Nextstrain Workshop
Trevor Bedford (@trvrb)
27 Mar 2019
AMD Training
CDC
Sequencing to reconstruct pathogen spread
Epidemic process
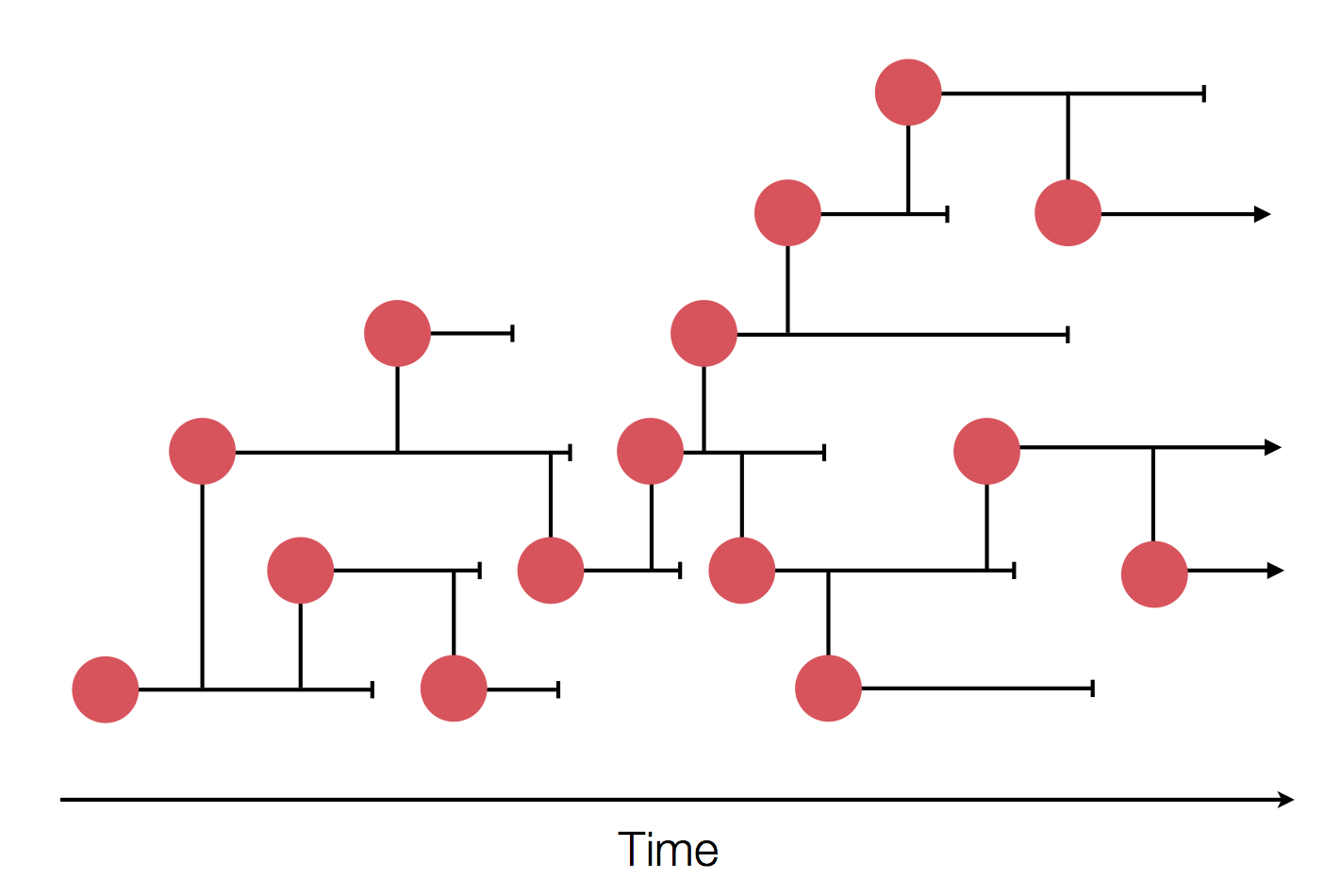
Sample some individuals
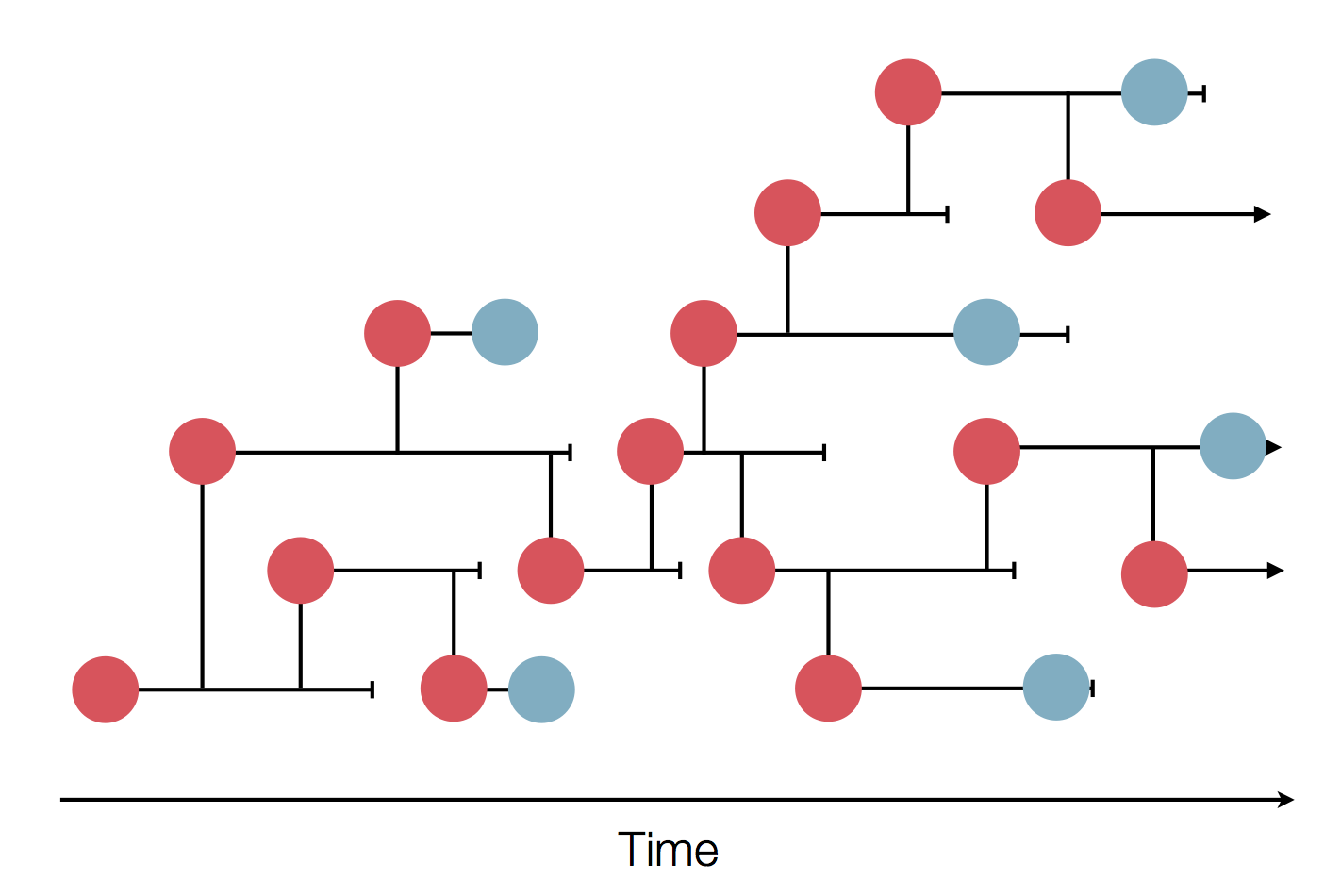
Sequence and determine phylogeny
Sequence and determine phylogeny
Pathogen genomes may reveal:
- Evolution of new adaptive variants
- Epidemic origins
- Patterns of geographic spread
- Animal-to-human spillover
- Transmission chains
Influenza: Forecasting spread of new variants for vaccine strain selection
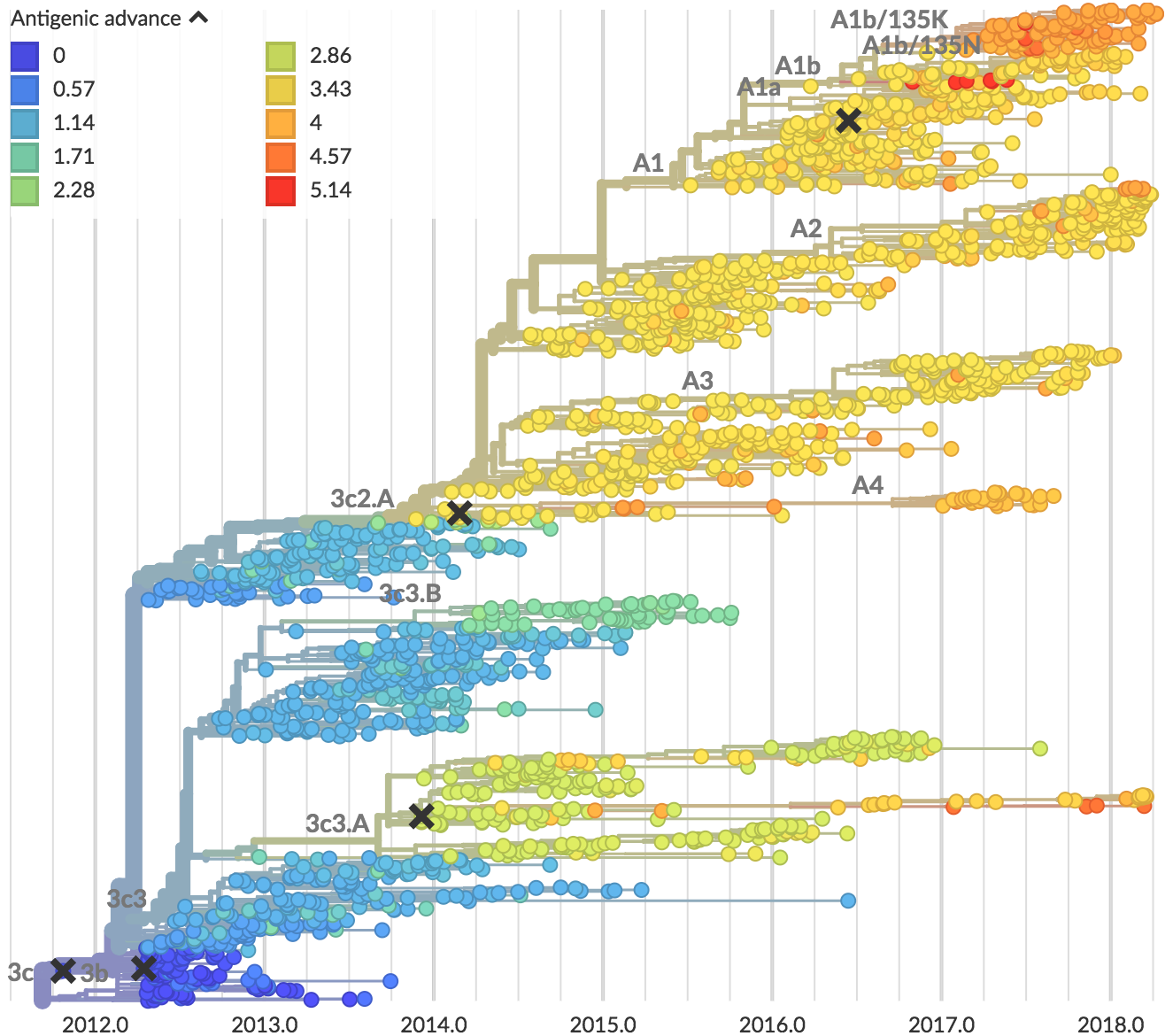
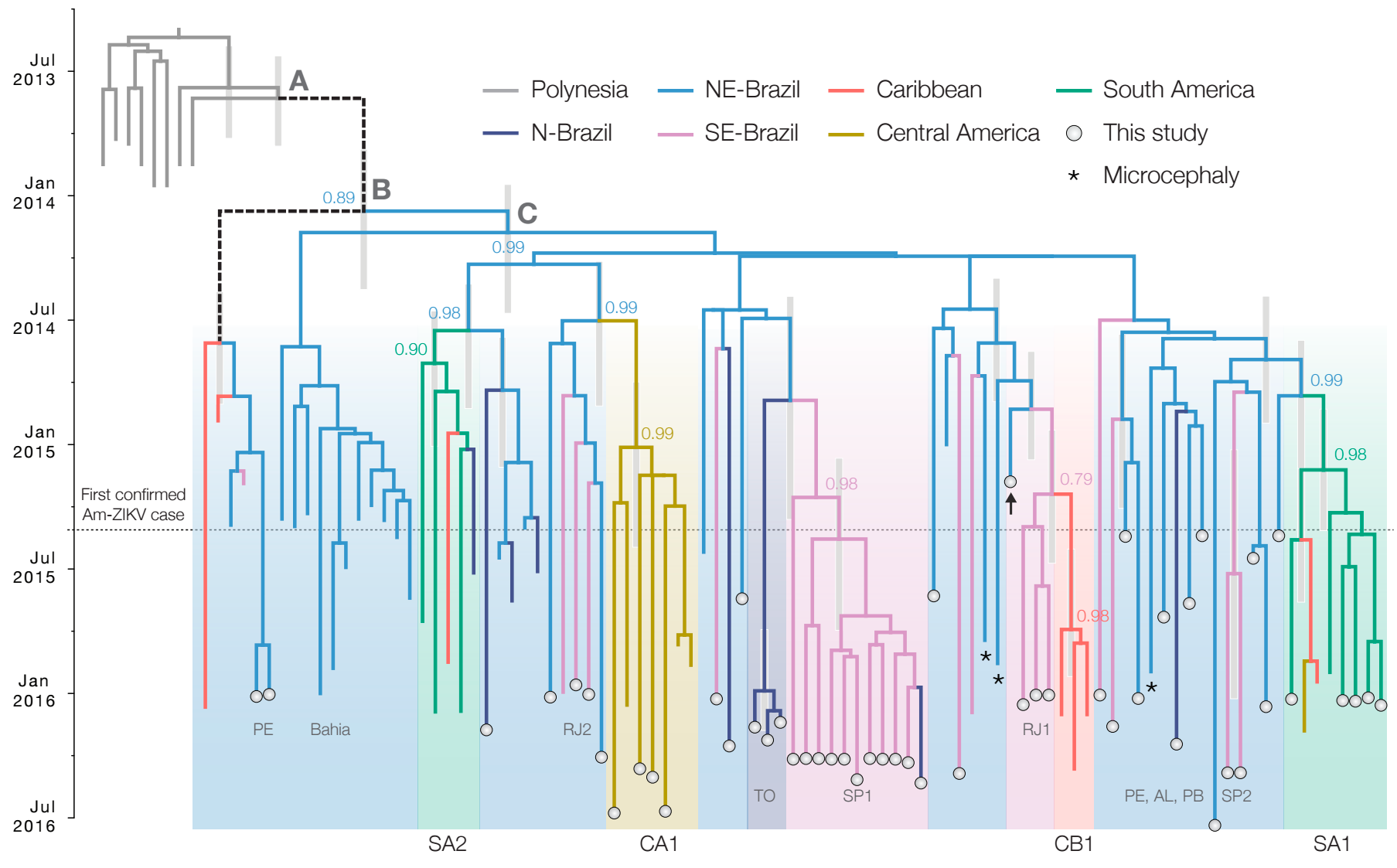
Zika: Uncovering origins of the epidemic in the Americas
Ebola: Revealing spatial spread and persistence in West Africa

