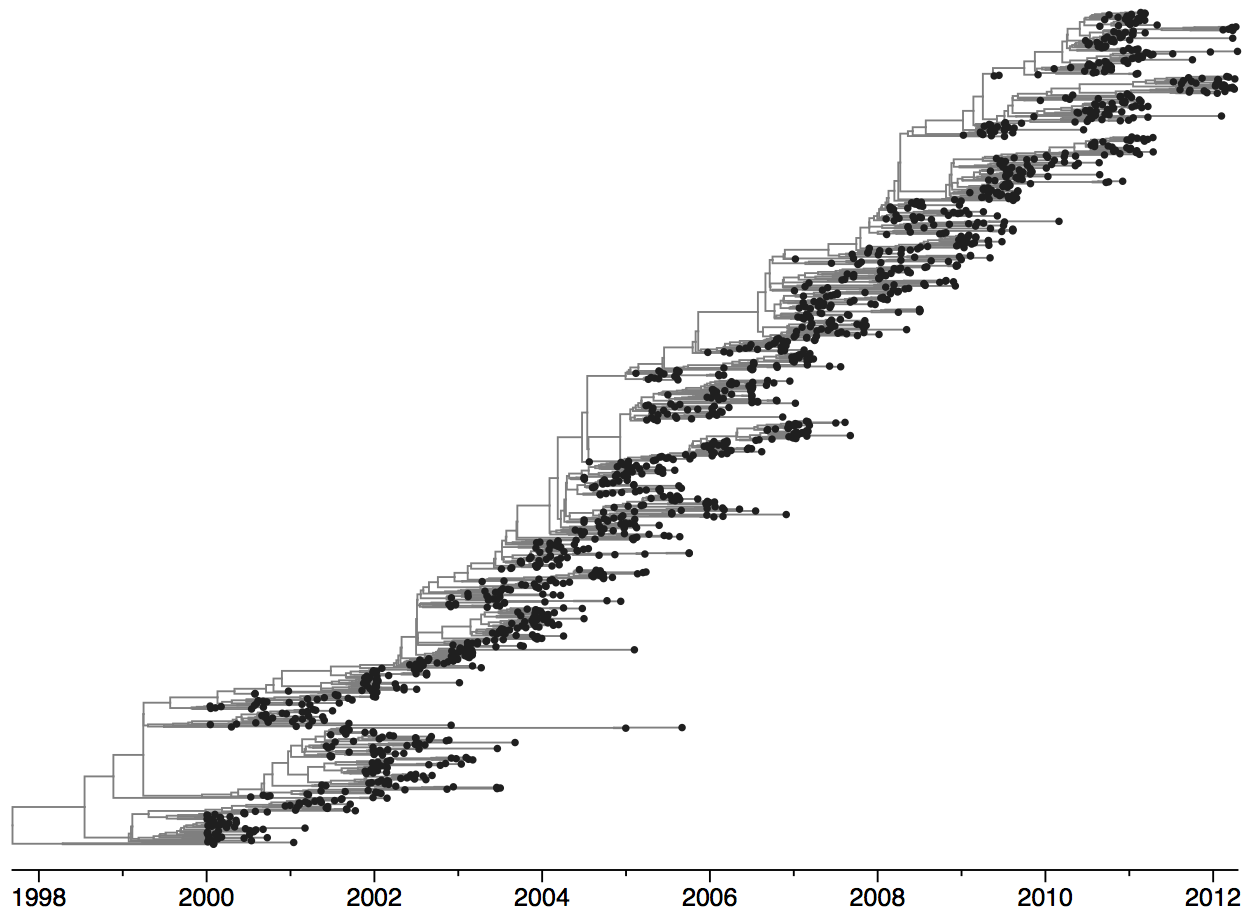
Clades emerge, die out and take over
Clades show rapid turnover

Dynamics driven by antigenic drift
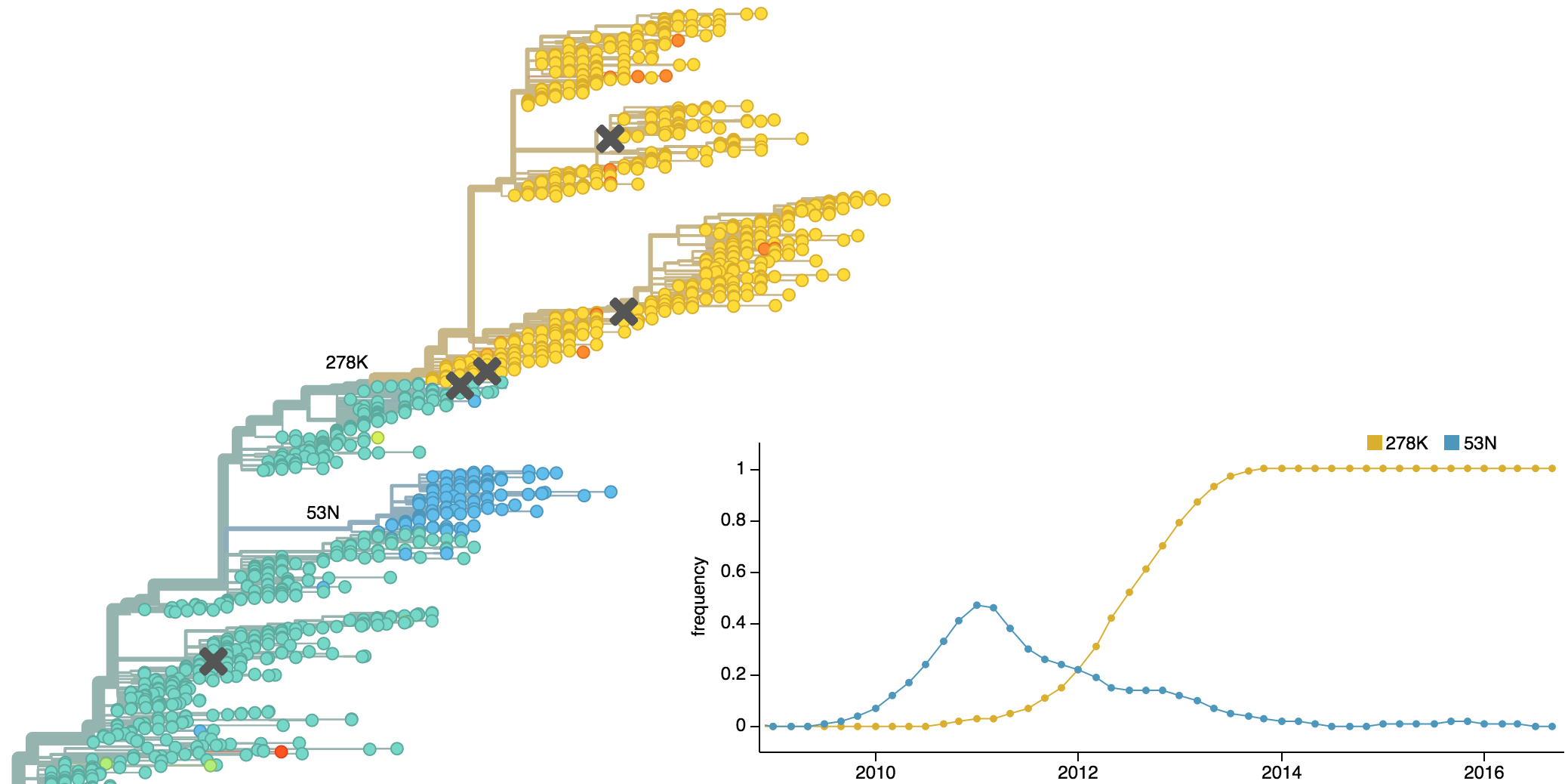
Drift variants emerge and rapidly take over in the virus population
This causes the side effect of evading existing vaccine formulations
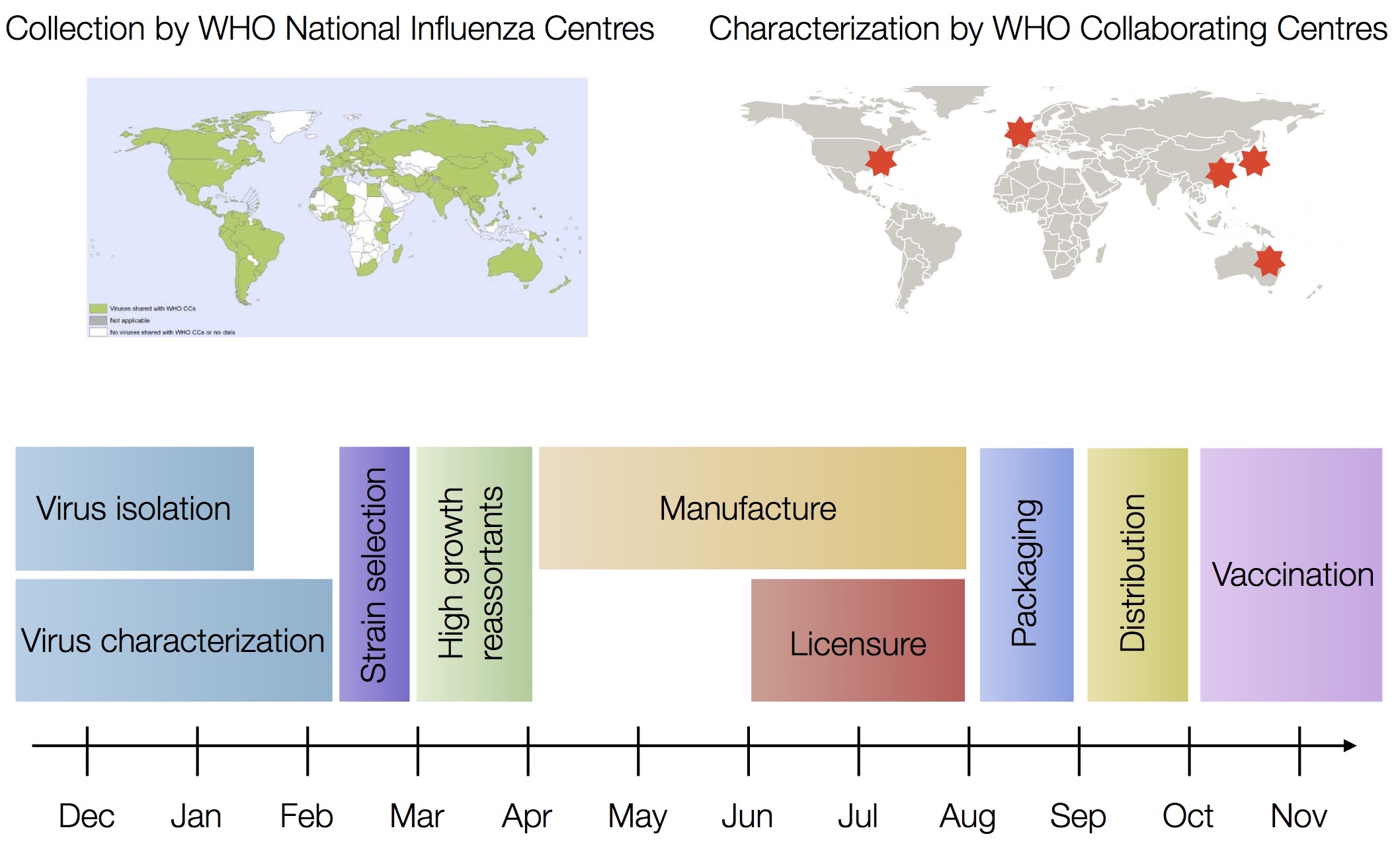
Drift necessitates vaccine updates
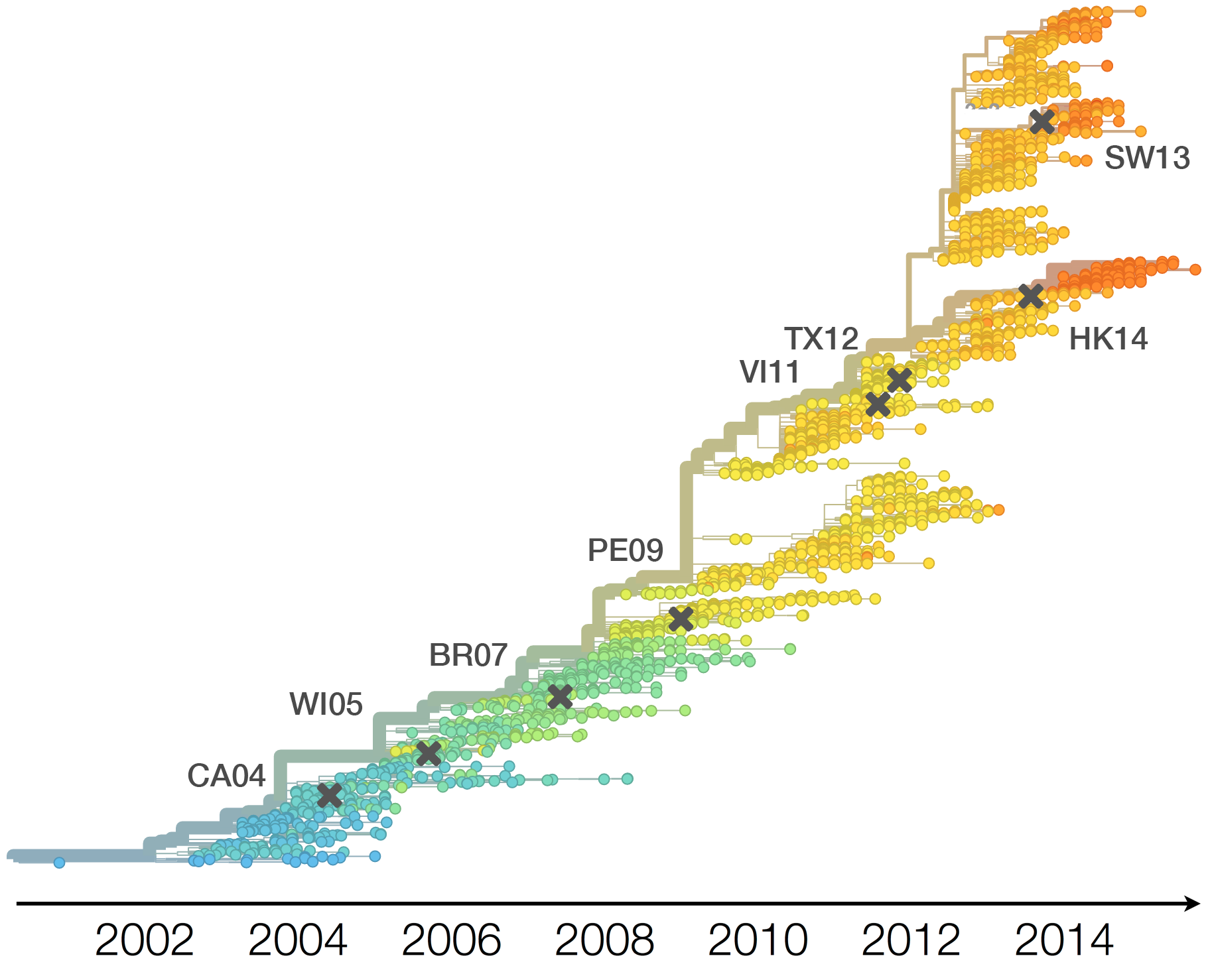
H3N2 vaccine updates occur every ~2 years
Timely surveillance and rapid analysis essential to vaccine strain selection
nextflu
Project to provide a real-time view of the evolving influenza population
nextflu
Project to provide a real-time view of the evolving influenza population
All in collaboration with Richard Neher

nextflu pipeline
- Download all recent HA sequences from GISAID
- Filter to remove outliers
- Subsample across time and space
