Genomic tracking of SARS-CoV-2 evolution and spread
Trevor Bedford (@trvrb)
20 May 2020
CCC Seminar
UT COVID-19 Modeling Consortium
Slides at: bedford.io/talks


Significant fog of war. Genomic approaches offer orthogonal data source to understand the pandemic.
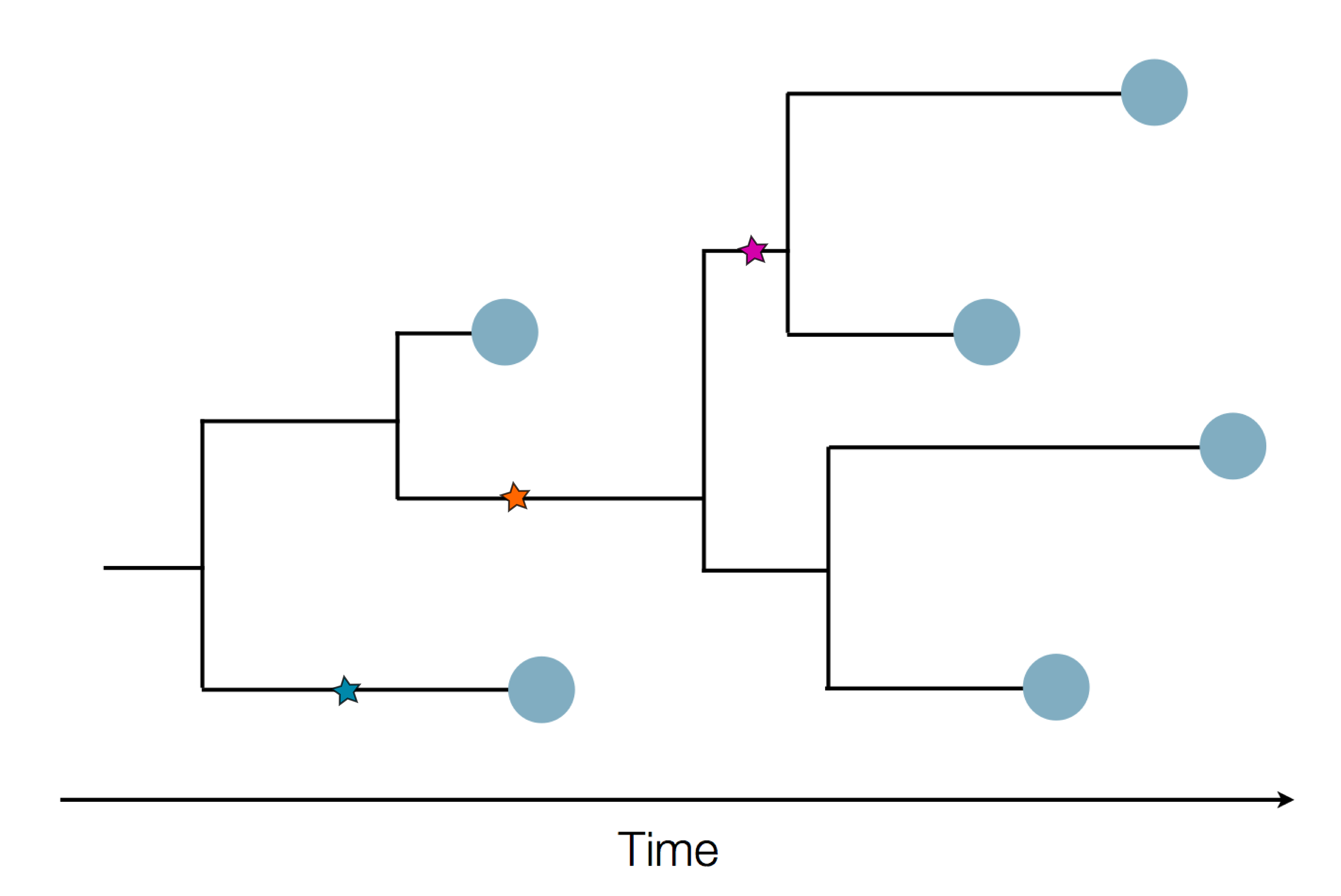
Epidemic process
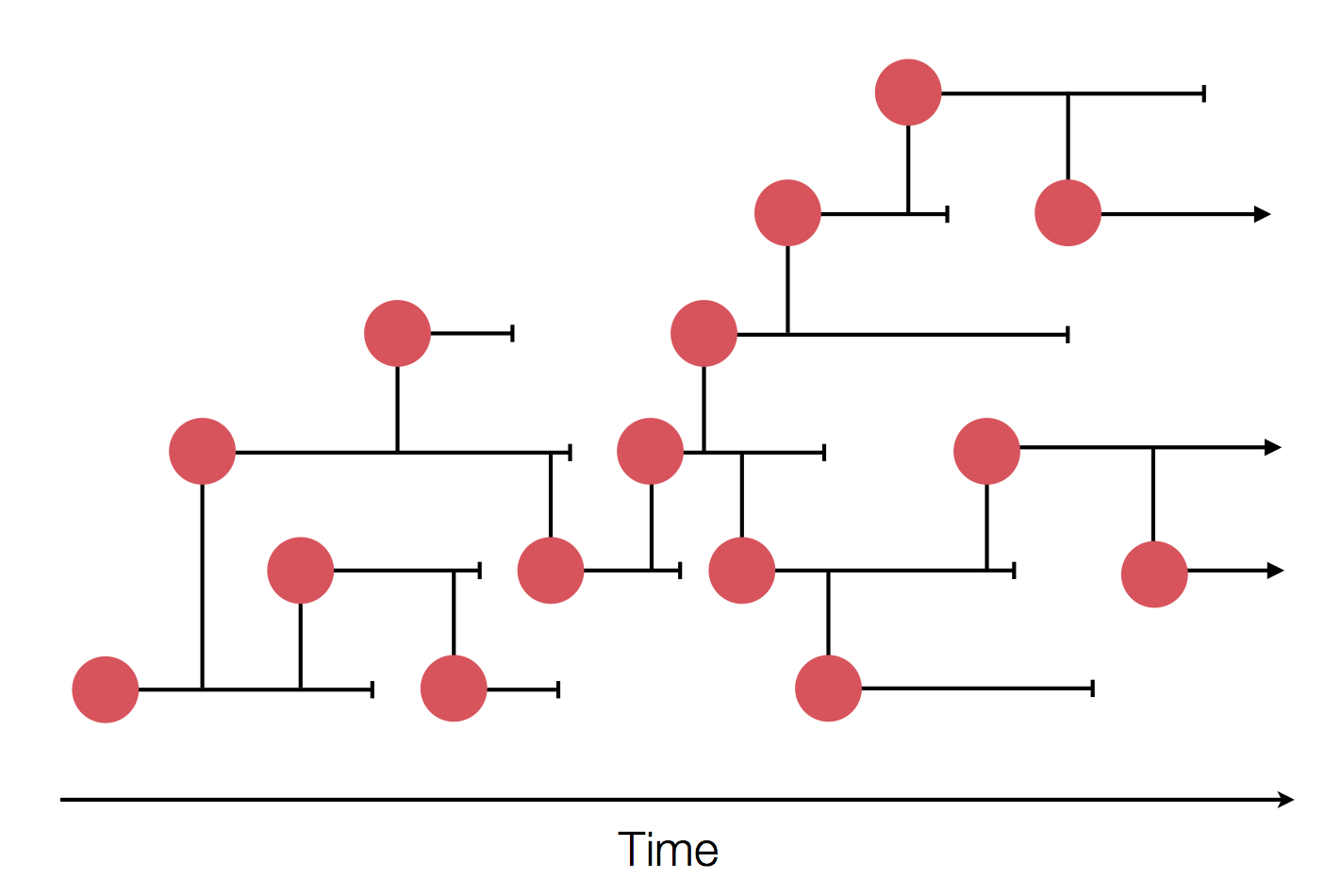
Sample some individuals
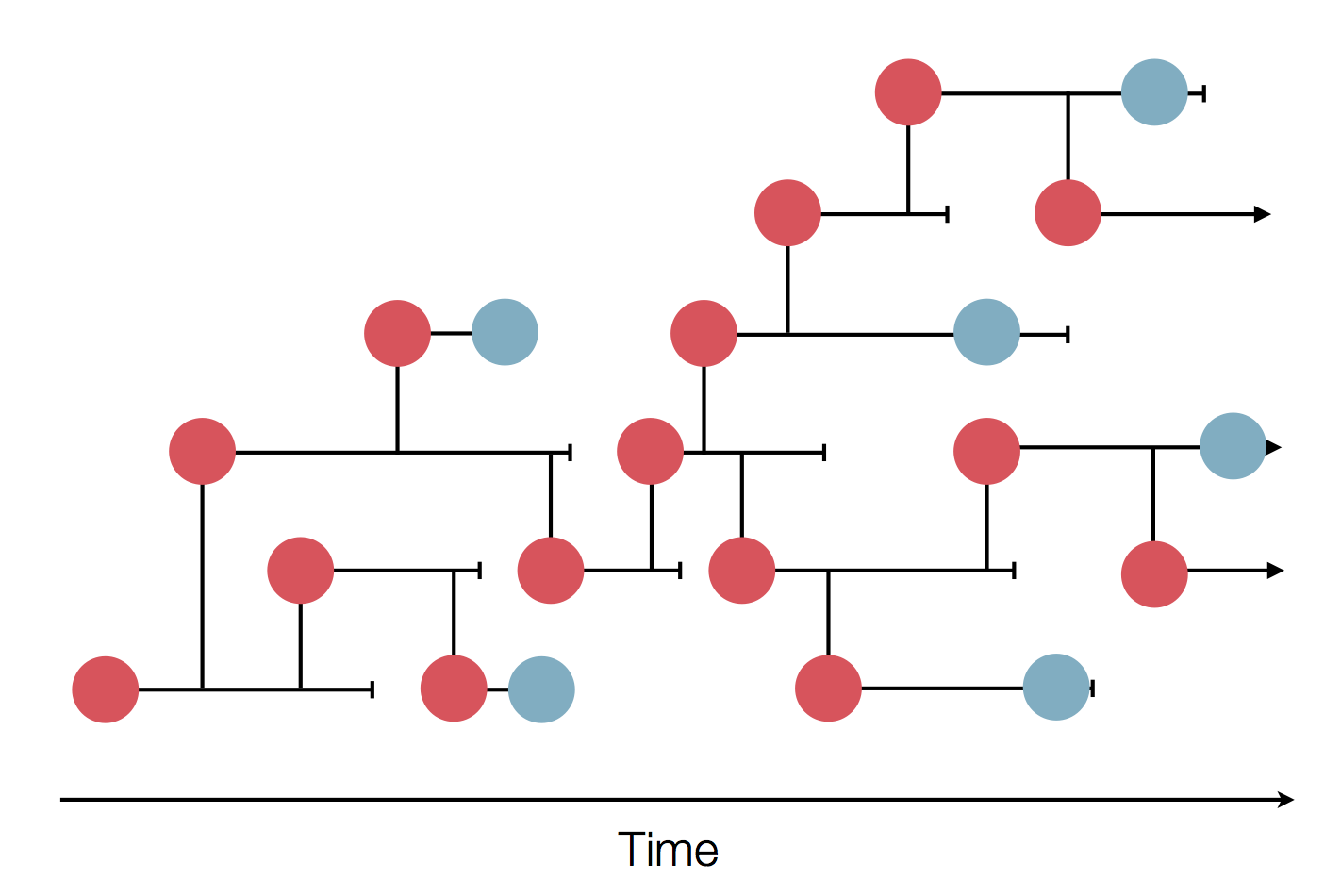
Sequence and determine phylogeny
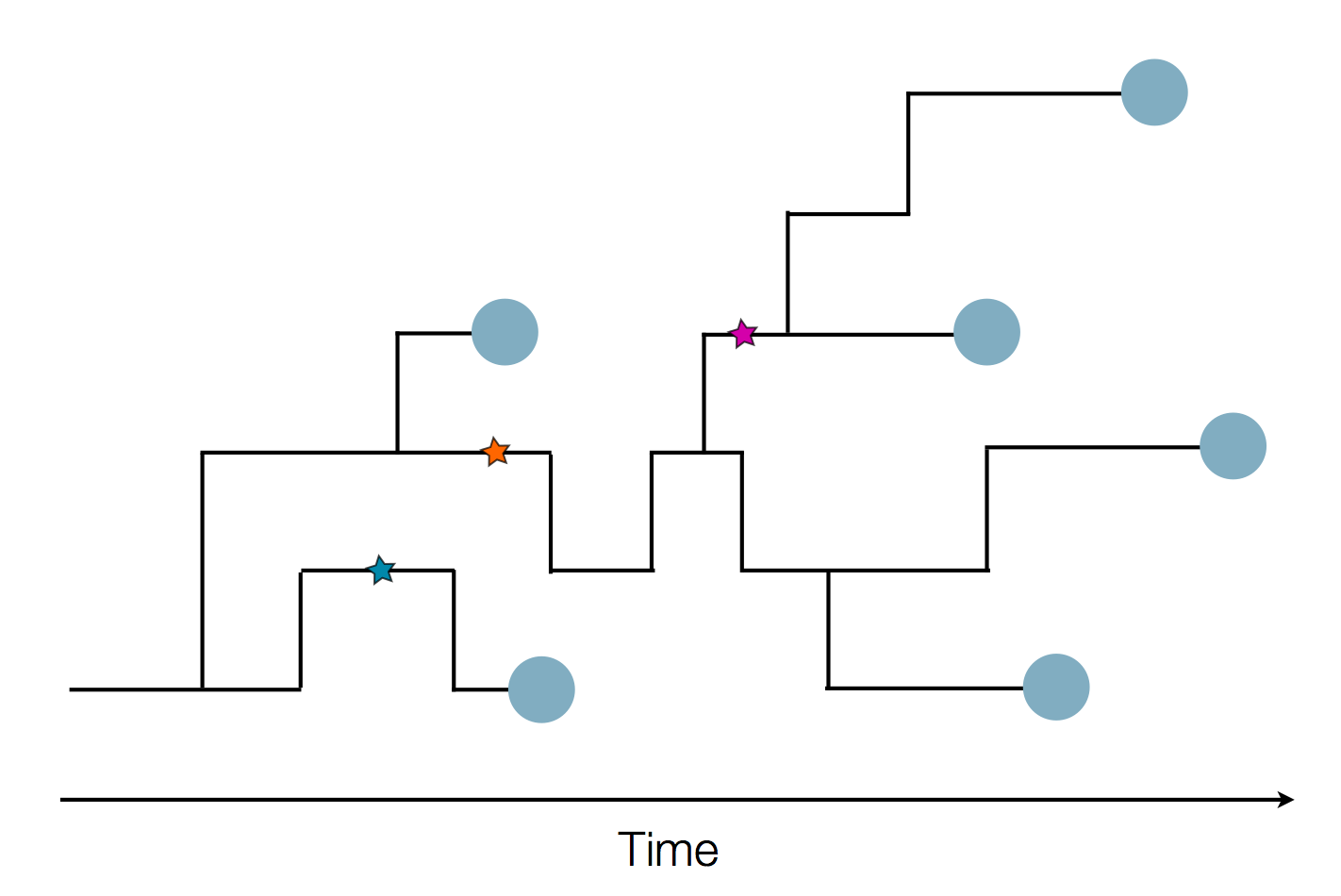