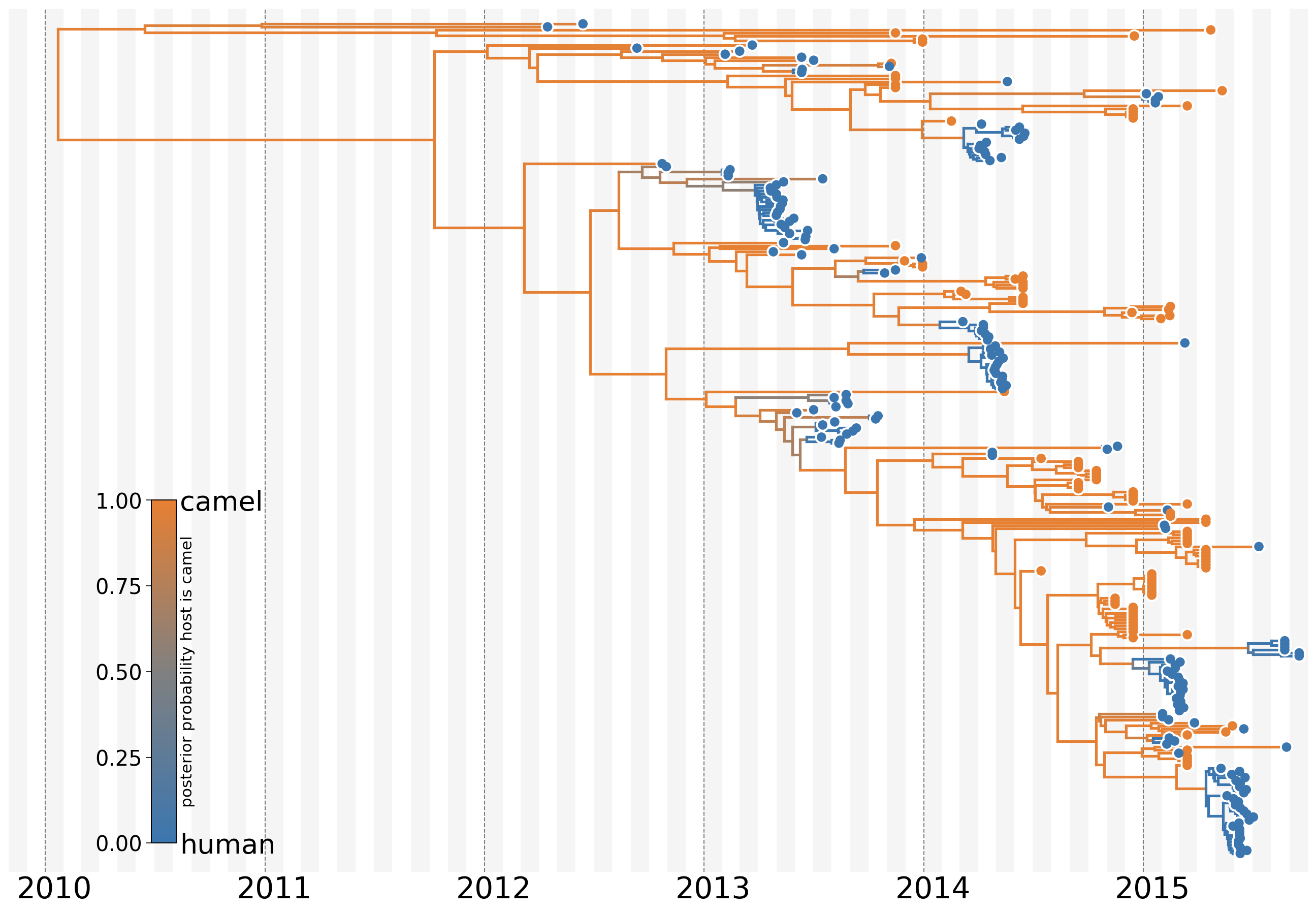
MERS: Quantifying camel-to-human spillover
TB: Tracking individual transmission chains
Actionable inferences
Genomic analyses are mostly done in a retrospective manner
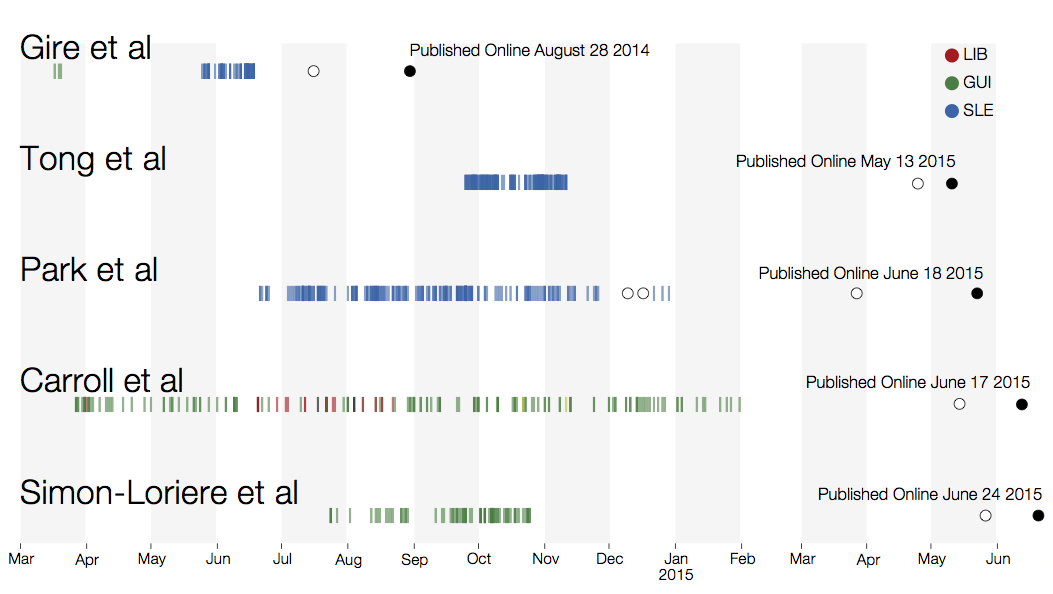
Key challenges to making genomic epidemiology actionable
- Timely analysis and sharing of results critical
- Dissemination must be scalable
- Integrate many data sources
- Results must be easily interpretable and queryable
Nextstrain
Project to conduct real-time molecular epidemiology and evolutionary analysis of emerging epidemics
with
![]() Richard Neher,
Richard Neher,
![]() James Hadfield,
James Hadfield,
![]() Emma Hodcroft,
Emma Hodcroft,
![]() Tom Sibley,
Tom Sibley,
![]() John Huddleston,
John Huddleston,