- Align sequences
- Build tree
- Estimate clade frequencies
- Infer antigenic phenotypes
- Export for visualization
nextflu.org
Forecasting
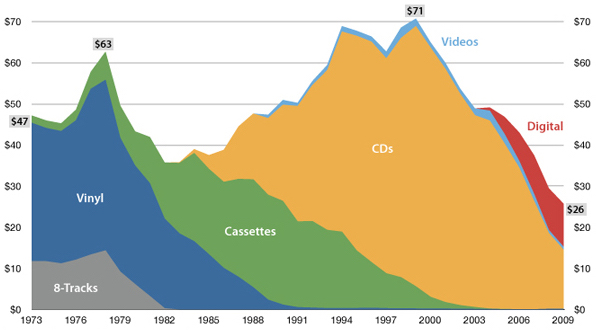
The future is here, it's just not evenly distributed yet
— William Gibson
USA music industry, 2011 dollars per capita
Influenza population turnover
Vaccine strain selection timeline
Seek to explain change in clade frequencies over 1 year
Fitness models can project clade frequencies
Clade frequencies $X$ derive from the fitnesses $f$ and frequencies $x$ of constituent viruses, such that
$$\hat{X}_v(t+\Delta t) = \sum_{i:v} x_i(t) \, \mathrm{exp}(f_i \, \Delta t)$$
This captures clonal interference between competing lineages
Predictive fitness models
A simple predictive model estimates the fitness $f$ of virus $i$ as
$$\hat{f}_i = \beta^\mathrm{ep} \, f_i^\mathrm{ep} + \beta^\mathrm{ne} \, f_i^\mathrm{ne}$$