Sequence and determine phylogeny
Nextstrain
Project to conduct real-time molecular epidemiology and evolutionary analysis of emerging epidemics
with
![]() Richard Neher,
Richard Neher,
![]() James Hadfield,
James Hadfield,
![]() Emma Hodcroft,
Emma Hodcroft,
![]() Thomas Sibley,
Thomas Sibley,
![]() John Huddleston,
John Huddleston,
![]() Louise Moncla,
Louise Moncla,
![]() Cassia Wagner,
Cassia Wagner,
![]() Miguel Paredes,
Miguel Paredes,
![]() Misja Ilcisin,
Misja Ilcisin,
![]() Kairsten Fay,
Kairsten Fay,
![]() Jover Lee,
Jover Lee,
![]() Allison Black,
Allison Black,
![]() Colin Megill,
Colin Megill,
![]() Sidney Bell,
Sidney Bell,
![]() Barney Potter,
Barney Potter,
![]() Charlton Callender
Charlton Callender
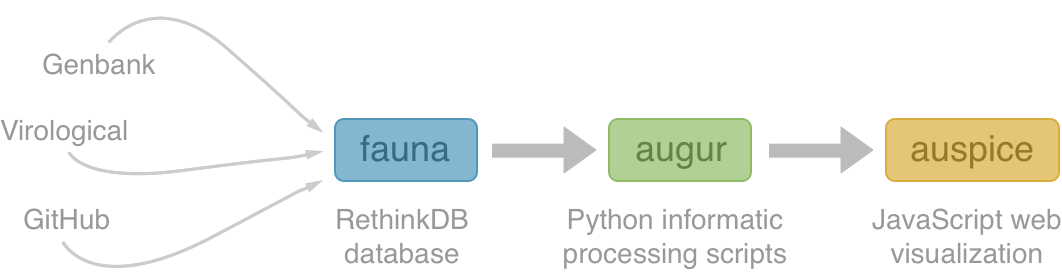
Nextstrain architecture
All code open source at github.com/nextstrain
Two central aims: (1) rapid and flexible phylodynamic analysis and
(2) interactive visualization
Rapid build pipeline for 3000 SARS-CoV-2 genomes (timings are for a laptop)
- Align with MAFFT (~20 min)
- Build ML tree with IQTREE (~40 min)
- Temporally resolve tree and geographic ancestry with TreeTime (~50 min)
- Total pipeline (~2 hr)
Current data flow for SARS-CoV-2
- Labs contribute directly to GISAID (now have >17k full genomes)
- Nextstrain pulls a complete dataset from GISAID every 60 minutes
- This triggers an automatic rebuild on Amazon Web Services
- We manually update new lat/longs, etc...