![]() Colin Megill,
Colin Megill,
![]() Sidney Bell,
Sidney Bell,
![]() Barney Potter,
Barney Potter,
![]() Charlton Callender
Charlton Callender
Nextstrain is two things
- a bioinformatics toolkit and visualization app, which can be used for a broad range of datasets
- a collection of real-time pathogen analyses kept up-to-date on the website nextstrain.org
Nextstrain architecture
All code open source at github.com/nextstrain
Two central aims: (1) rapid and flexible phylodynamic analysis and
(2) interactive visualization
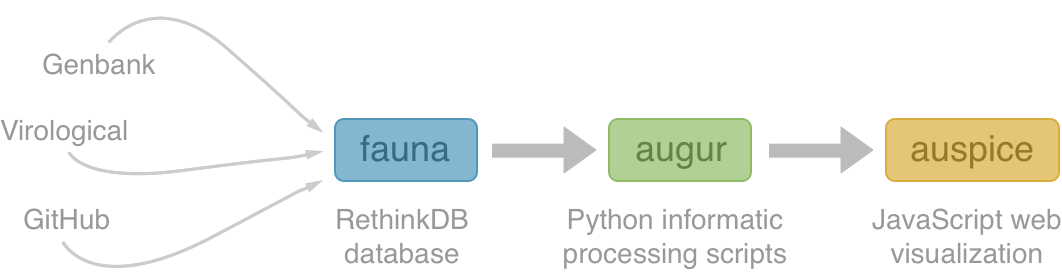
Fauna
Rethink database of virus and titer data
- Harmonizes data from different sources
- Integrates different types of data (serology, sequences, case details)
- Provides an interface for downstream analysis