where $f_i^\mathrm{ep}$ measures cross-immunity via substitutions at epitope sites and $f_i^\mathrm{ep}$ measures mutational load via substitutions at non-epitope sites
We implement a similar model based on two predictors
- Clade frequency change
- Antigenic advancement
Project frequencies forward,
growing clades have high fitness
Calculate antigenic difference from ancestor via serological data
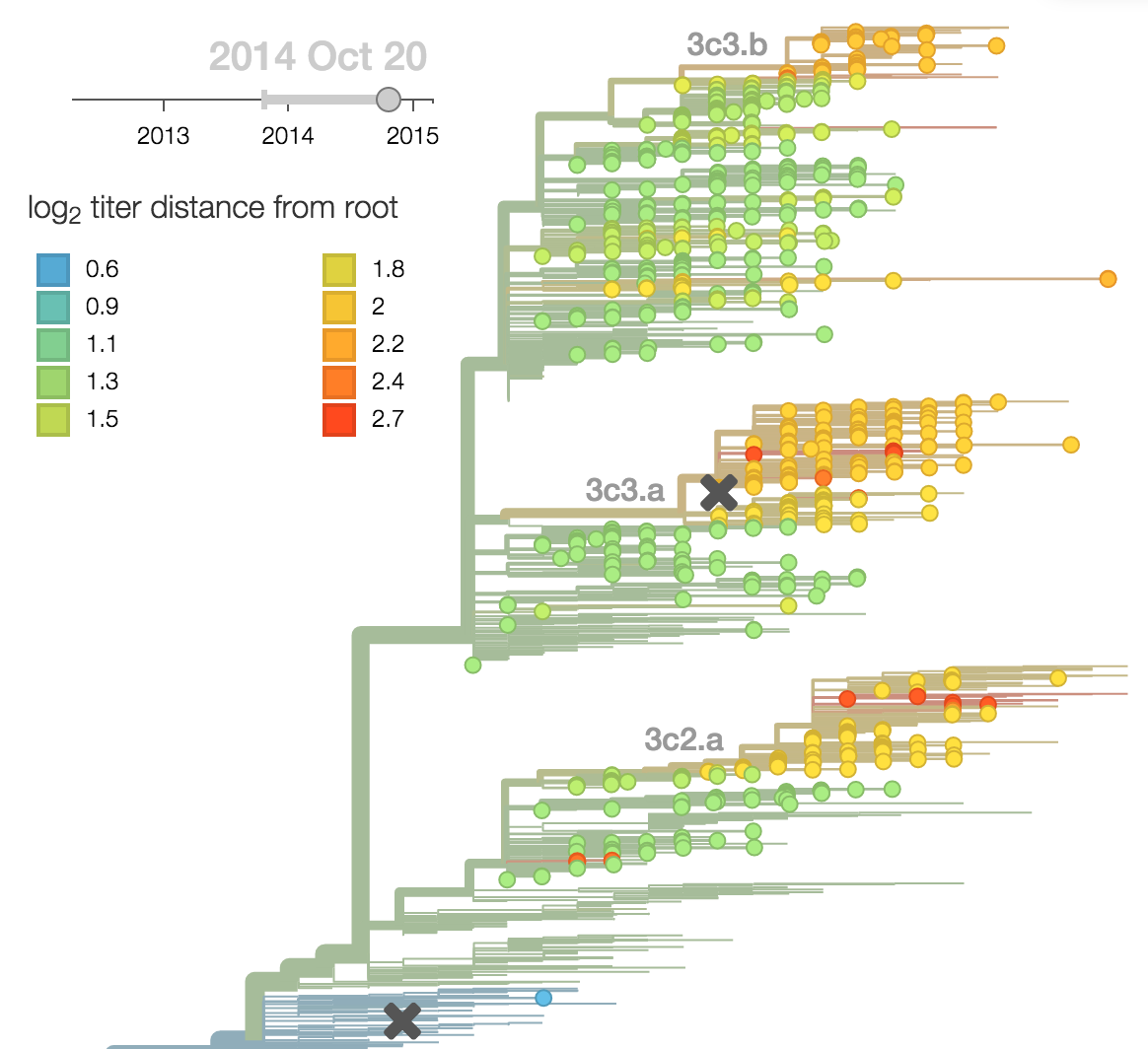
Fitness model parameterization
Our predictive model estimates the fitness $f$ of virus $i$ as
$$\hat{f}_i = \beta^\mathrm{freq} \, f_i^\mathrm{freq} + \beta^\mathrm{HI} \, f_i^\mathrm{HI}$$
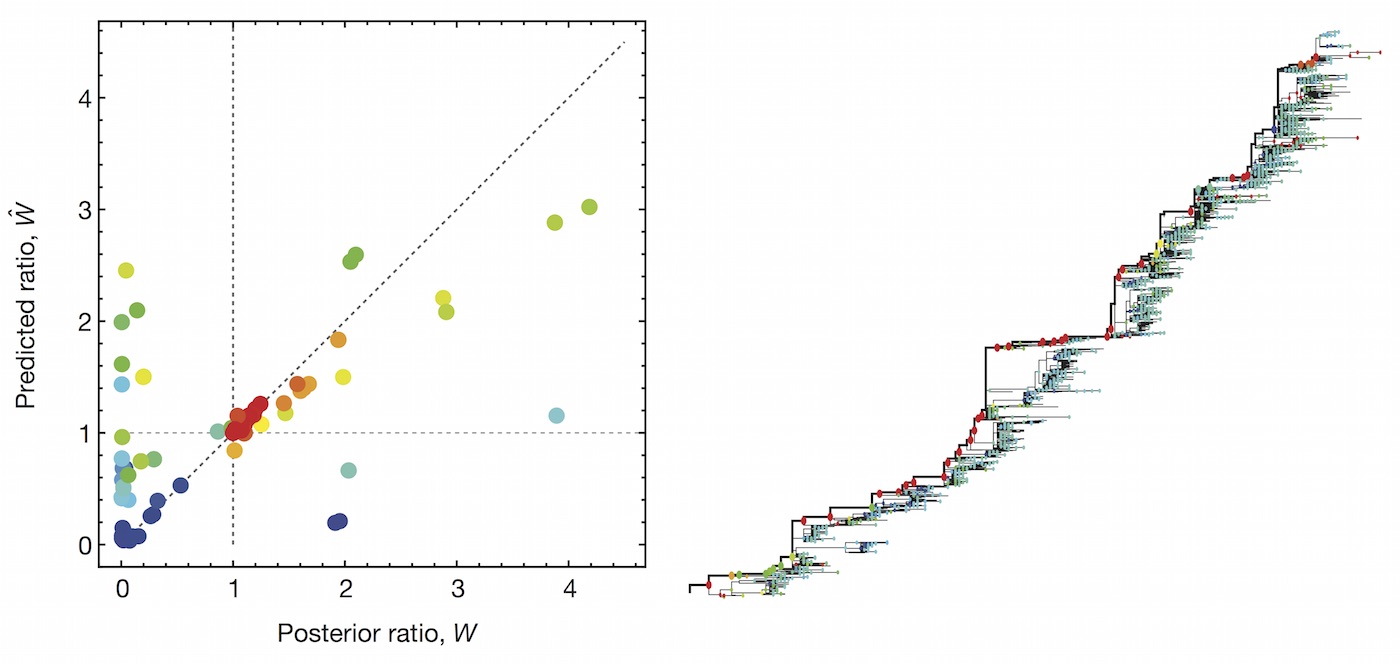
We learn coefficients and validate model based on previous 15 H3N2 seasons
Clade growth rate is well correlated (ρ = 0.66)
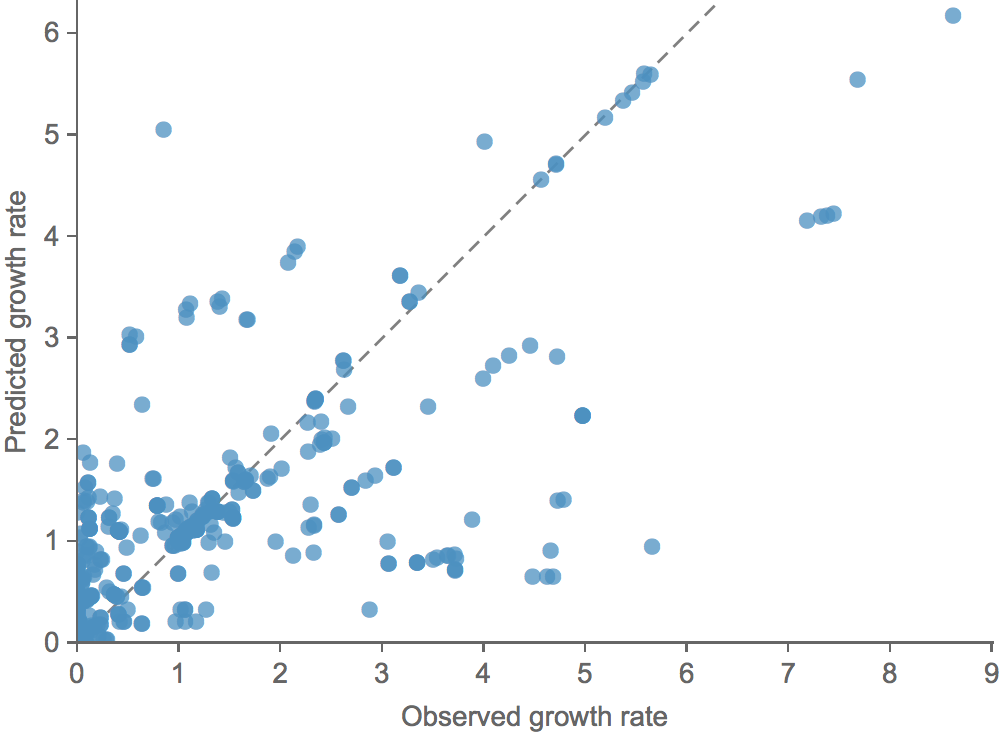
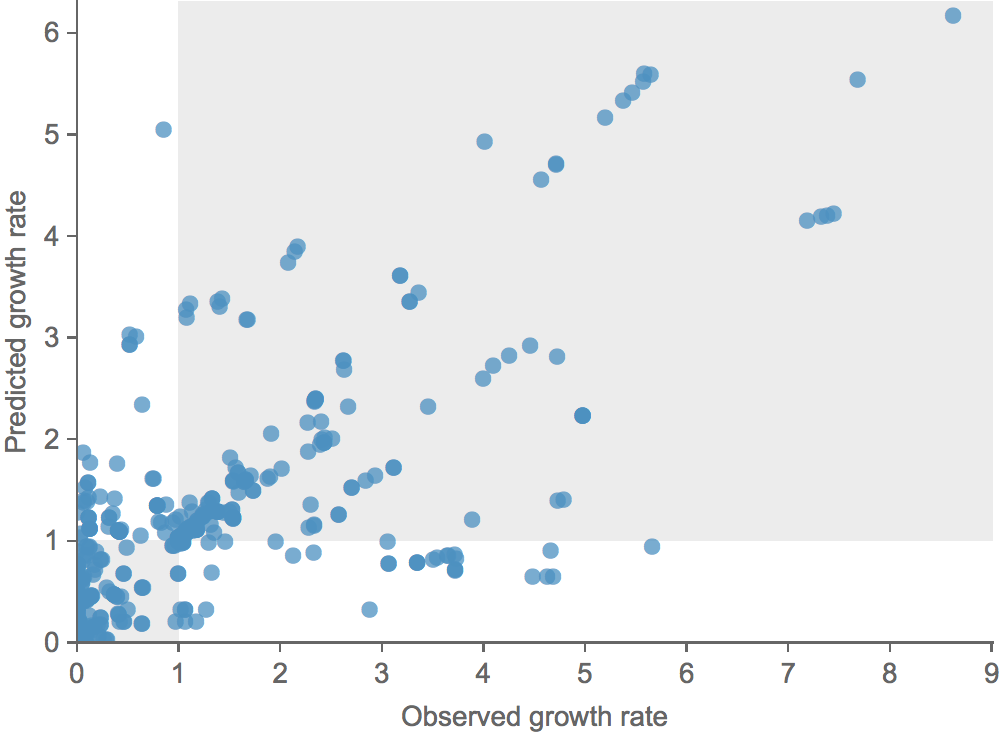