- We push this build online to nextstrain.org and tweet the update from @nextstrain
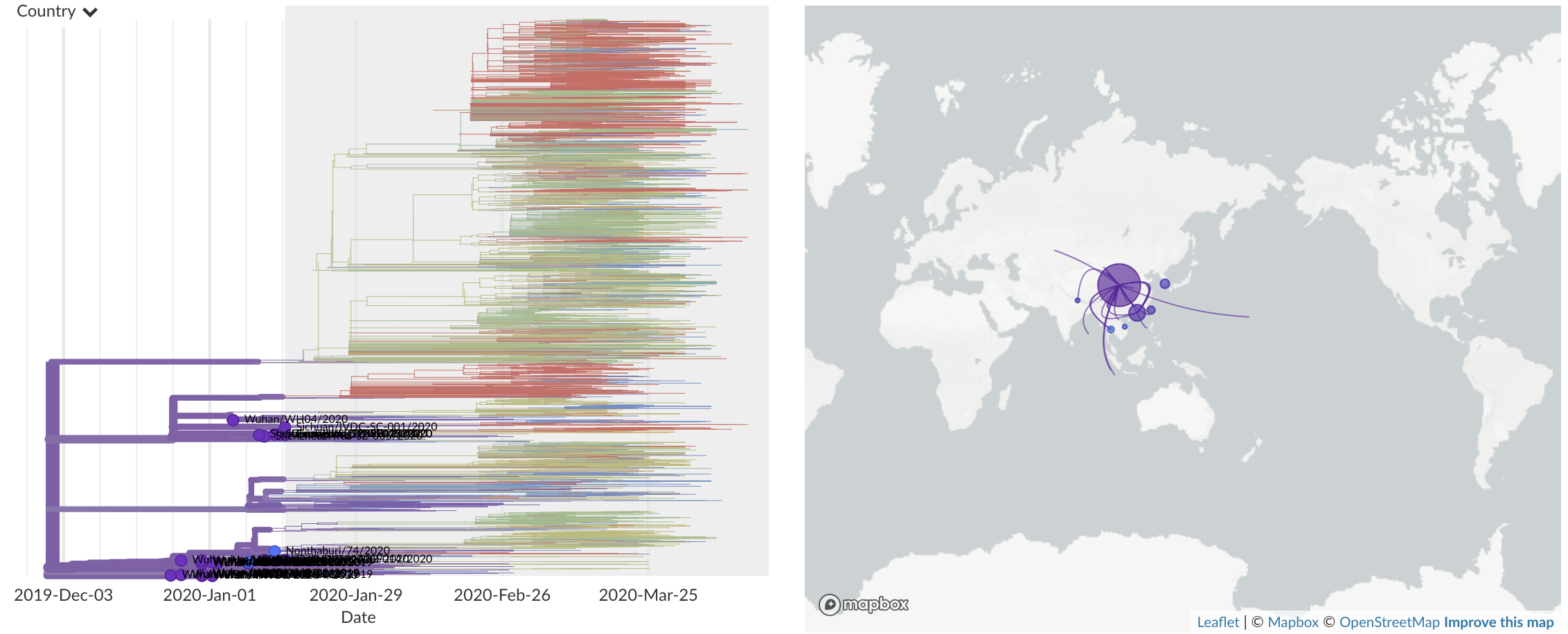
Dec/Jan: Emergence of SARS-CoV-2 