Augur
Build scripts to align sequences, build trees and annotate
- Flexible build scripts to incorporate different viruses and analyses
- Constructs time-resolved phylogenies
- Annotates with geographic transitions and mutation events
Example augur pipeline for 1600 Ebola genomes
- Align with MAFFT (34 min)
- Build ML tree with RAxML (54 min)
- Temporally resolve tree and geographic ancestry with TreeTime (16 min)
- Total pipeline (1 hr 46 min)
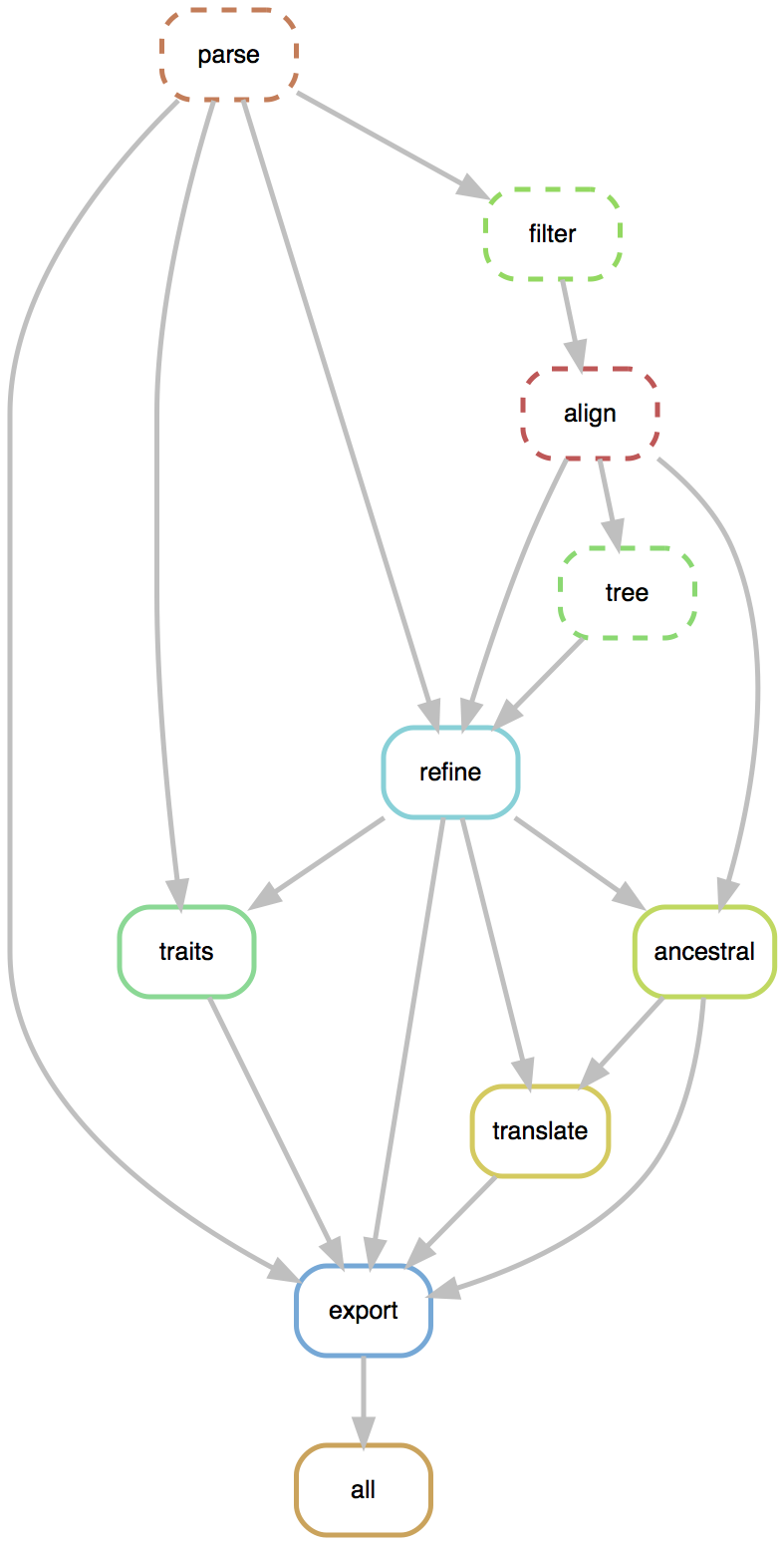
Pipeline consists of Unix-like command line modules
- Modules called via
augur filter,augur tree,augur traits, etc... - Designed to be composable across pathogen builds
- Uses Snakemake to define a pipeline, making steps obvious
- Provides depedency graph for fast recomputation