Growth vs decline correct in 84% of cases
Trajectories show more detailed congruence
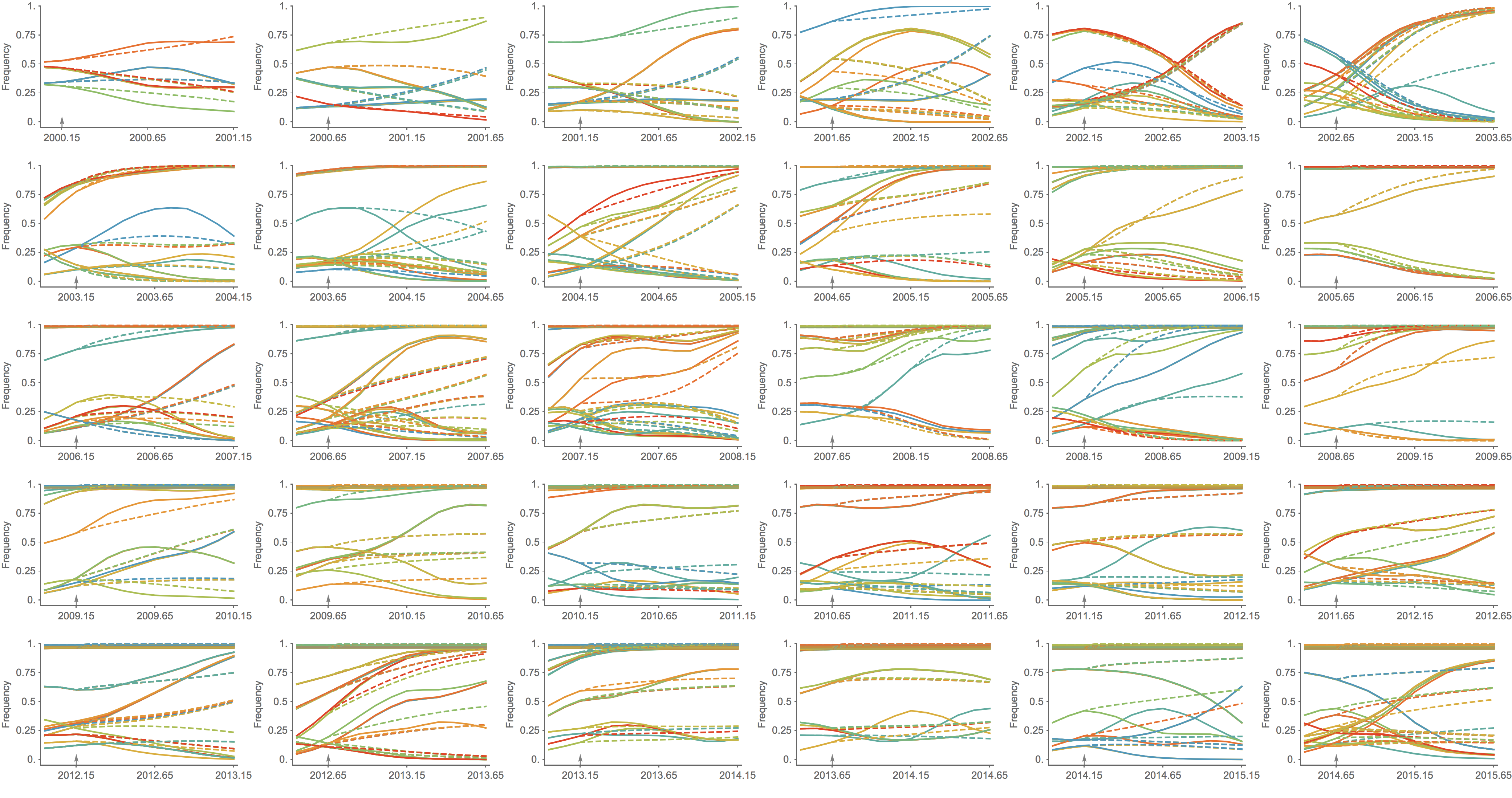
Real-time analyses are actionable and may inform influenza vaccine strain selection
Ebola
Tracking geographic spread of the Ebola epidemic
with Gytis Dudas, Luiz Carvalho, Marc Suchard, Philippe Lemey, Andrew Rambaut
and many others
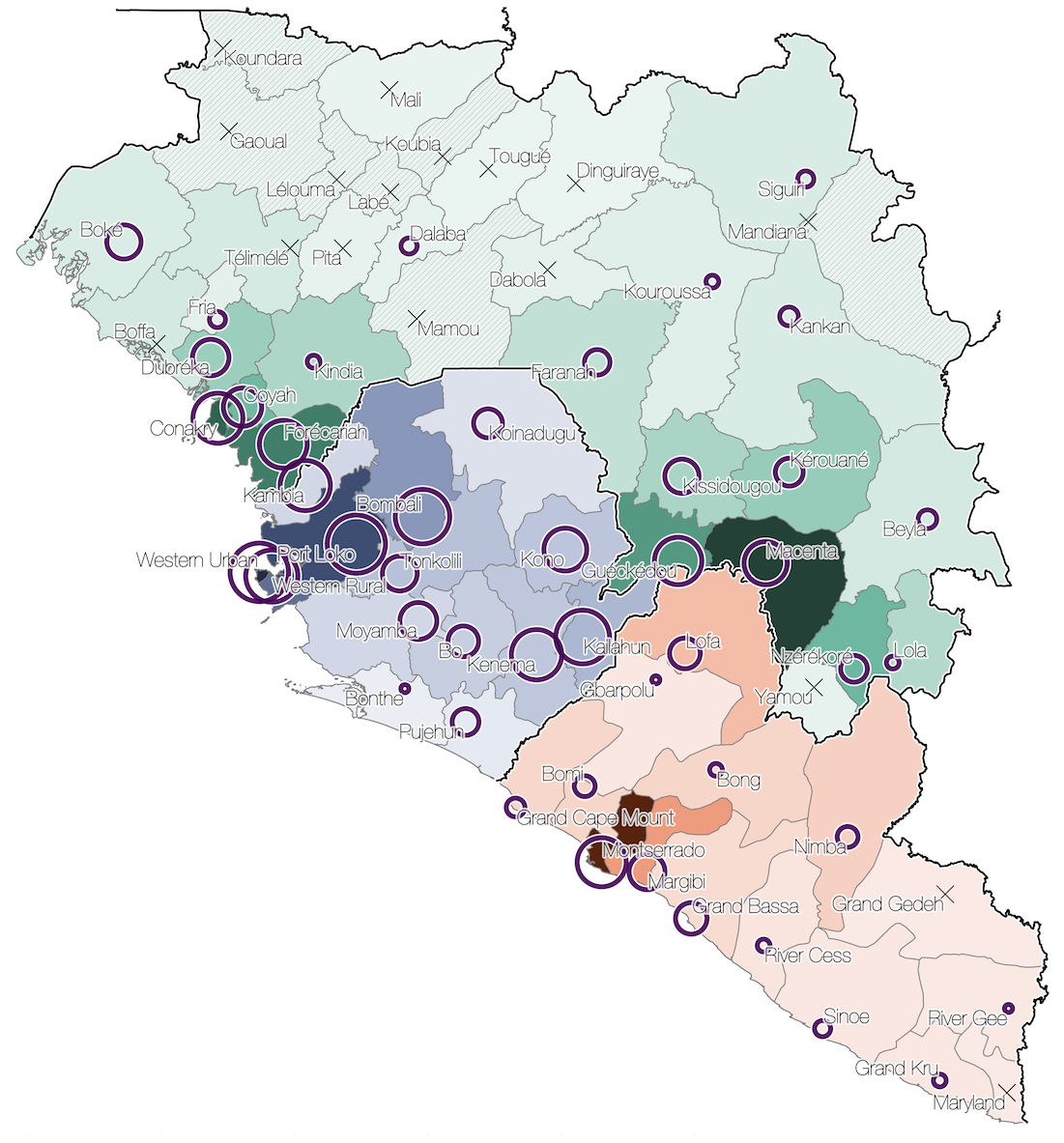
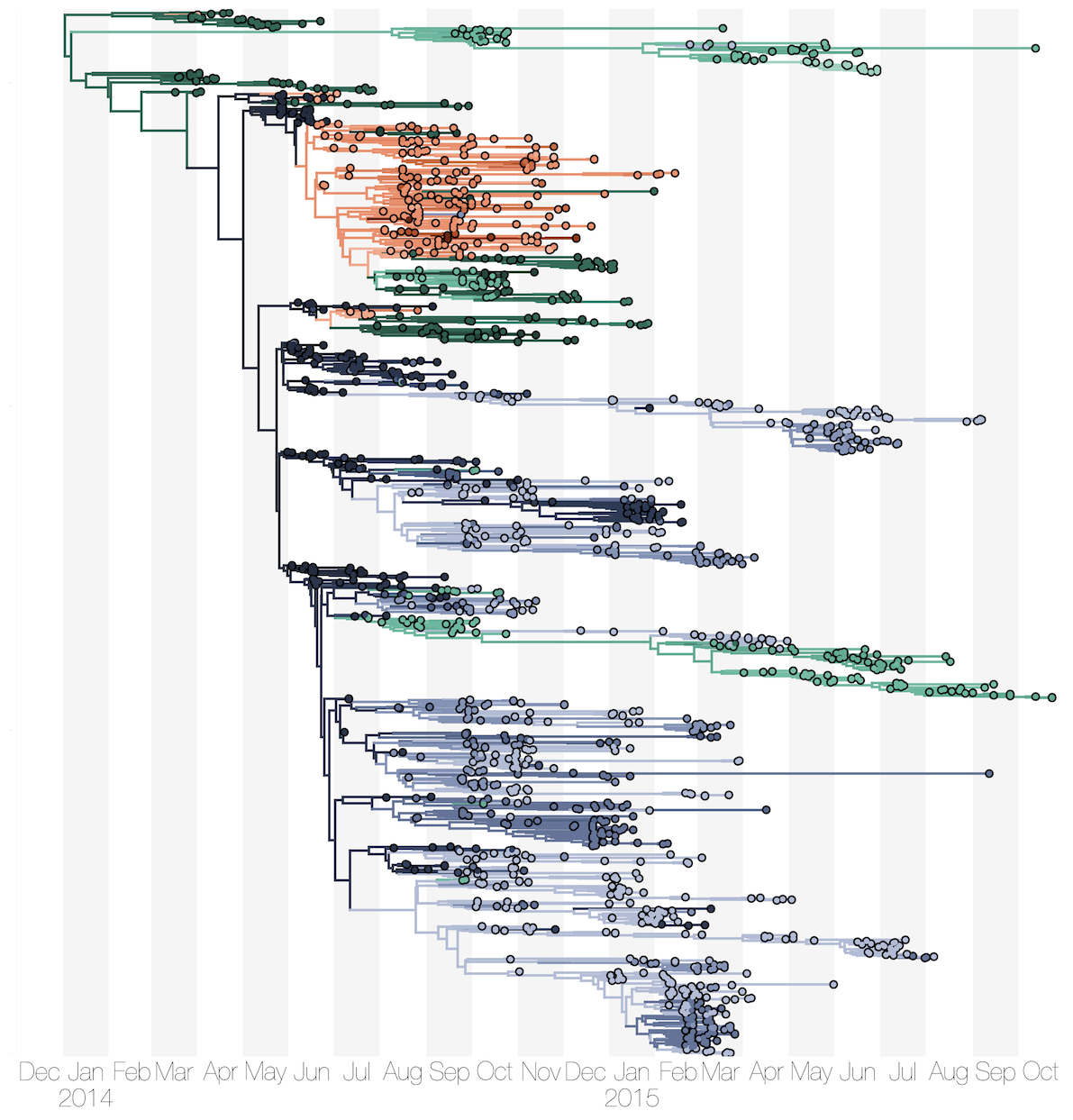
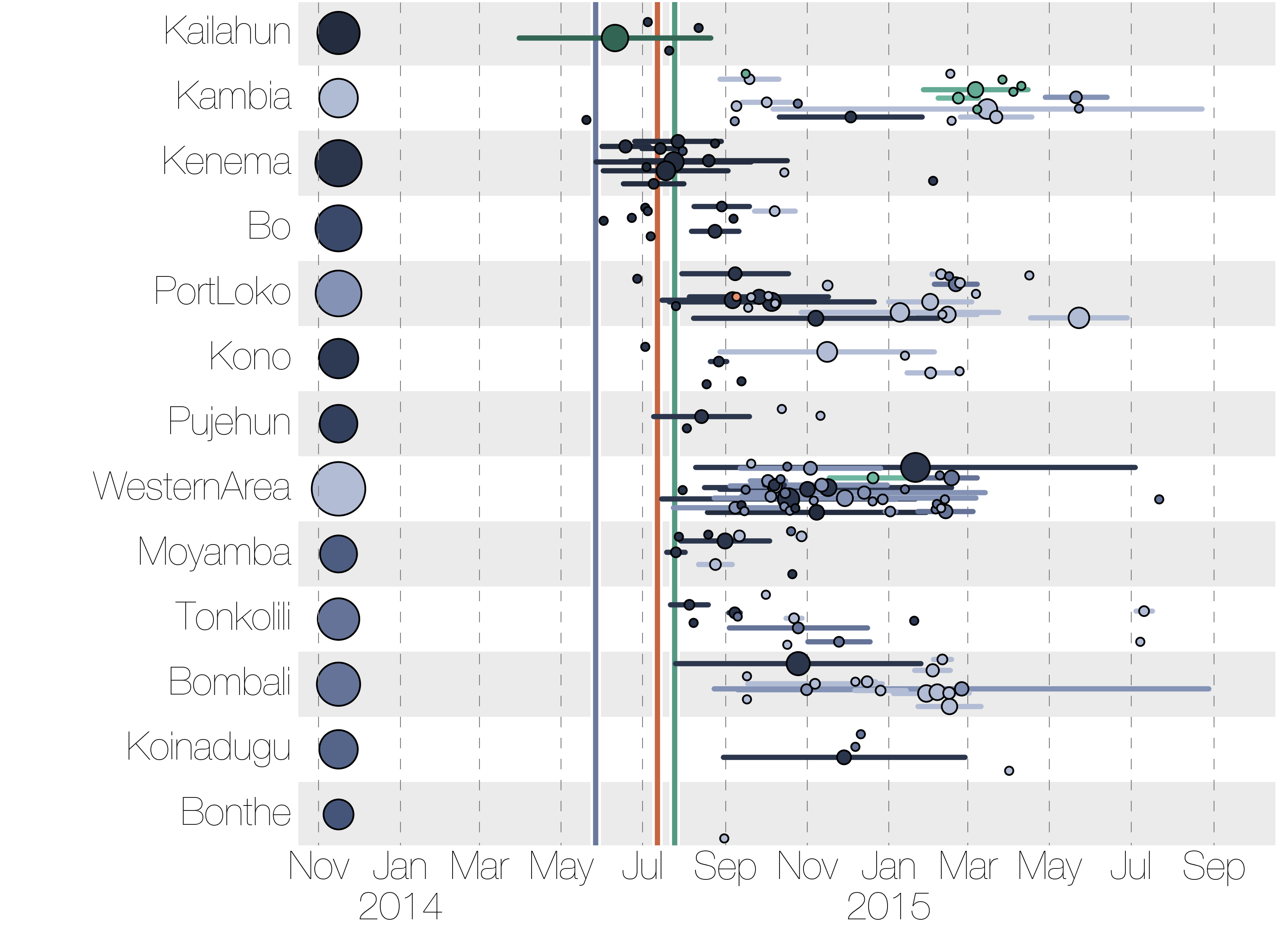
Sequencing of 1610 Ebola virus genomes collected during the 2013-2016 West African epidemic
Phylogenetic reconstruction of evolution and spread
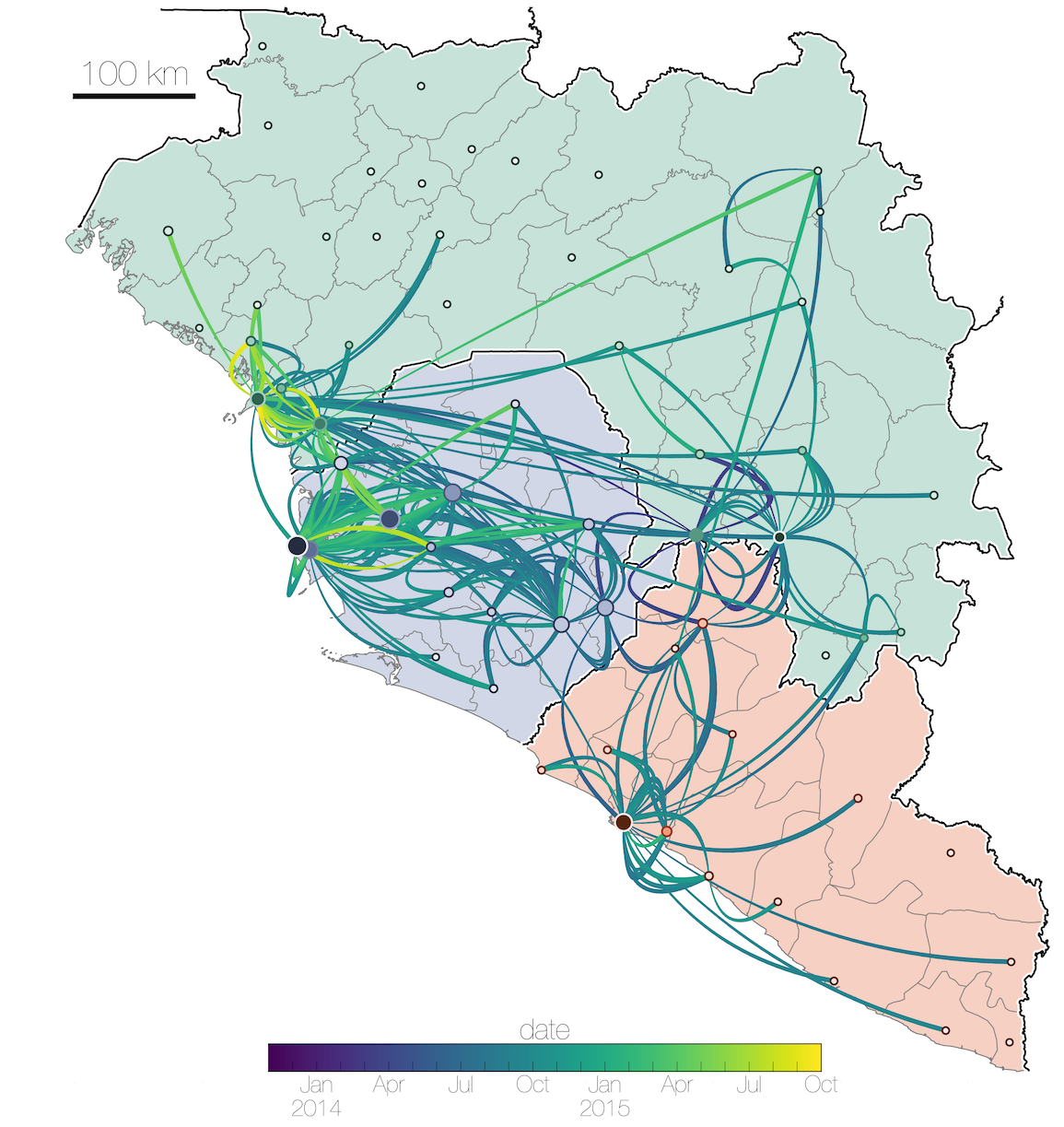
Tracking migration events
Factors influencing migration rates
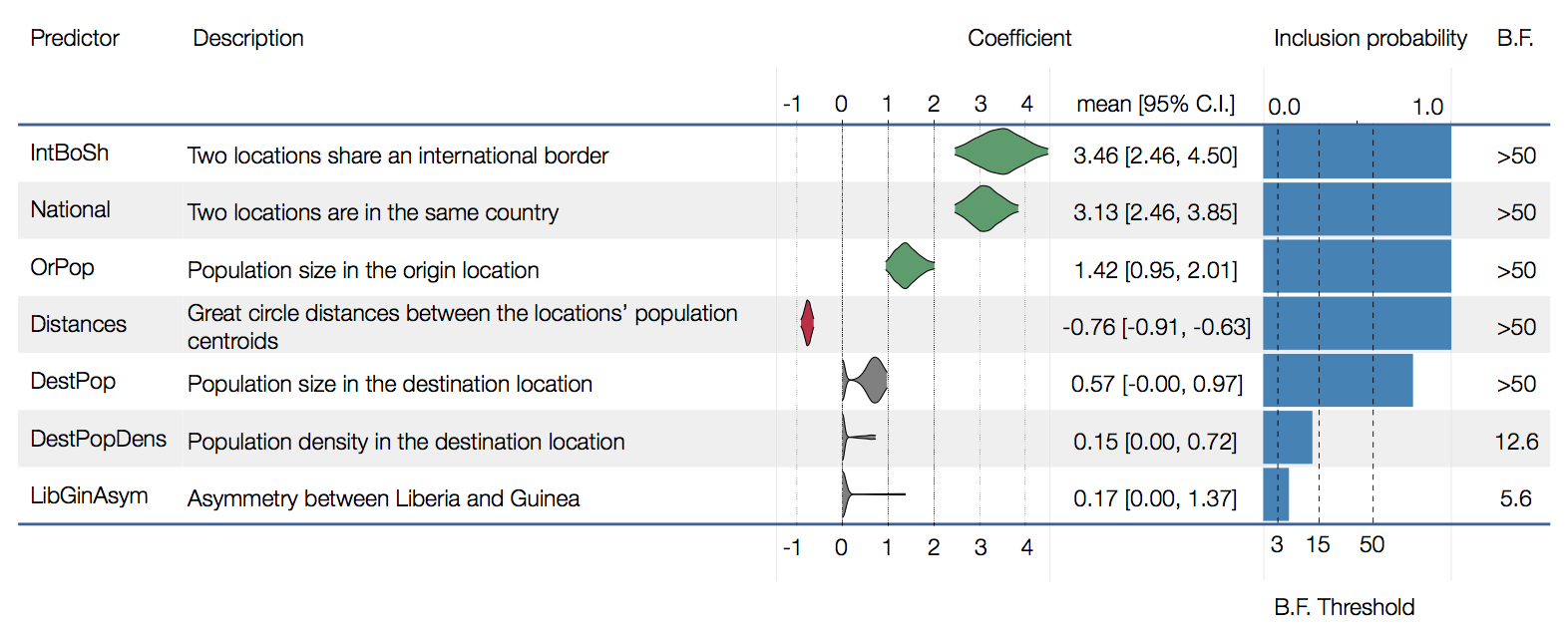
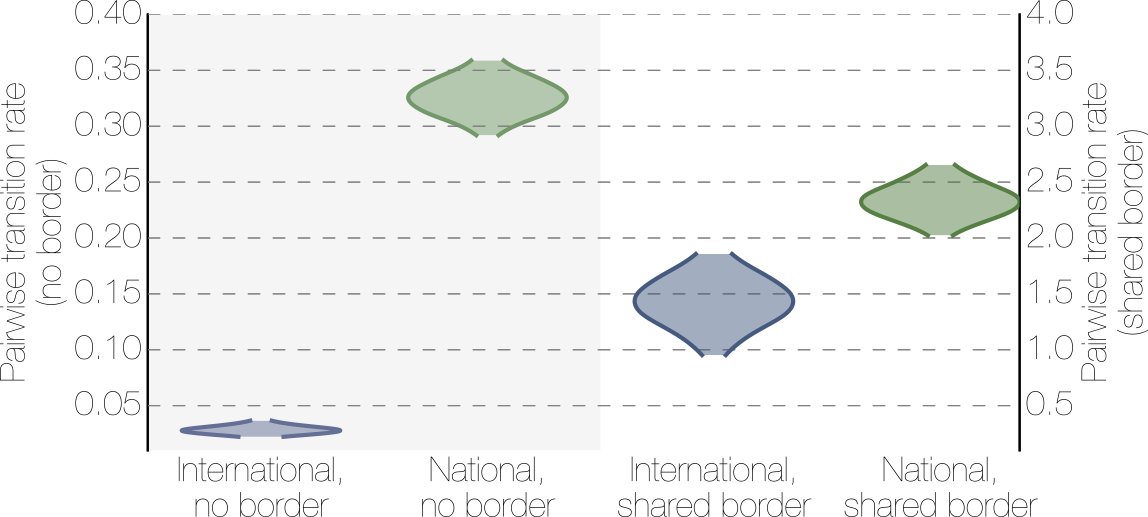
Effect of borders on migration rates
Spatial structure at the country level