from Wuhan in ~Nov 2019
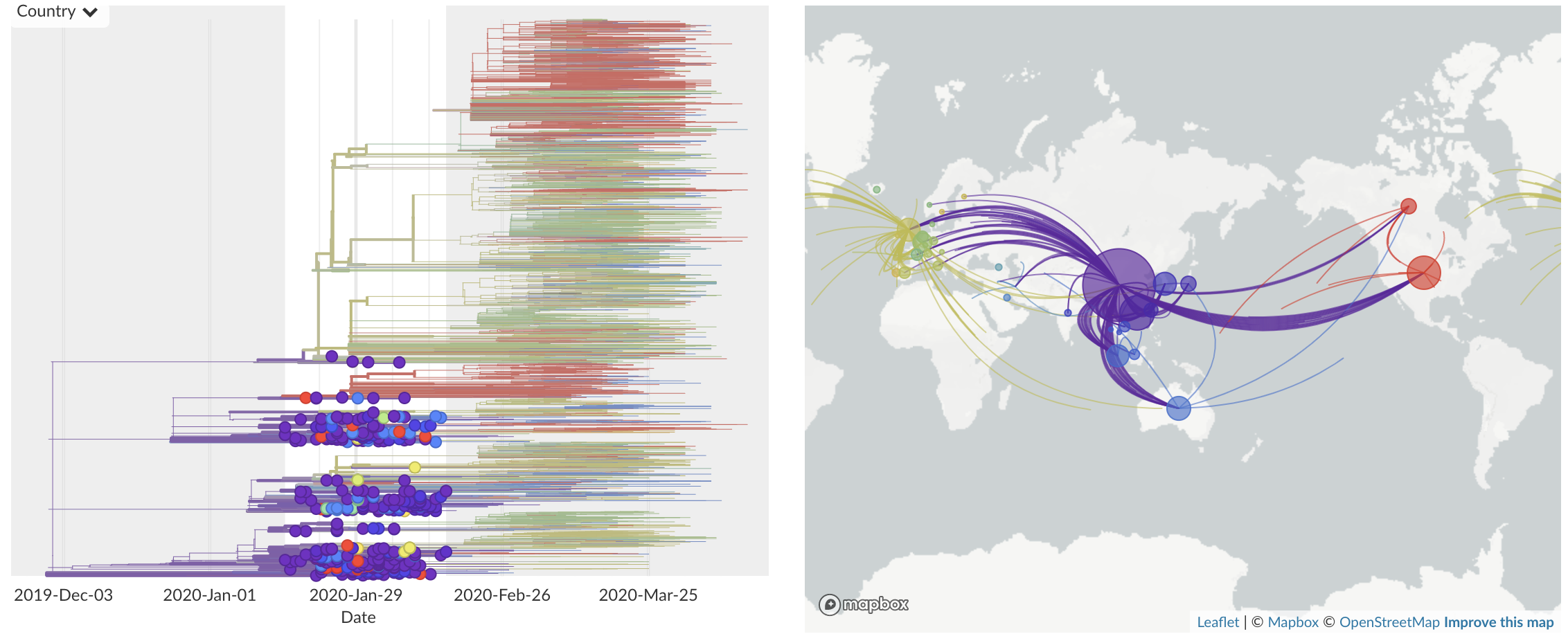
Jan/Feb: Spread within China and seeding elsewhere
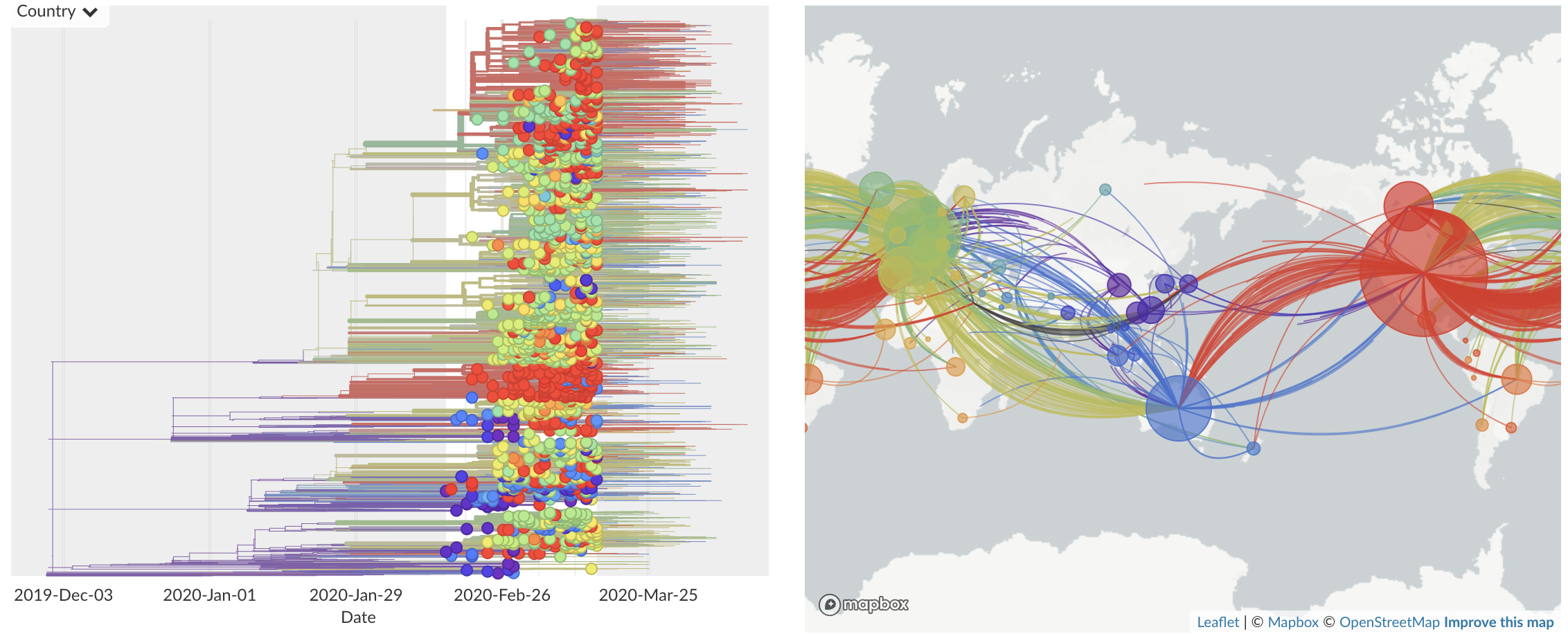