- Pathogen-specific repos give users an obvious foundation to build off of
Auspice
Web visualization of resulting trees
- Interactive data exploration and filtering
- Framework through React / D3
- Connects phylogeny, geography and genotypes
Demo nextstrain.org focusing on visualization features
Todays' workshop
- Brief background on phylodynamics
- Focus of running augur and constructing snakefiles
- Running auspice
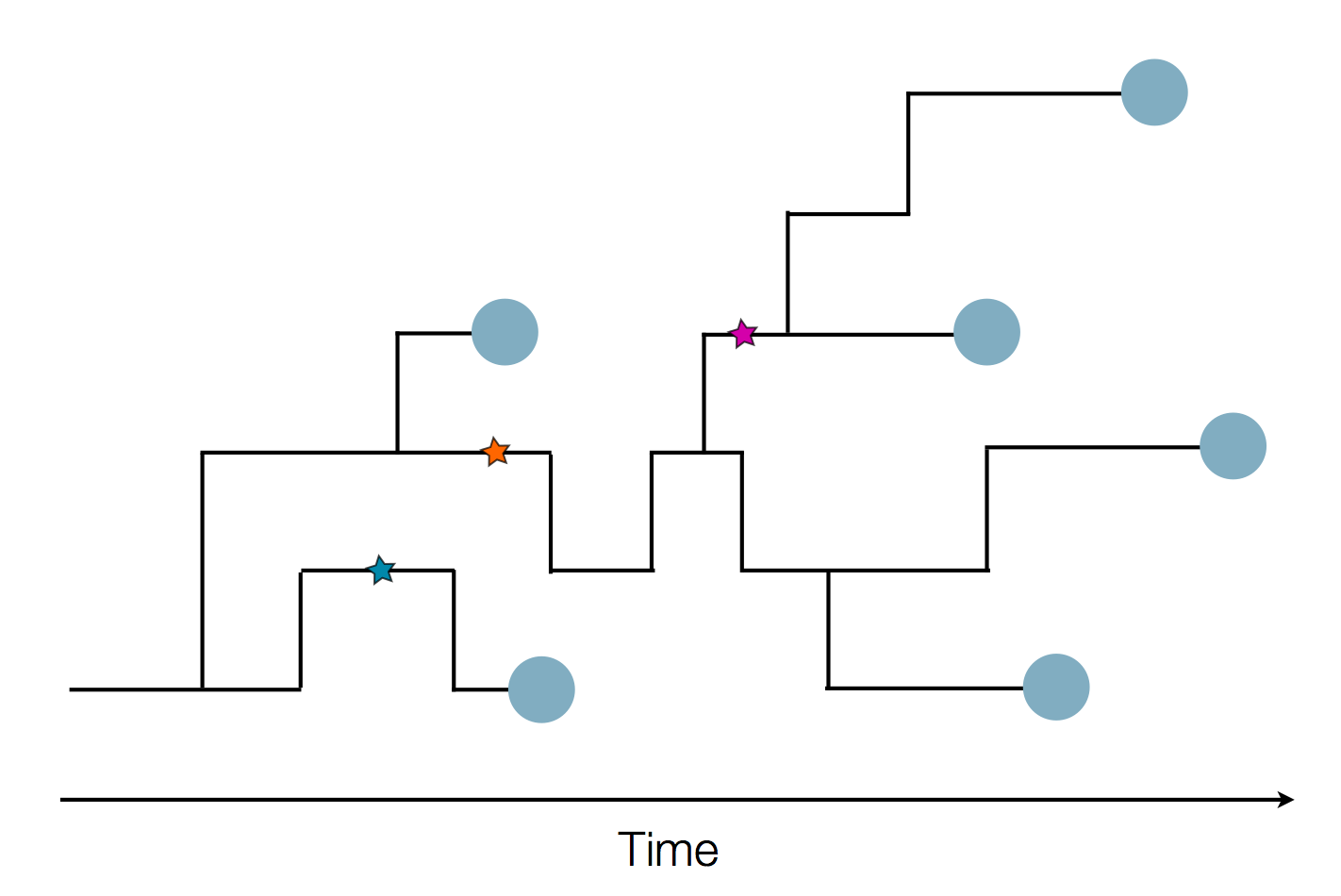
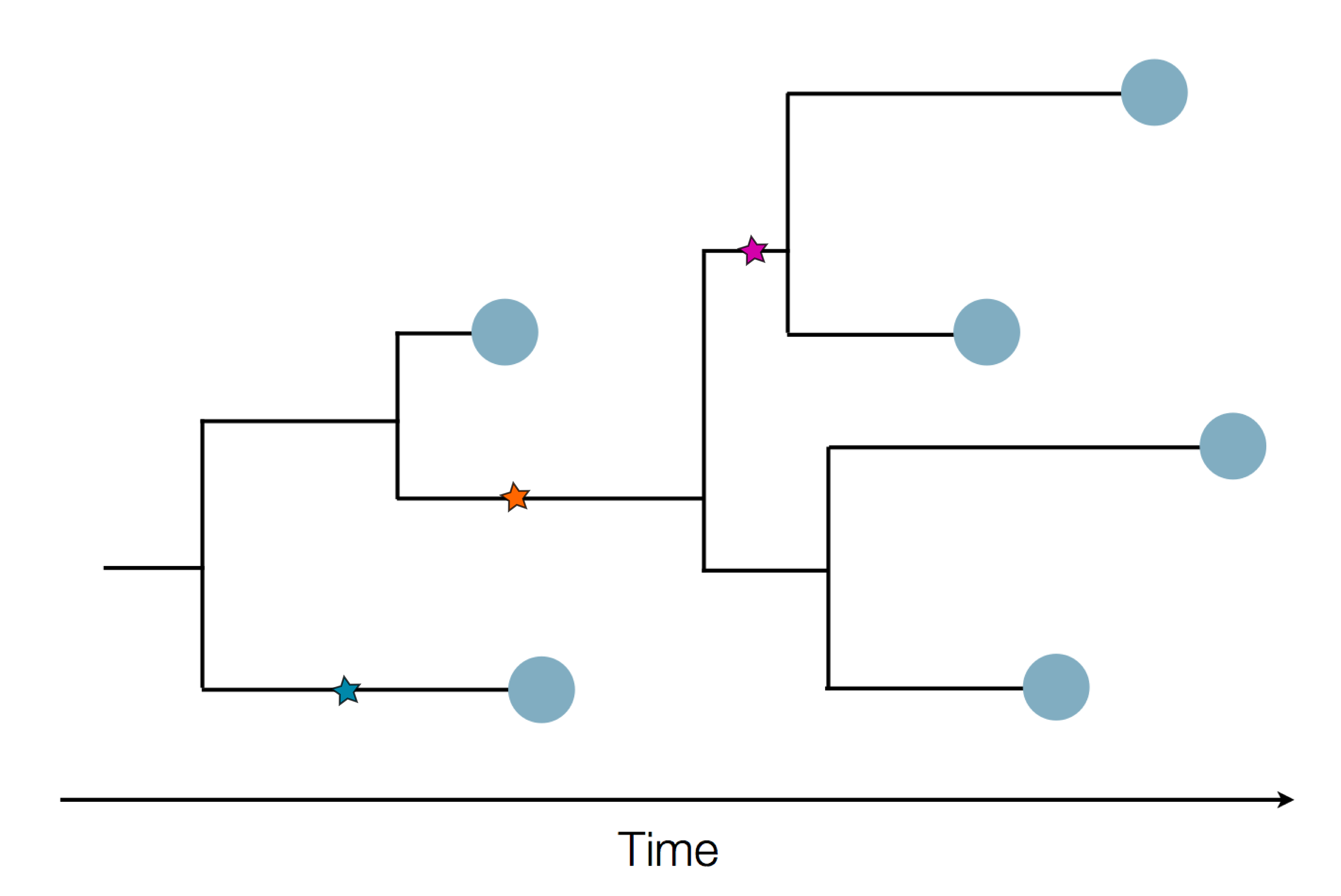
Phylogeny describes evolutionary relationships
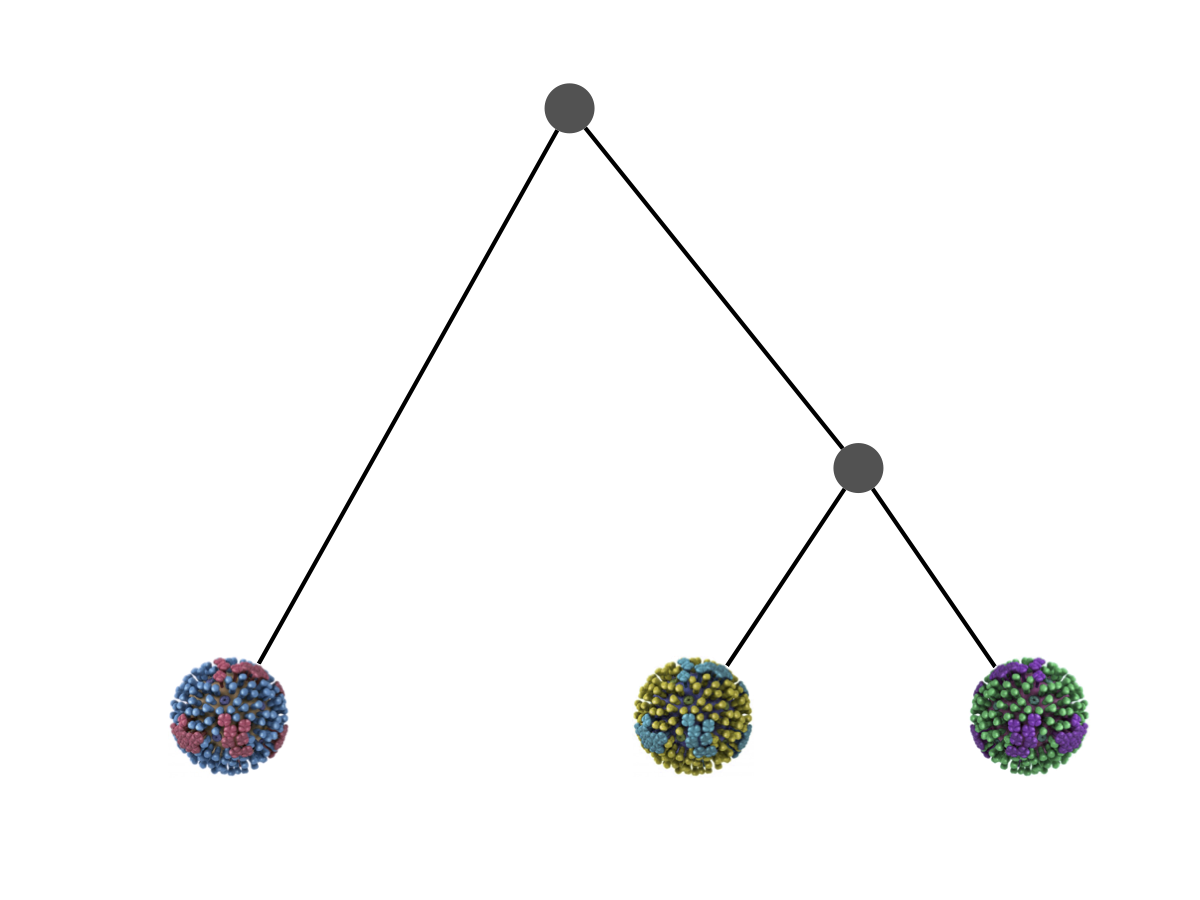
Phylogeny is usually a hypothesis based on characteristics of sampled taxa
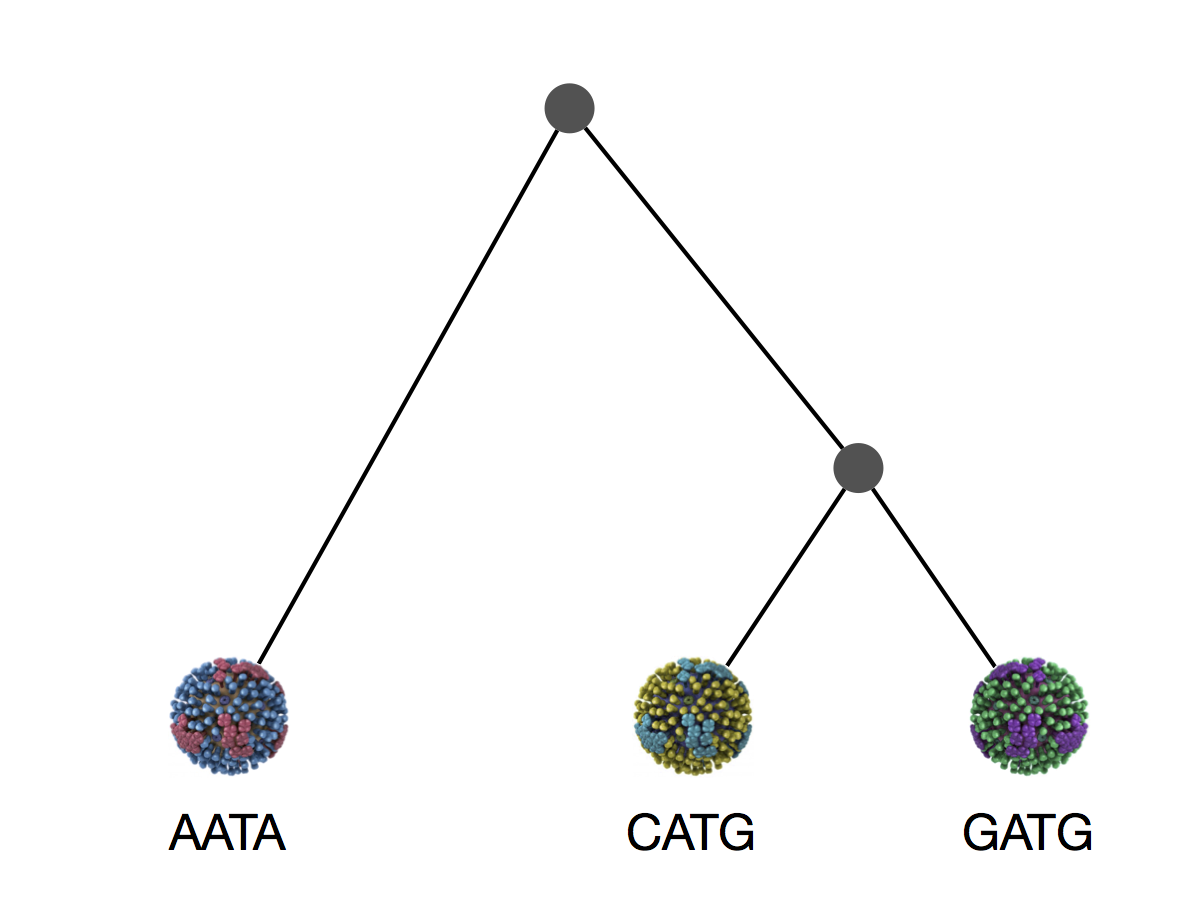