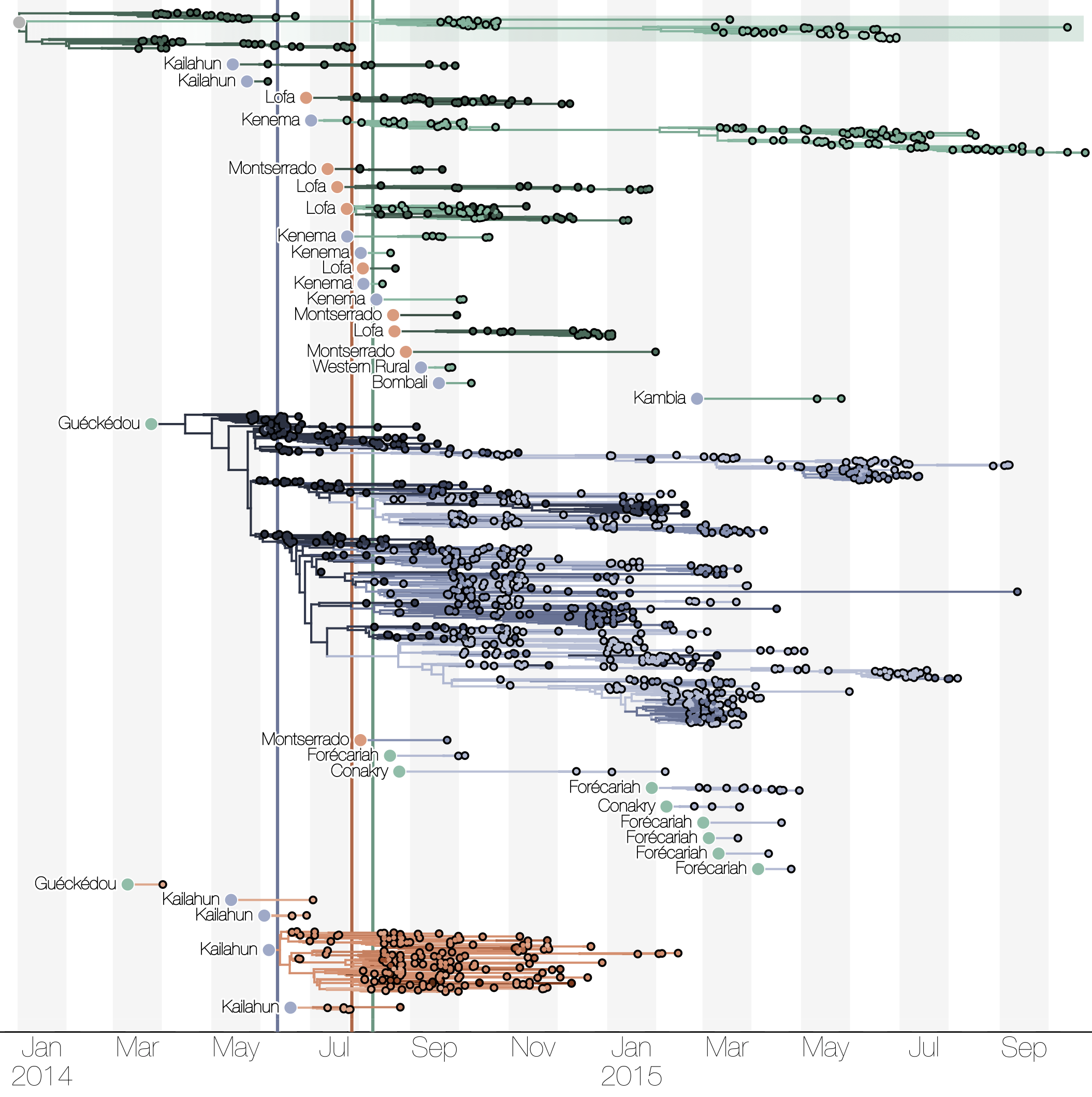
Substantial mixing at the regional level
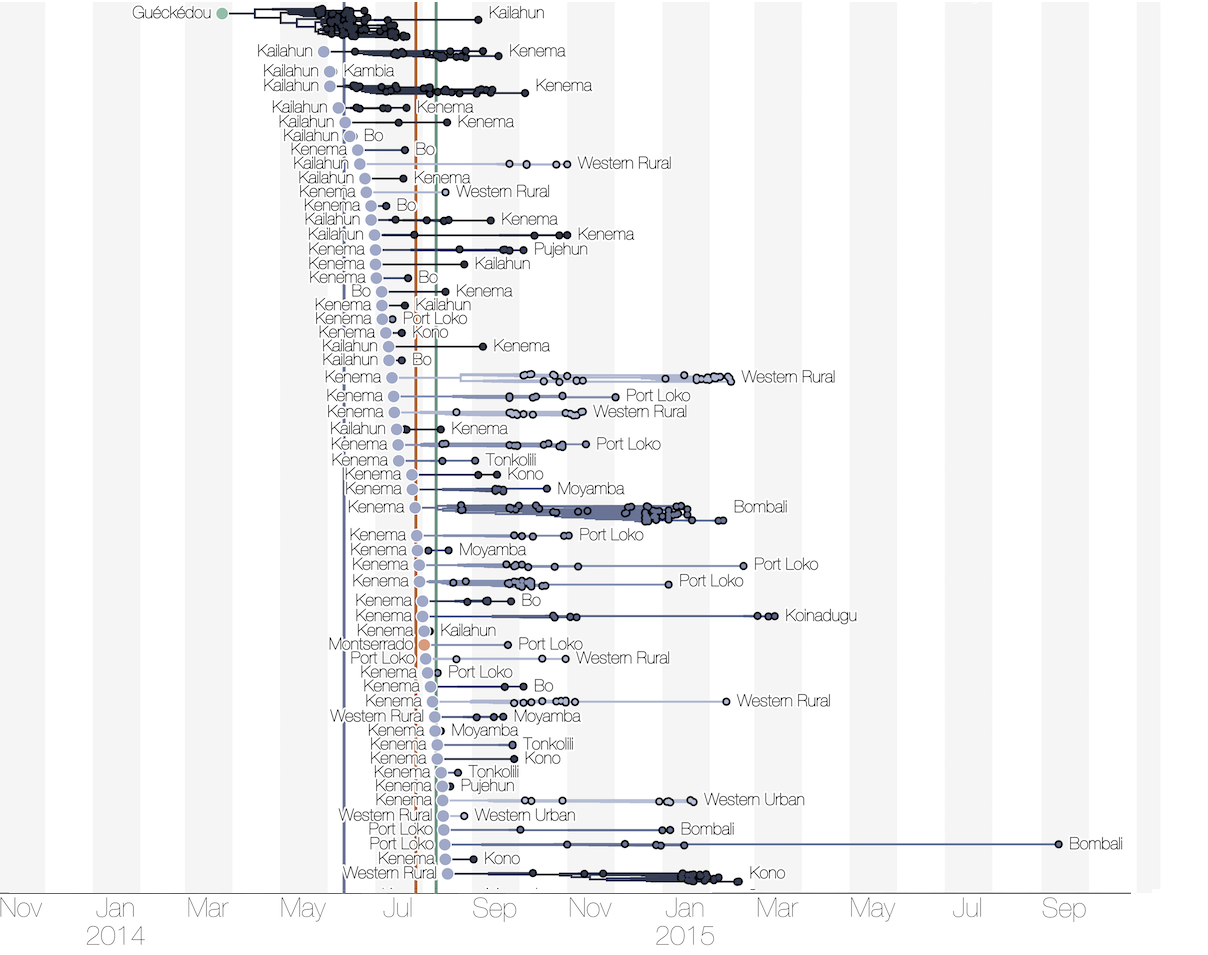
Regional outbreaks due to multiple introductions
Each introduction results in a minor outbreak
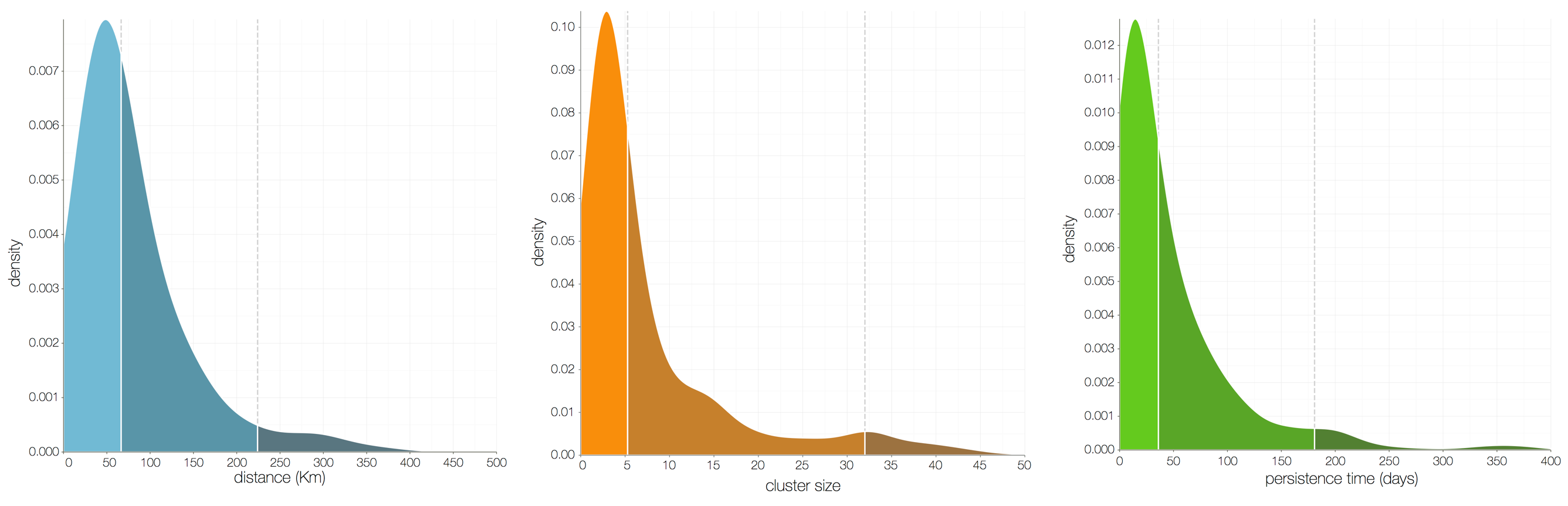
Ebola spread in West Africa followed a gravity model with moderate slowing by international borders, in which spread is driven by short-lived migratory clusters
Zika
Zika's arrival and spread in the Americas
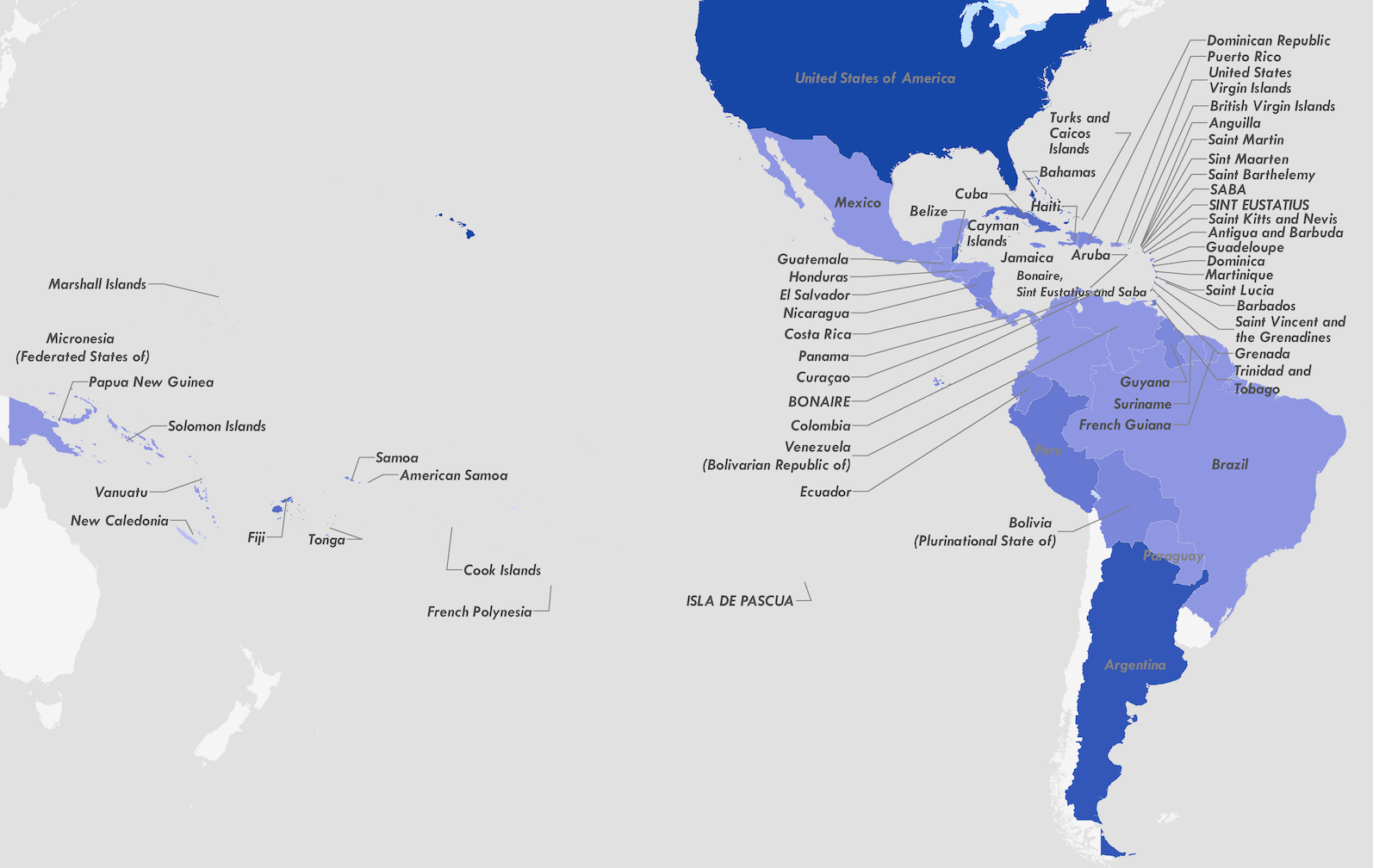
Tracking origins of the Zika epidemic
with Nuno Faria, Nick Loman, Oli Pybus, Luiz Alcantara, Ester Sabino, Josh Quick, Allison Black, Ingra Morales, Julien Thézé, Marcio Nunes, Jacqueline de Jesus, Marta Giovanetti, Moritz Kraemer, Sarah Hill and many others
Road trip through northeast Brazil to collect samples and sequence
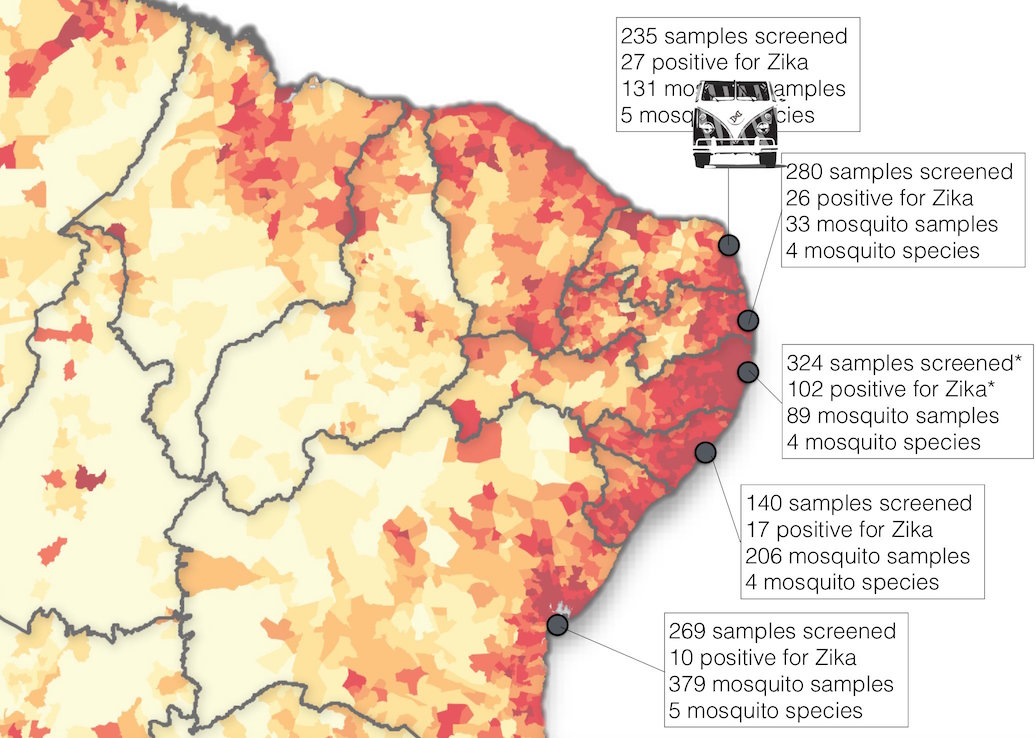