Phylogeny implies a series of mutational events leading to observed tip states
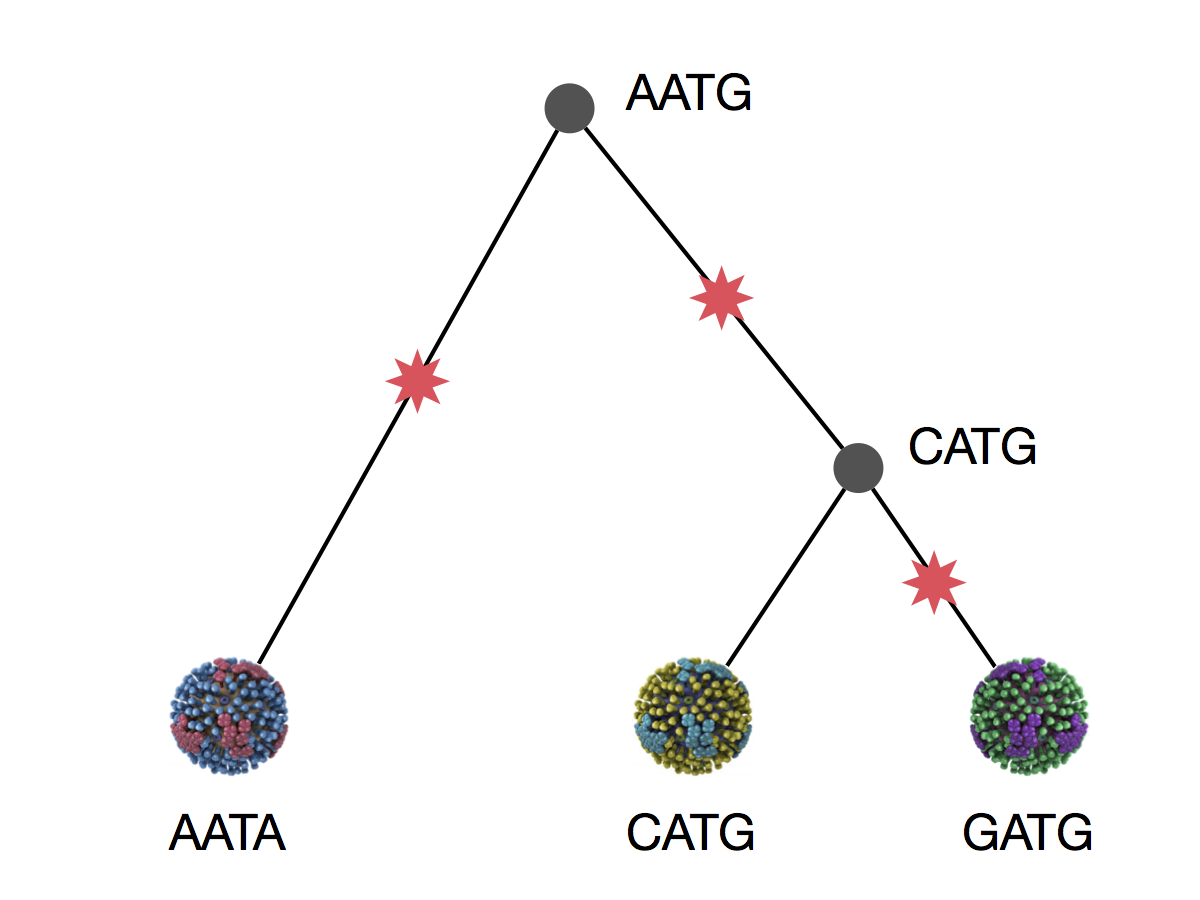
Phylogenetic inference
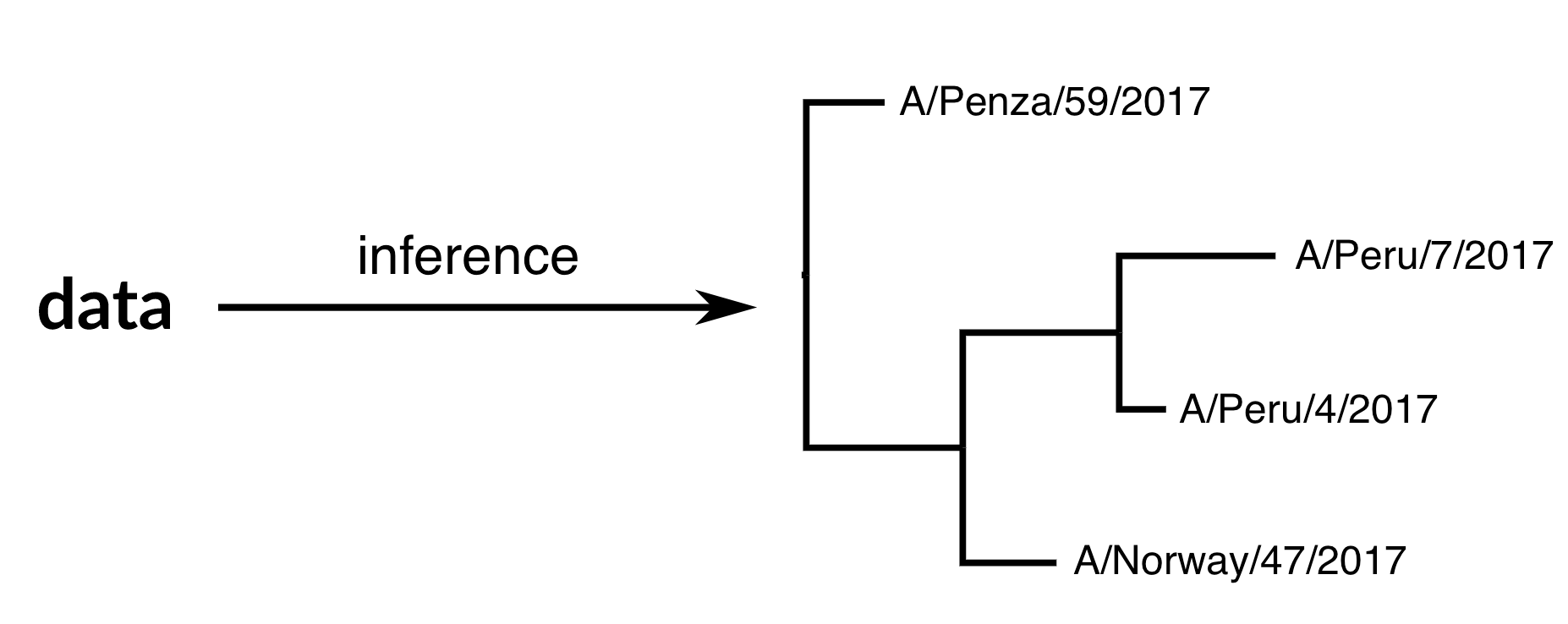
"Data" is generally a sequence alignment

Phylogeny structures site patterns
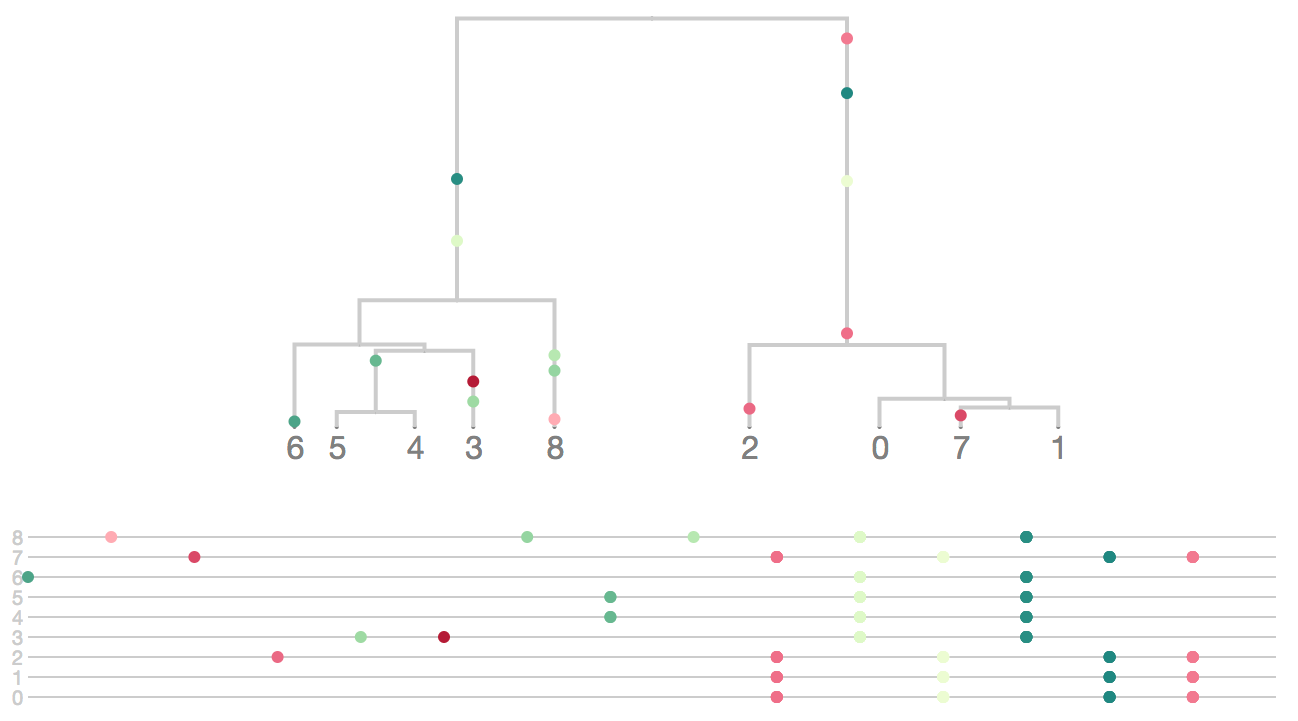
Tree space is vast
There are (2n-3)!! rooted trees for n taxa
- 3 taxa: 3 trees
- 5 taxa: 105 trees
- 10 taxa: 34,459,425 trees
- 20 taxa: 8.2 × 1021 trees
- 50 taxa: 2.8 × 1076 trees
- 100 taxa: 3.3 × 10184 trees
Solution space is rugged
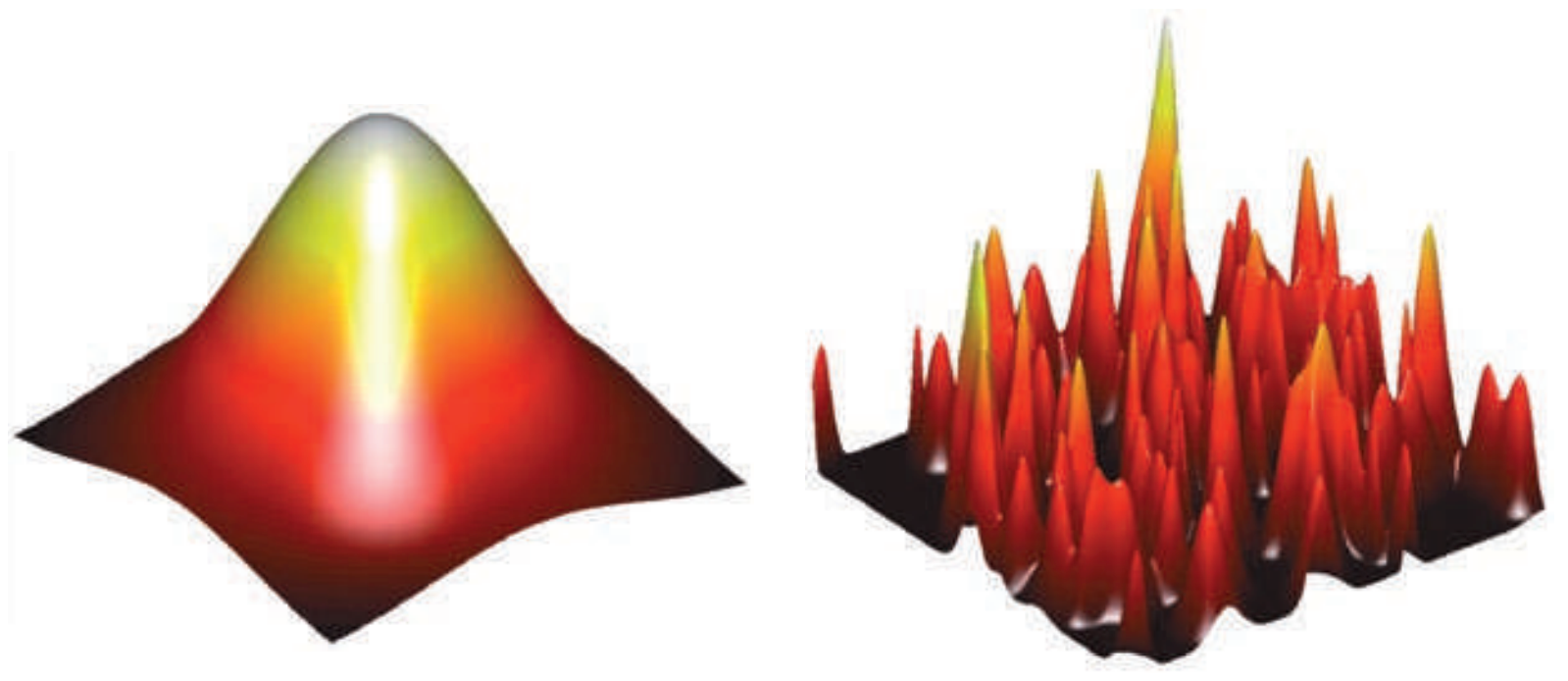
Types of phylogenetic inference methods
- Distance-based (neighbor-joining, fast, heuristic)
- Parsimony (fast, "model-free")
- Maximum likelihood (infers model of mutation, accurate, examples: FastTree, RAxML)