Case reports and diagnostics suggest initiation in northeast Brazil
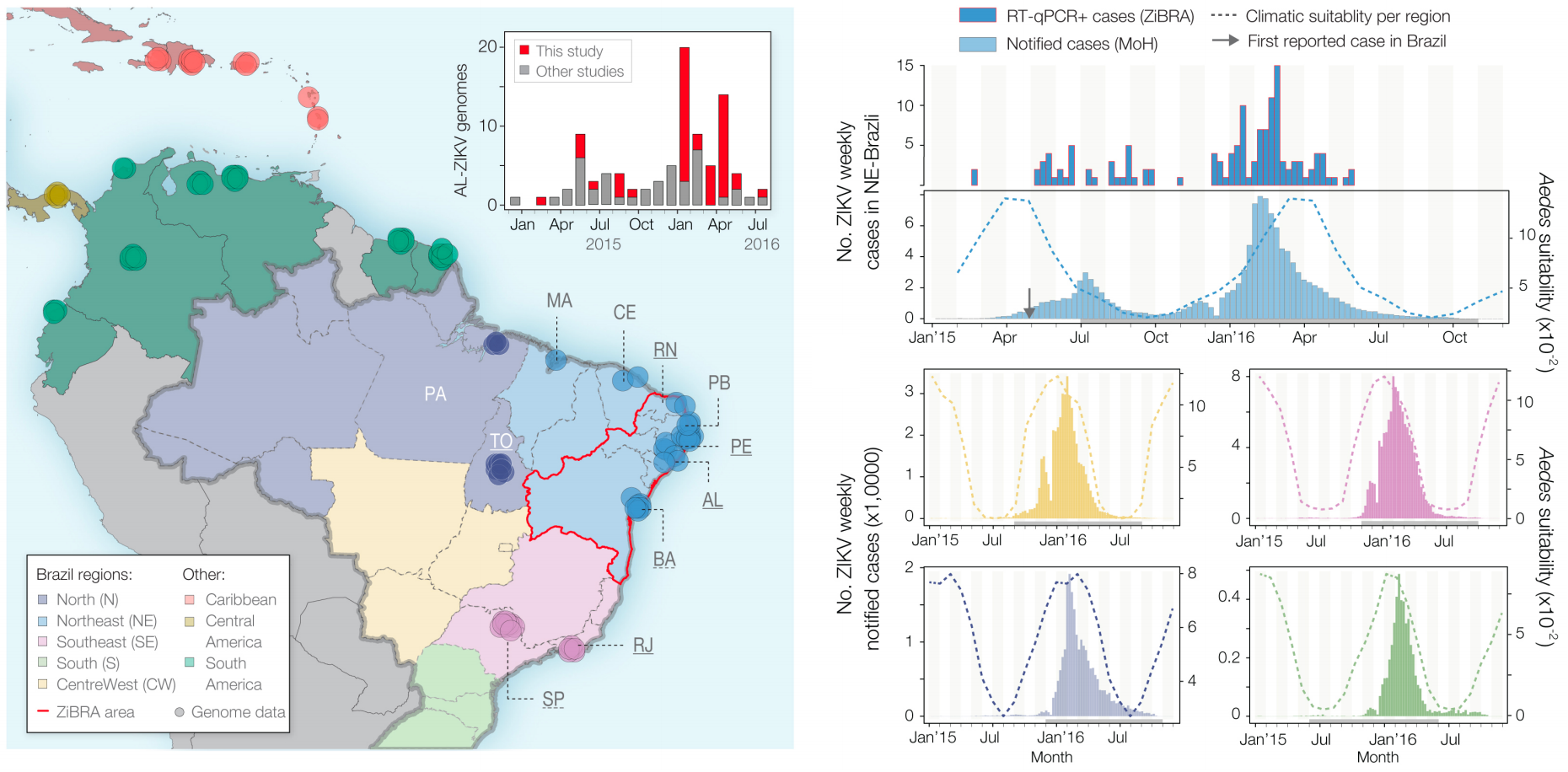
Sequencing shows accumulating genetic diversity
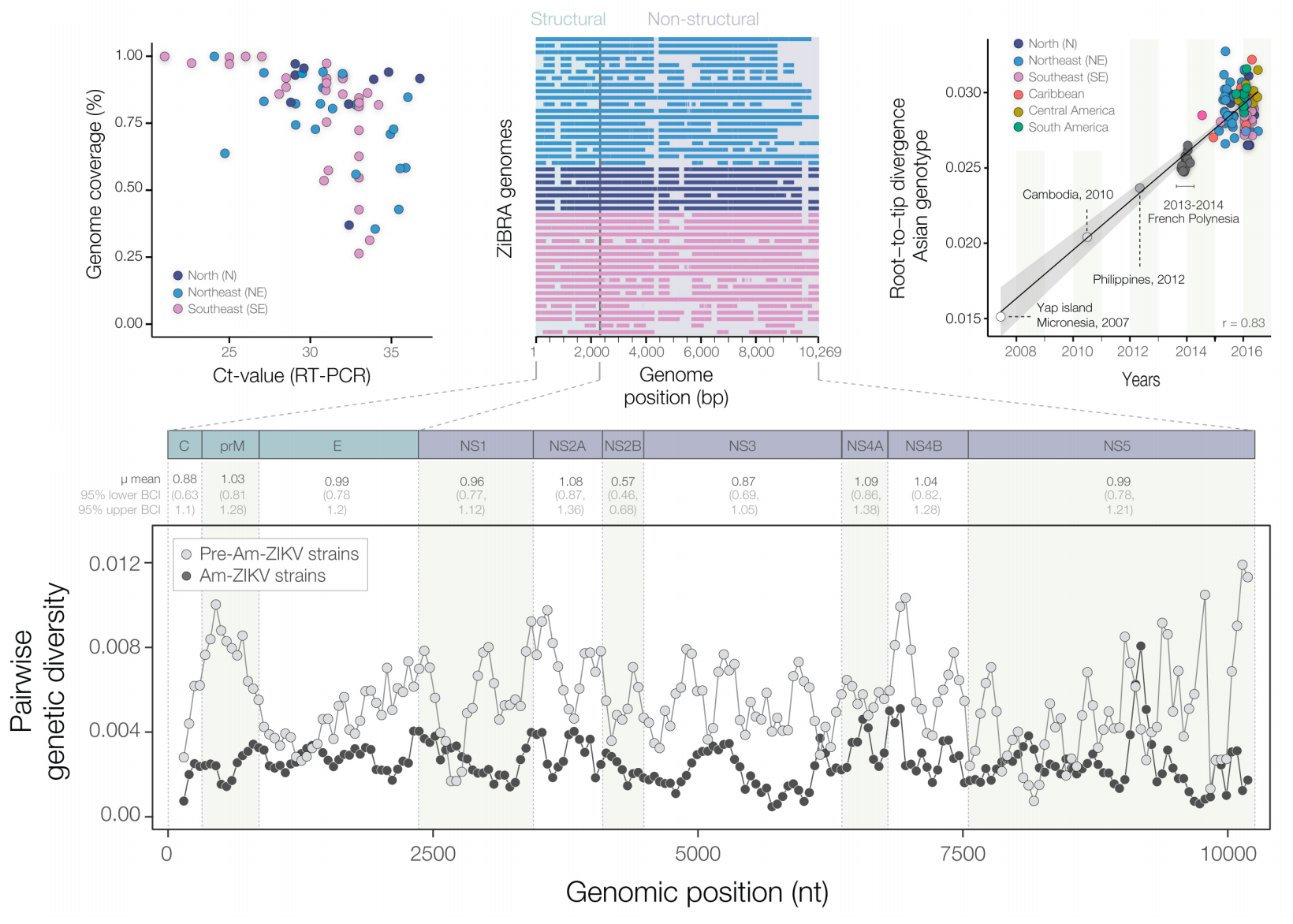
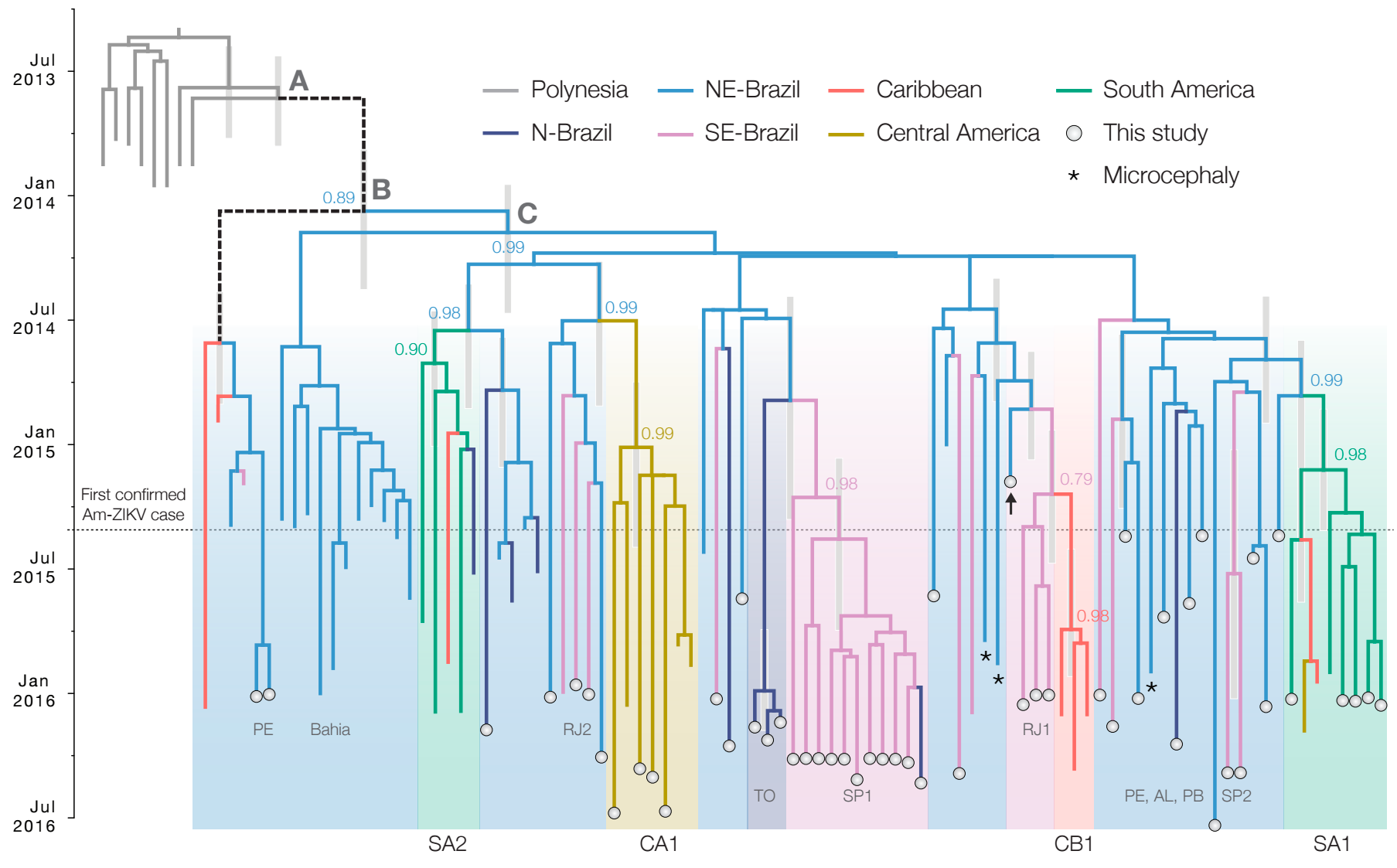
Phylogeny infers an origin in northeast Brazil
Local spread of Zika in Florida
with Kristian Andersen, Nathan Grubaugh, Jason Ladner, Gustavo Palacios, Sharon Isern, Oli Pybus, Moritz Kraemer, Gytis Dudas, Amanda Tan, Karthik Gangavarapu, Michael Wiley, Stephen White, Julien Thézé, Scott Michael, Leah Gillis, Pardis Sabeti, and many others
Outbreak of locally-acquired infections focused in Miami-Dade county
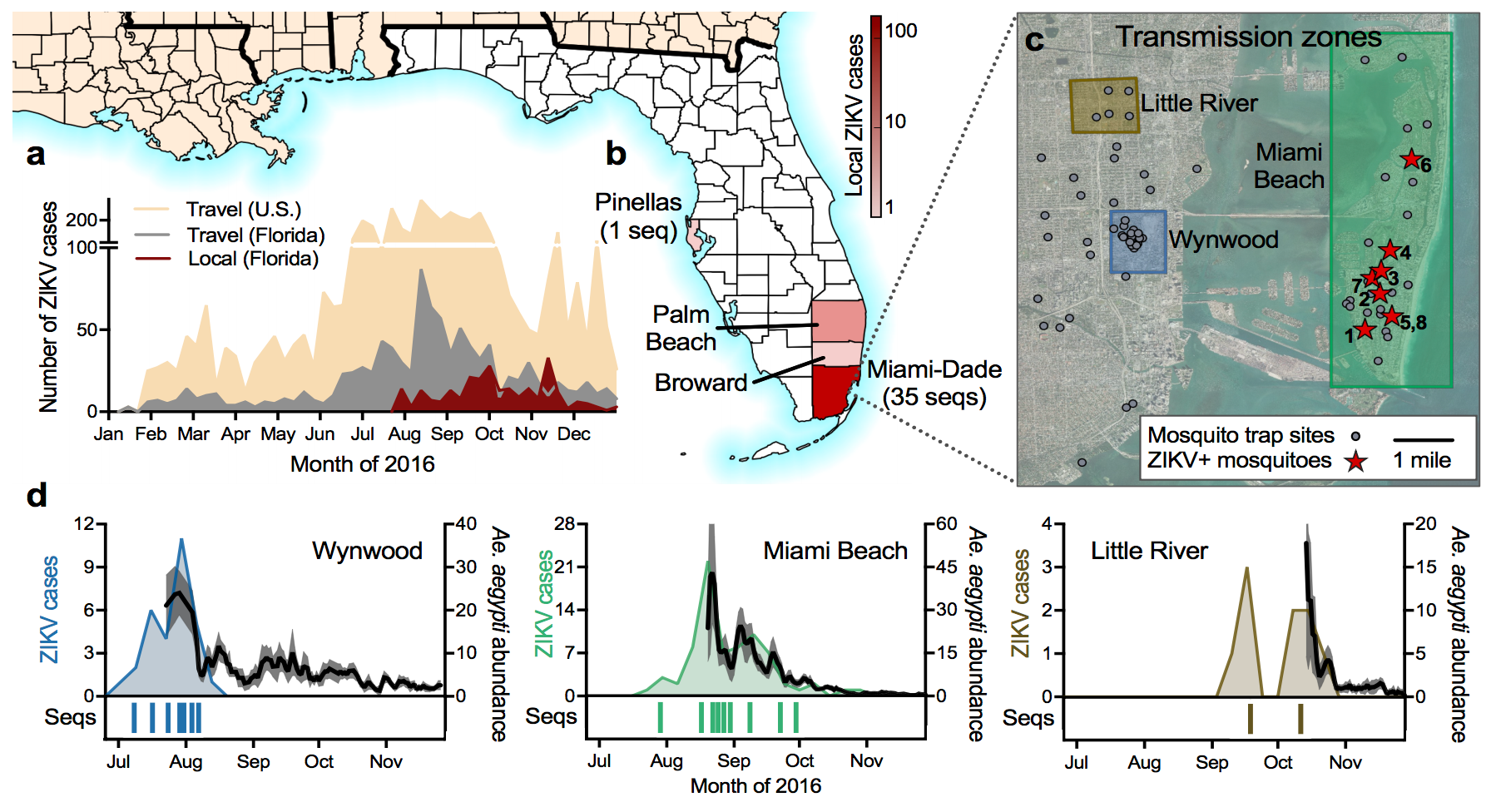
Phylogeny shows a surprising degree of clustering
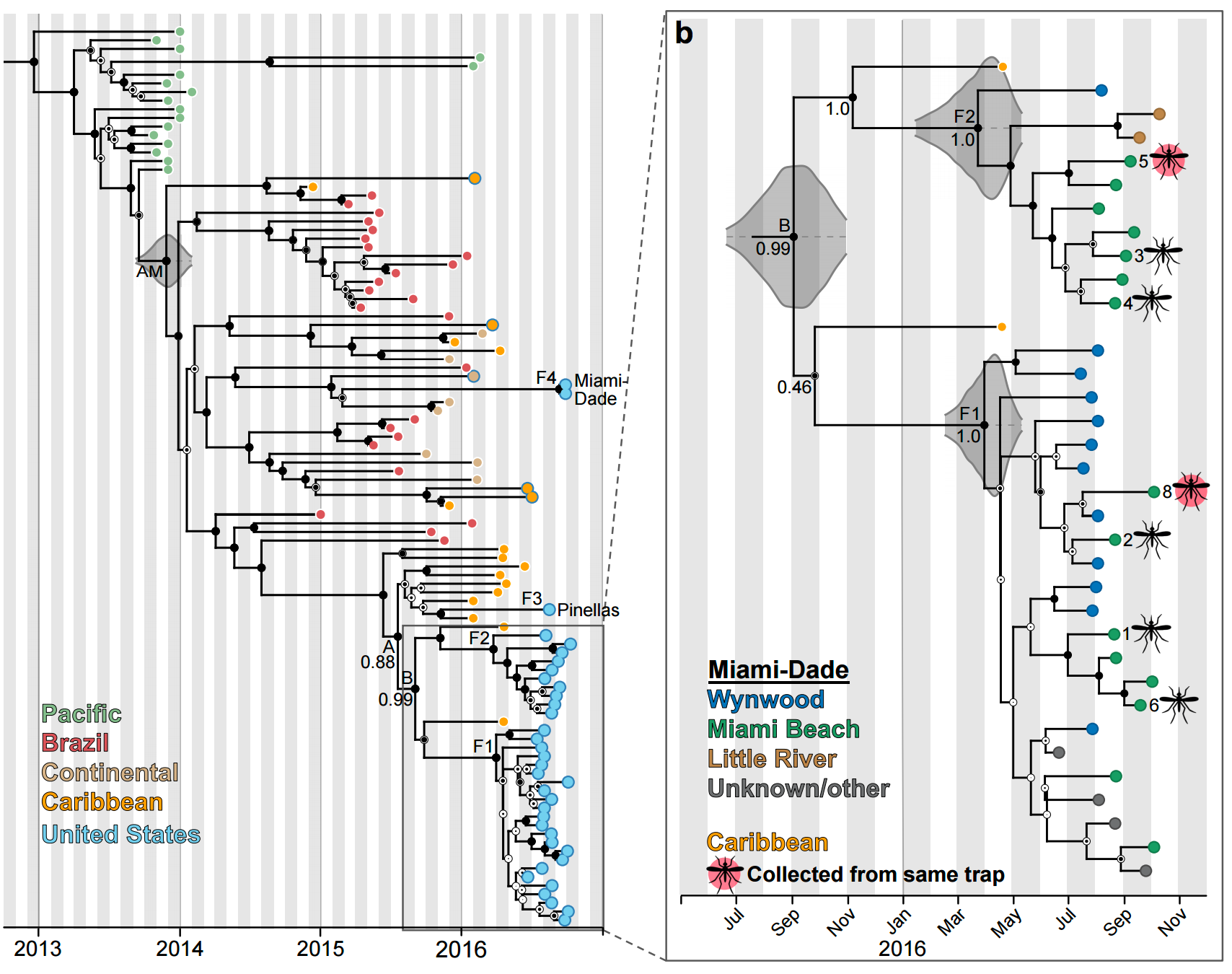
Clustering suggests fewer, longer transmission chains and higher R0
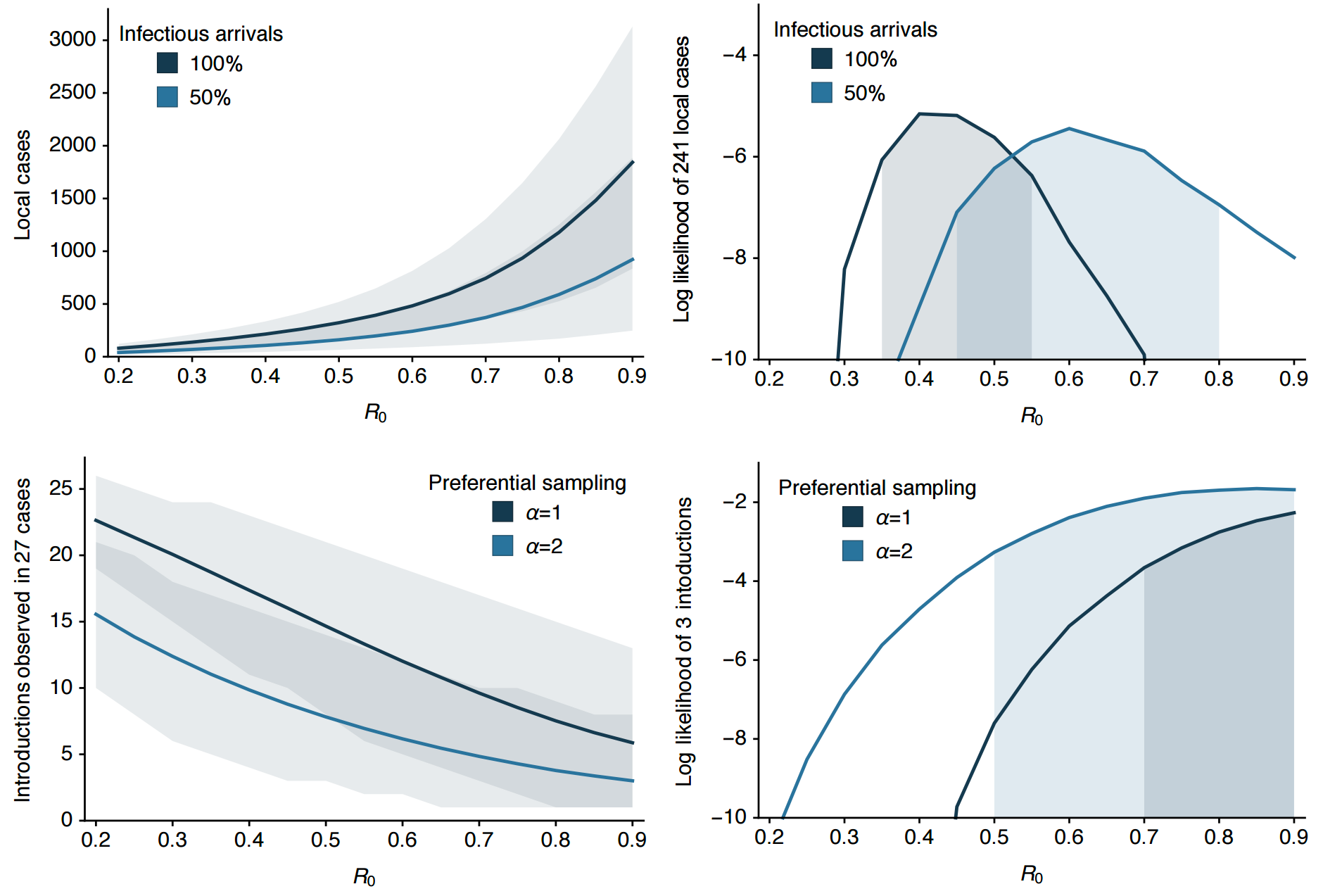
Extrapolate R0 to predict introduction counts driving outbreak
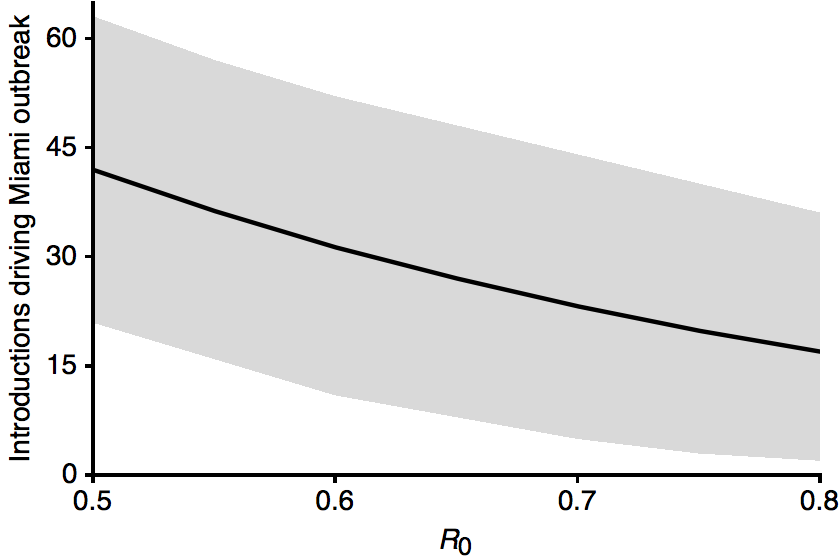
Flow of infected travelers greatest from Caribbean
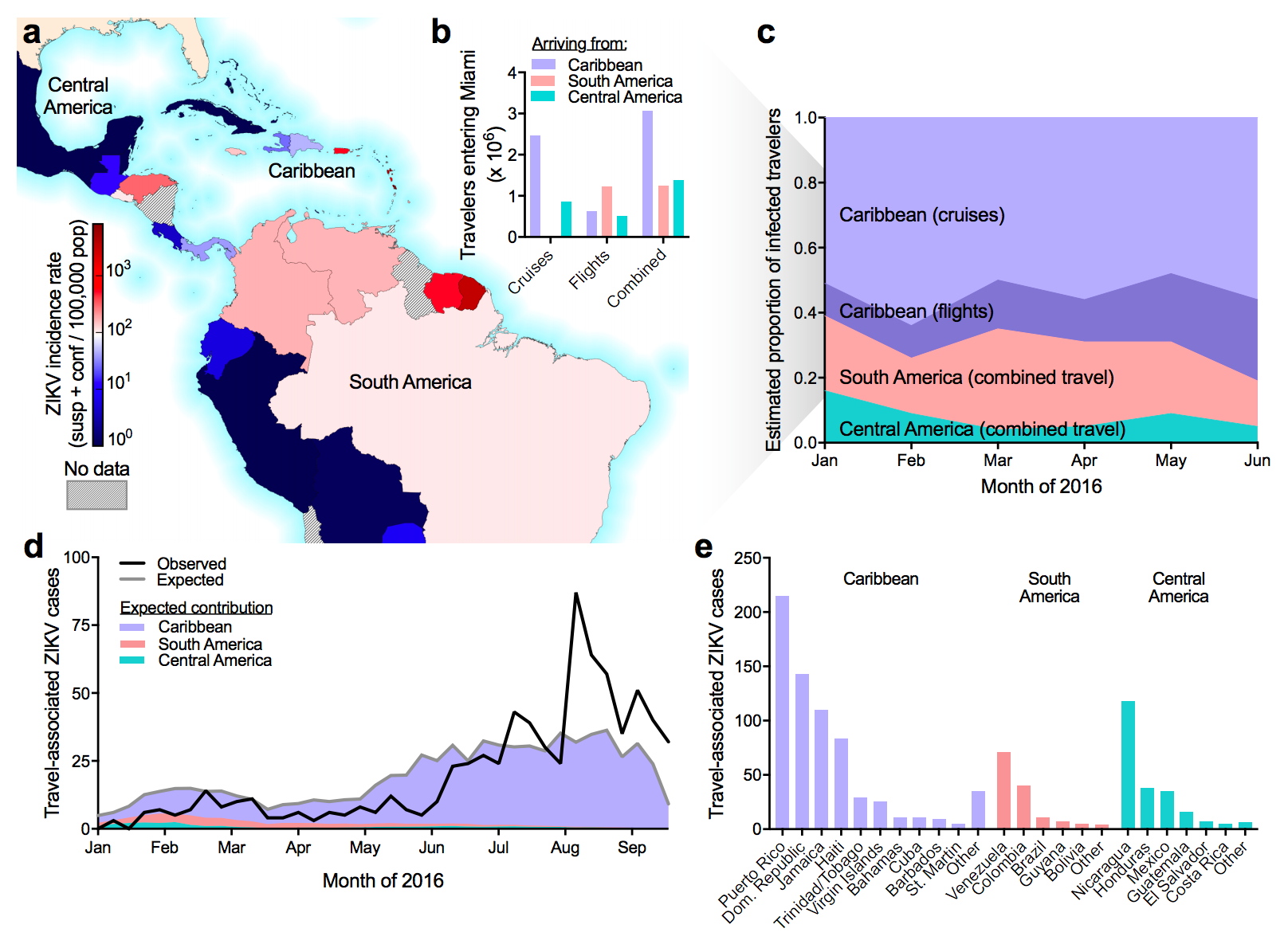
Southern Florida has high potential for Aedes borne outbreaks
Important analyses, let's make them more rapid and more automated
Key challenges
- Timely analysis and sharing of results critical
- Dissemination must be scalable
- Integrate many data sources
- Results must be easily interpretable and queryable
nextstrain
Project to conduct real-time molecular epidemiology and evolutionary analysis of emerging epidemics
 Richard Neher,
Richard Neher,
 Trevor Bedford,
Trevor Bedford,
 Colin Megill,
Colin Megill,
 James Hadfield,
James Hadfield,
 Charlton Callender,
Charlton Callender,
 Sidney Bell,
Sidney Bell,
 Barney Potter,
Barney Potter,
 Sarah Murata,
Sarah Murata,