- Bayesian (like ML, but requires prior, produces estimates of uncertainty, examples: MrBayes, BEAST)
Inference is a tree topology, branch lengths and ancestral states
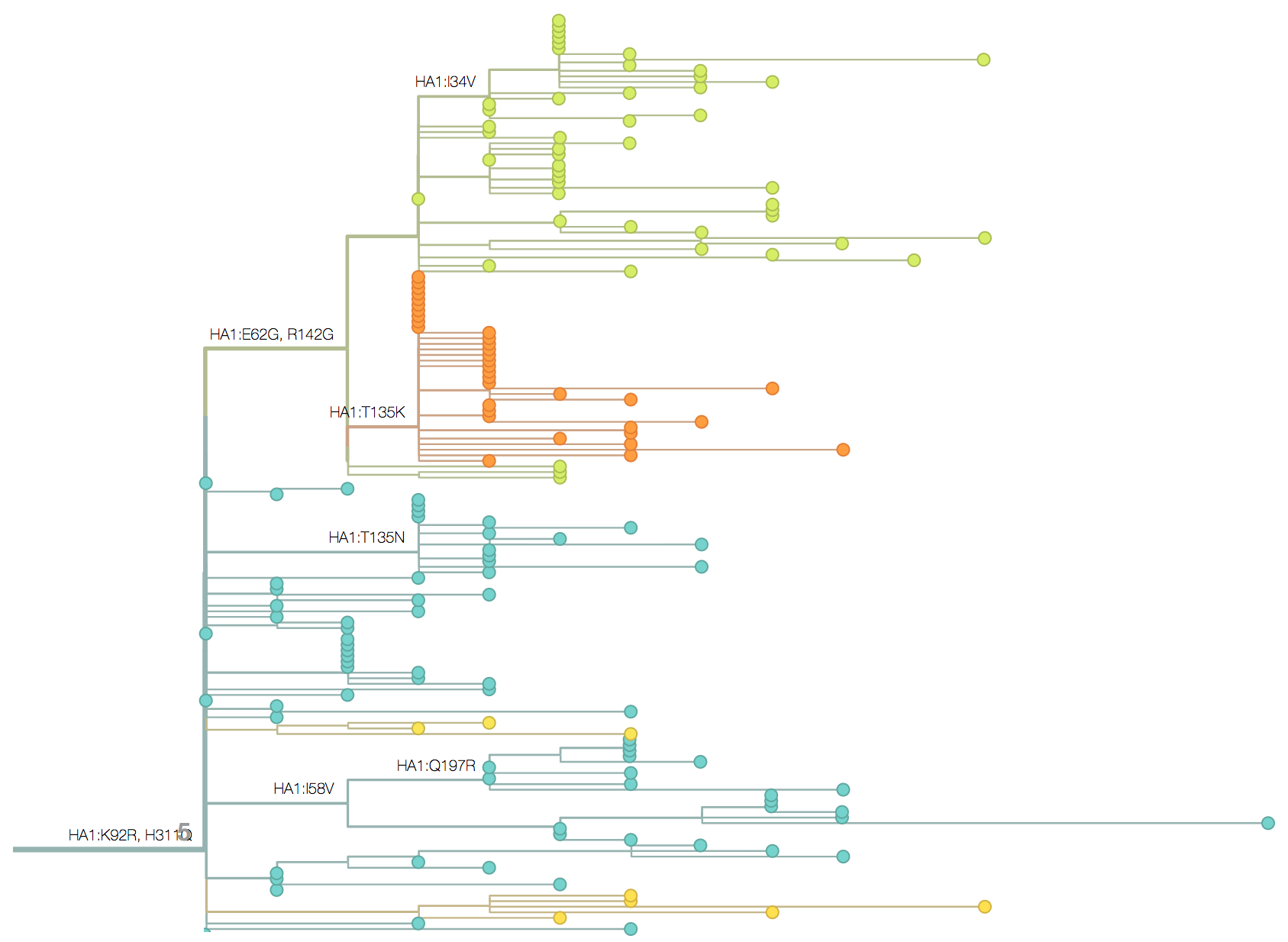
Molecular clocks and dated phylogenies
Mutations tend to accumulate in a clock-like fashion
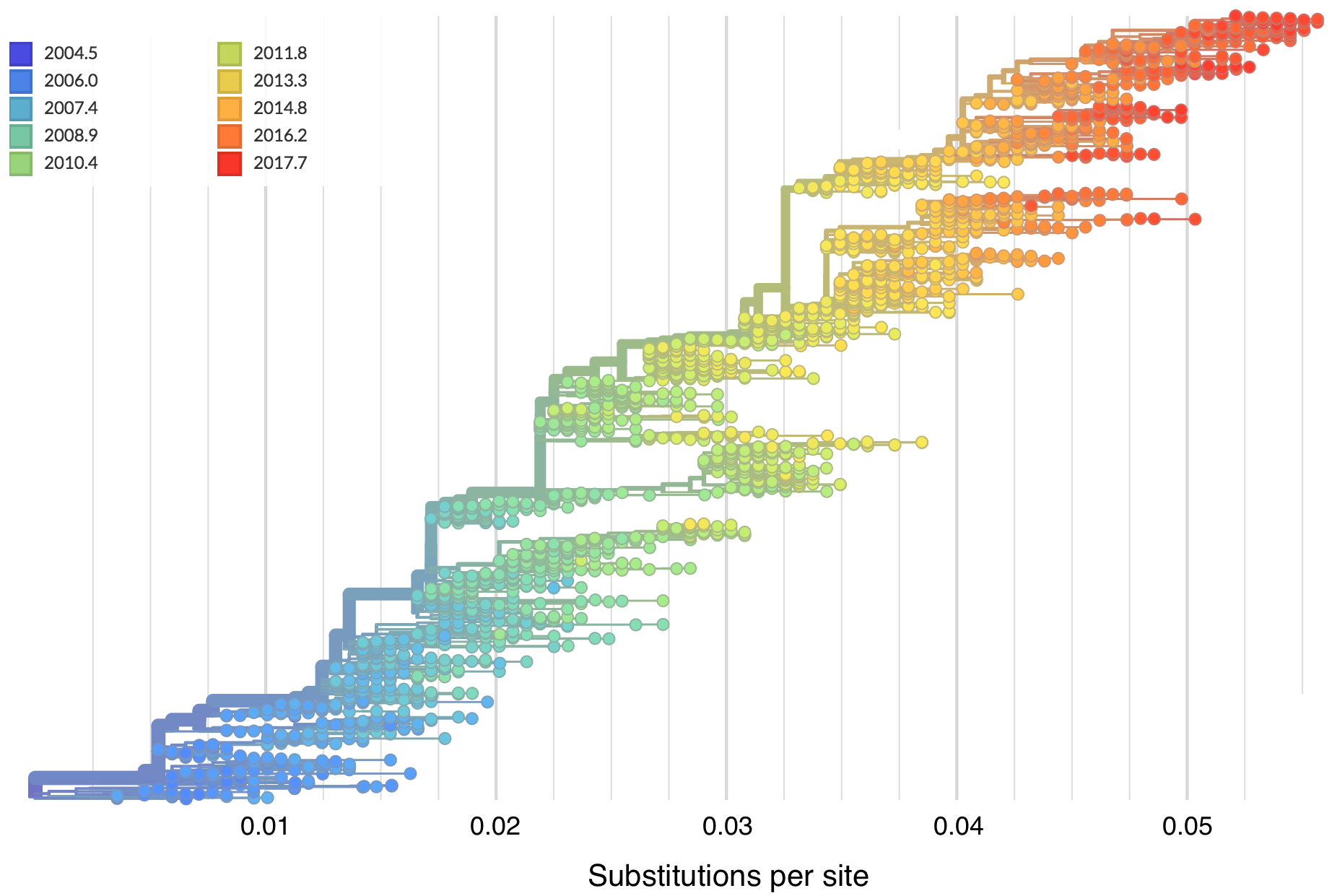
"Root-to-tip" plots show temporal signal
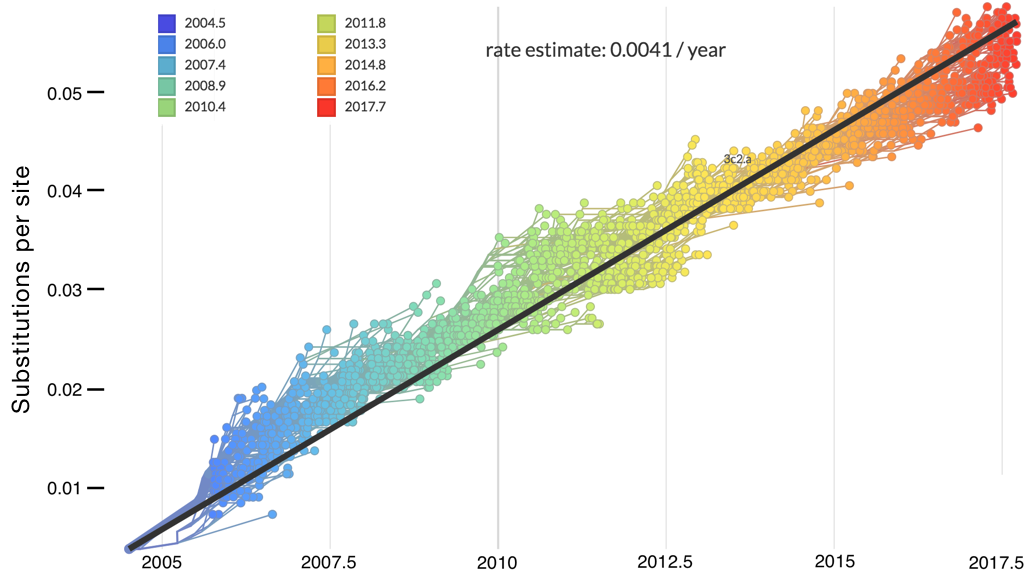
Allows conversion between branch length and time
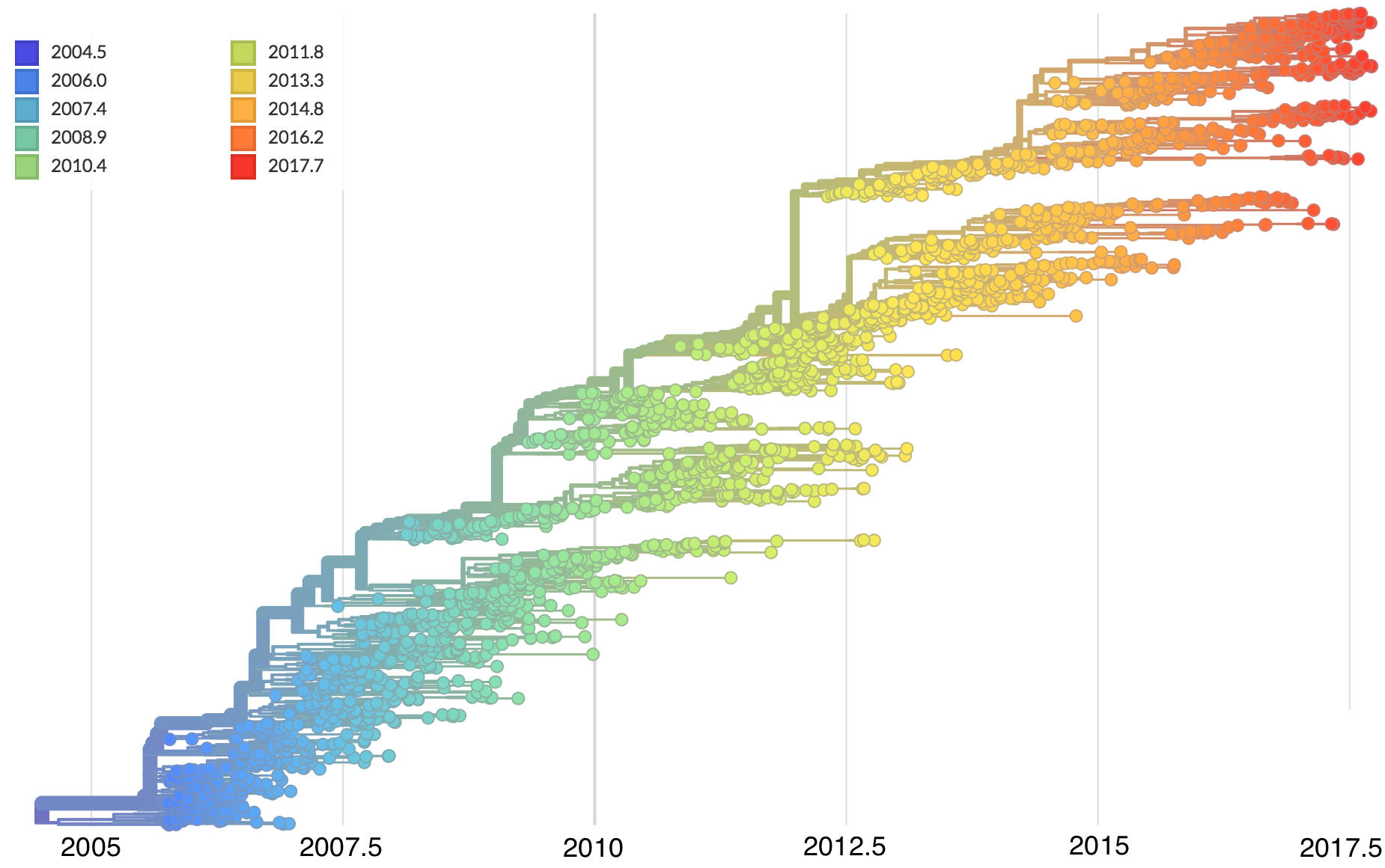
Dated phylogenies provide real-world context
Inference of discrete traits
"Data" is a phylogeny and tip states
States include nucleotides, amino acids, geo locations, hosts, etc...
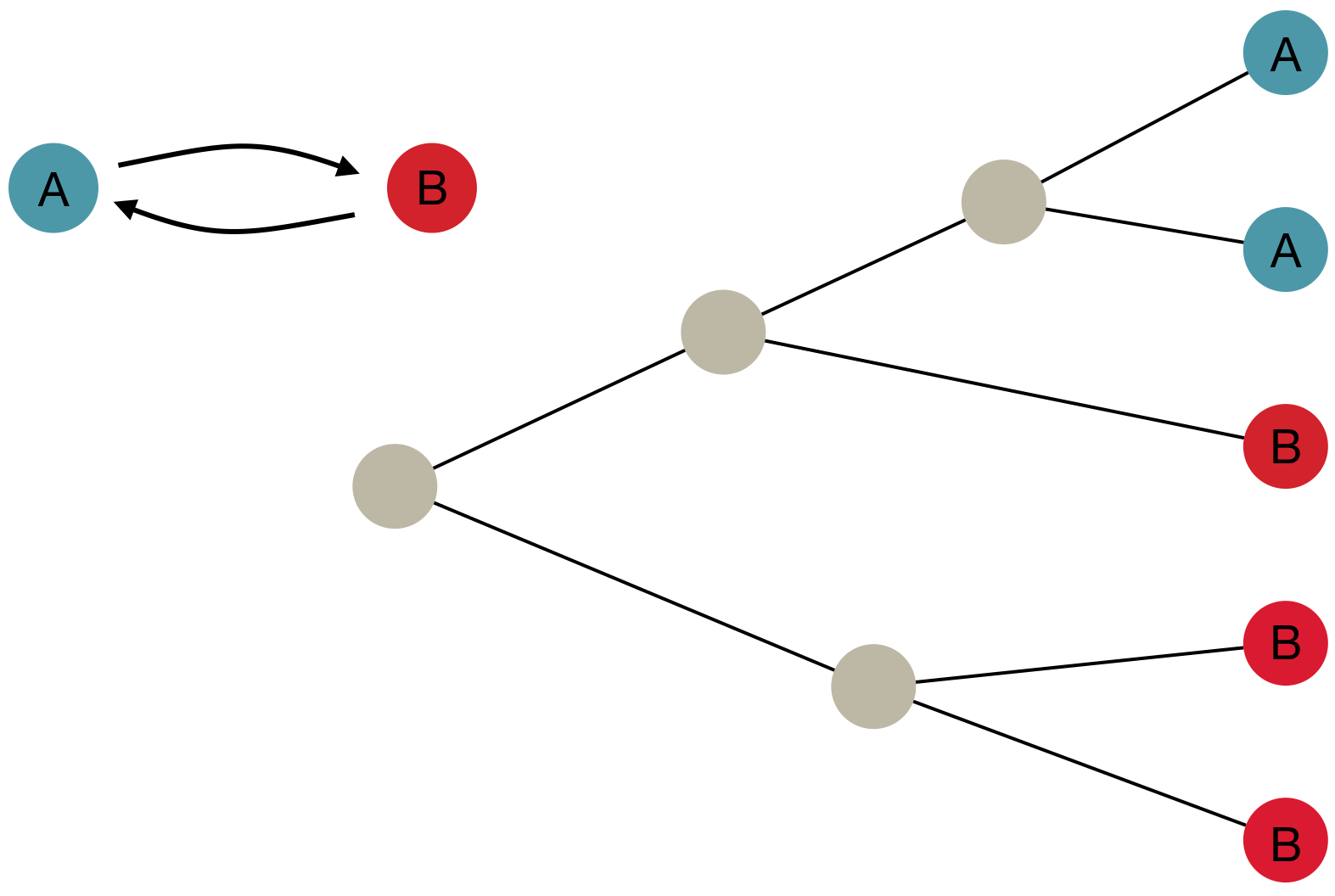