Nextstrain architecture
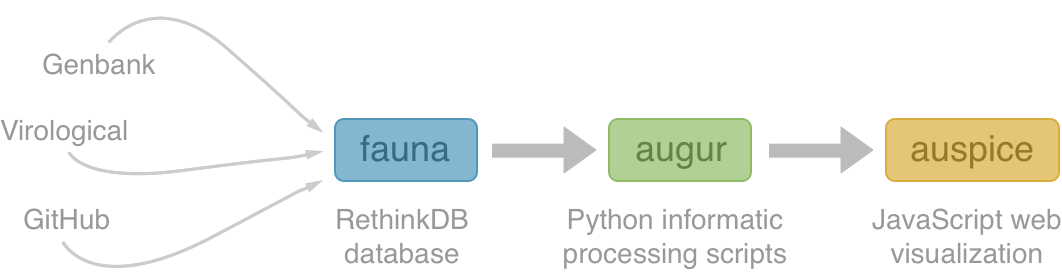
nextstrain.org
Rapid on-the-ground sequencing by Ian Goodfellow, Matt Cotten and colleagues
Desired analytics are pathogen specific and tied to response measures
Acknowledgements
Influenza: WHO Global Influenza Surveillance Network, GISAID, Worldwide Influenza Centre at the Francis Crick Institute, Richard Neher, Colin Russell, Boris Shraiman
Ebola: data producers, Gytis Dudas, Andrew Rambaut, Luiz Carvalho, Philippe Lemey, Marc Suchard, Andrew Tatem, Nick Loman, Ian Goodfellow, Matt Cotten, Paul Kellam, Kristian Andersen, Pardis Sabeti, many others
Zika: data producers, Nick Loman, Nuno Faria, Oliver Pybus, Josh Quick, Allison Black, Kristian Andersen, Nathan Grubaugh, Gytis Dudas, many others
Nextstrain: Richard Neher, Colin Megill, James Hadfield, Charlton Callender, Sarah Murata, Sidney Bell, Barney Potter