Model infers transition matrix and ancestral states
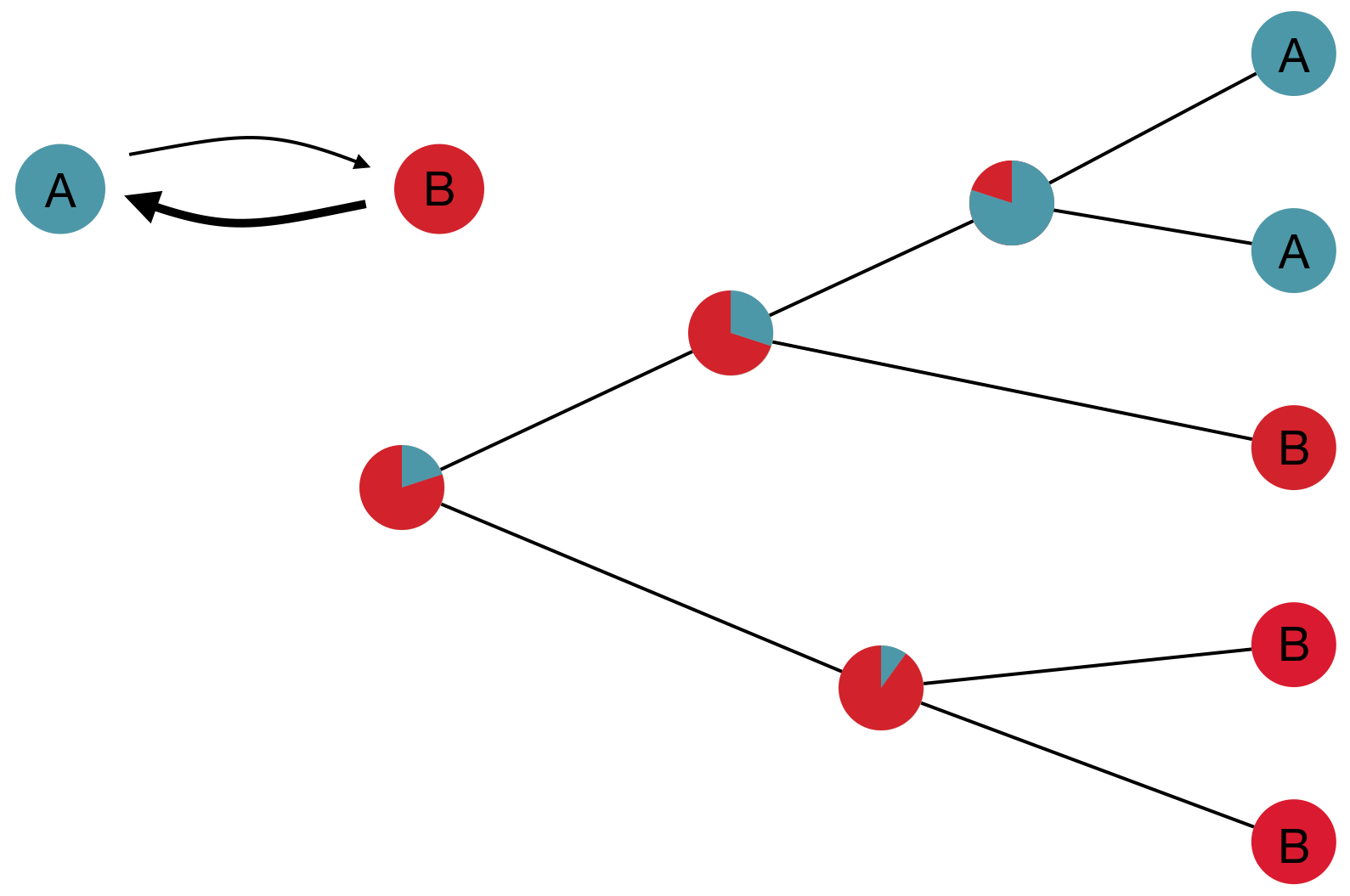
Rare transitions, short branches and many taxa increase confidence
Phylogeography
Nesting patterns are informative
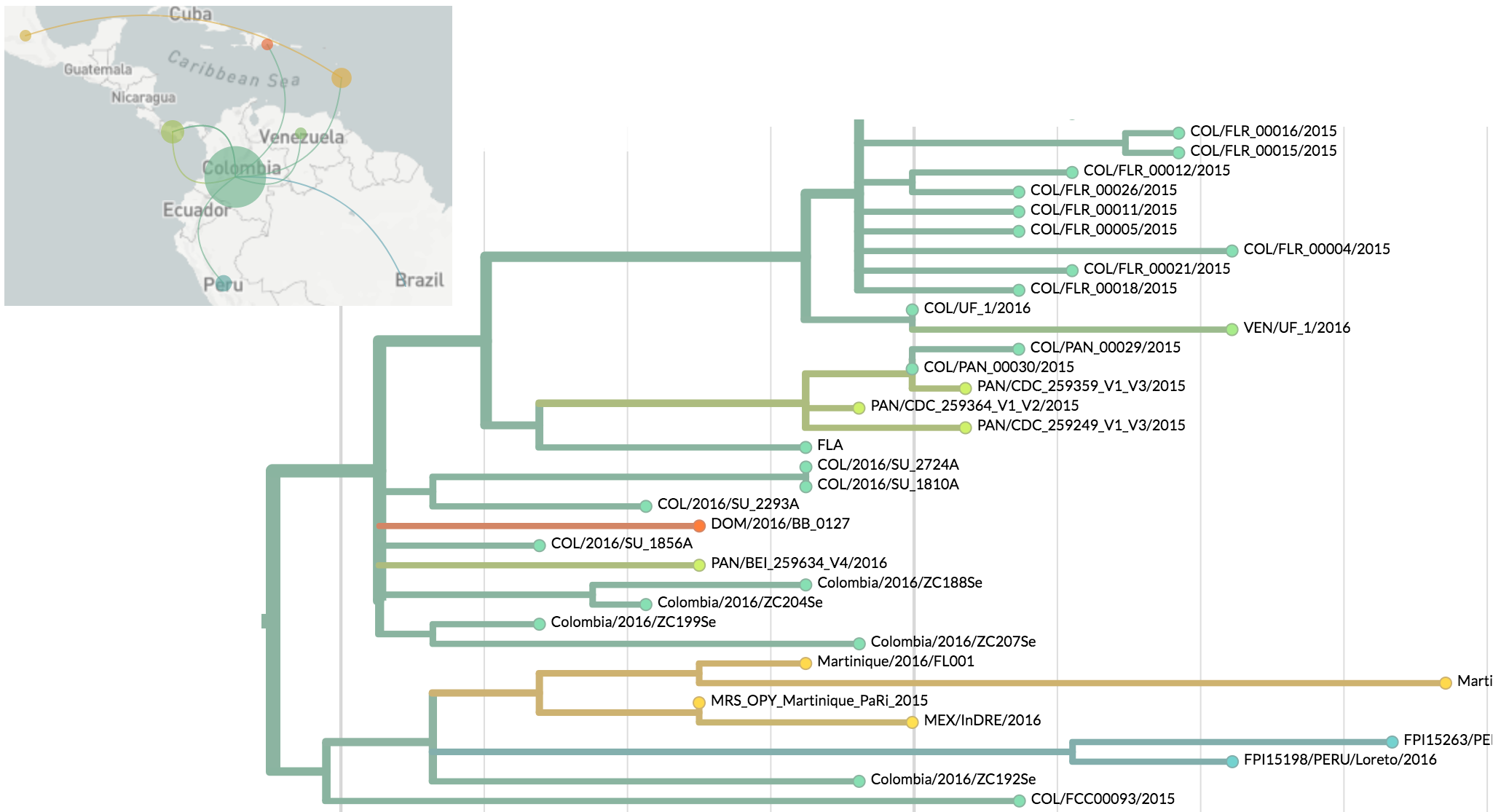
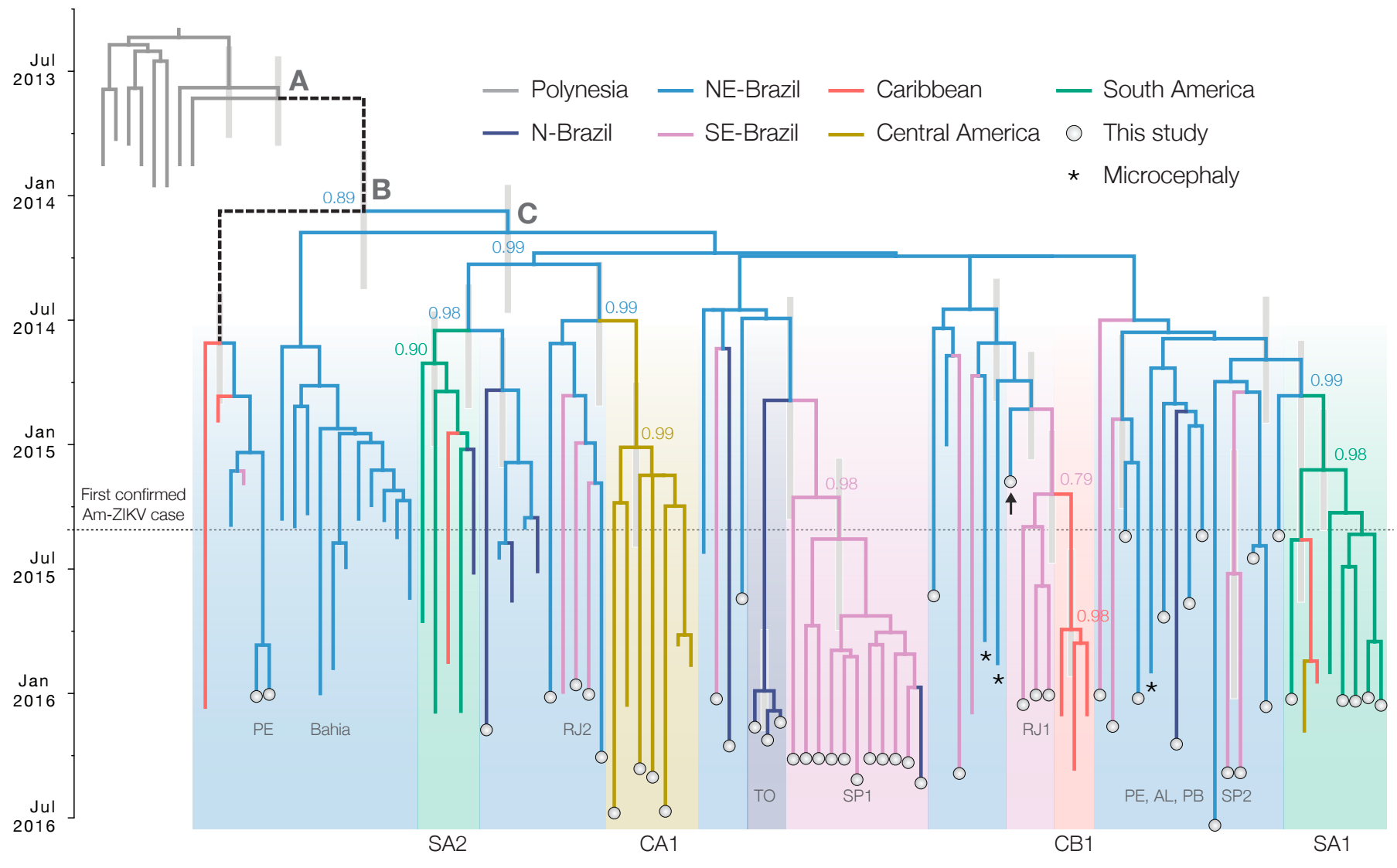
Zika phylogeny infers an origin in northeast Brazil
nextstrain.org/docs
Acknowledgements
Bedford Lab:
 Alli Black,
Alli Black,
 John Huddleston,
John Huddleston,
 Barney Potter,
Barney Potter,
 James Hadfield,
James Hadfield,
 Louise Moncla,
Louise Moncla,
 Tom Sibley,
Tom Sibley,
 Maya Lewinsohn,
Maya Lewinsohn,
 Katie Kistler
Katie Kistler
Nextstrain: Richard Neher, James Hadfield, Emma Hodcroft, Tom Sibley, John Huddleston, Sidney Bell, Barney Potter, Colin Megill, Charlton Callender