Feb/Mar: Epidemic spread within North America and Europe
Mar/Apr: Continued growth, but decreasing transmission with social distancing measures
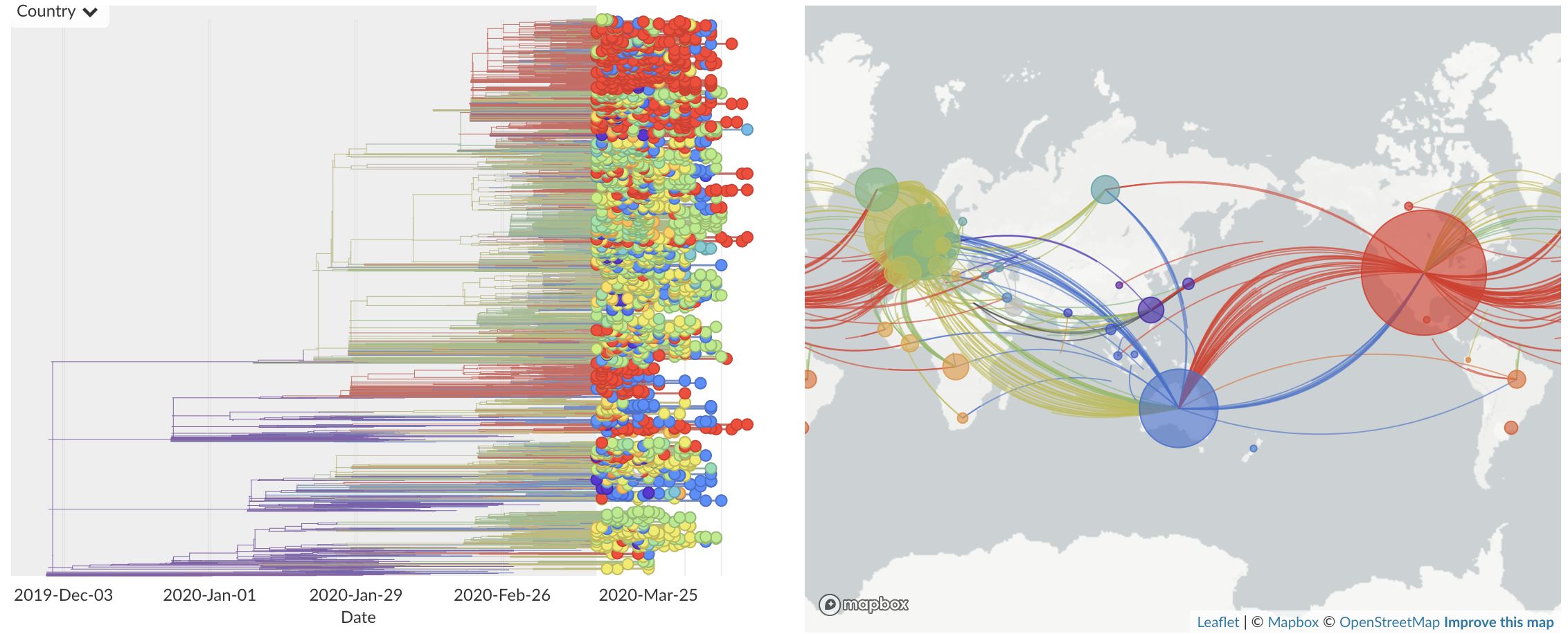
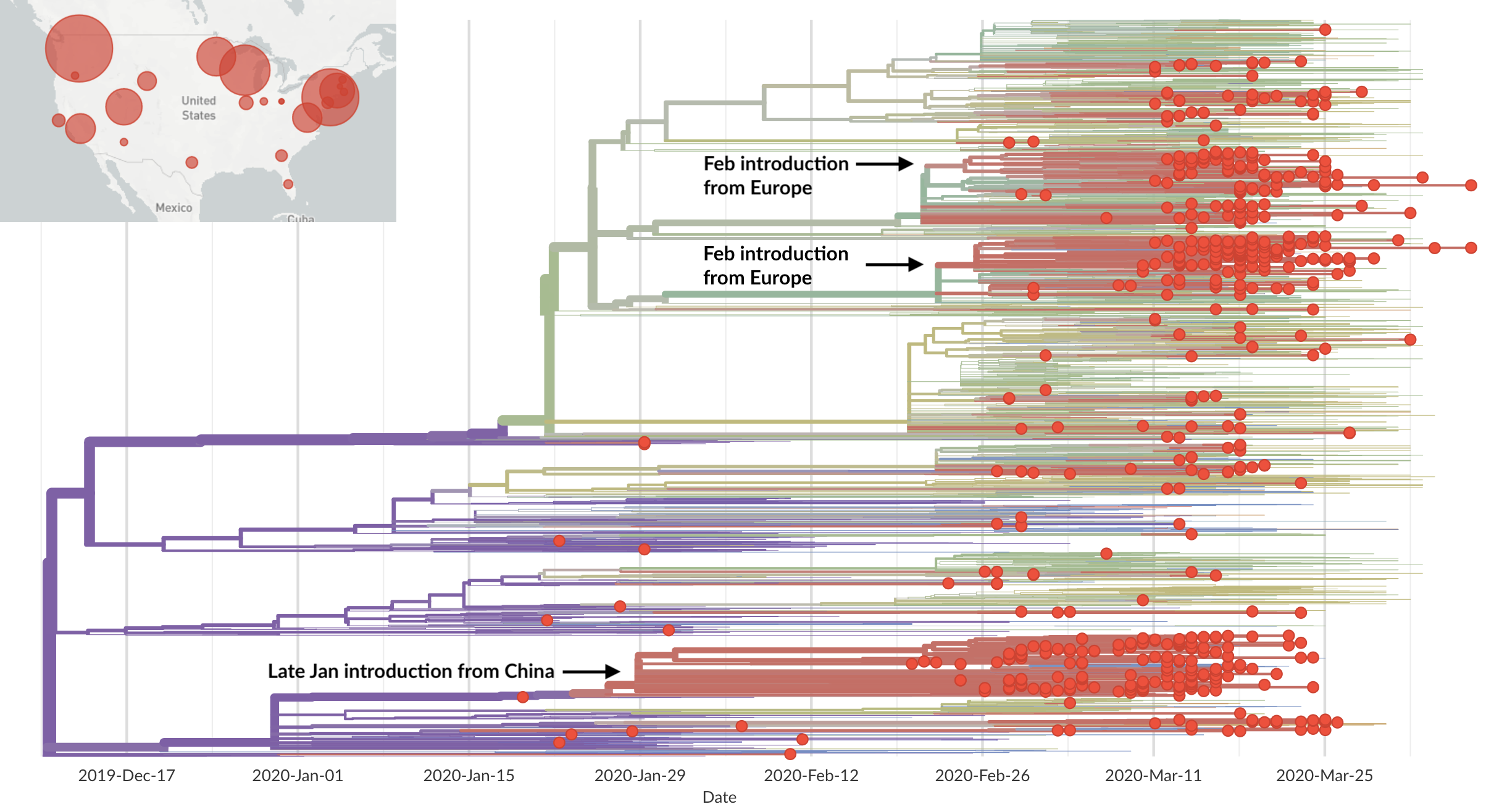
Epidemic in the USA was introduced from China in late Jan and from Europe during Feb
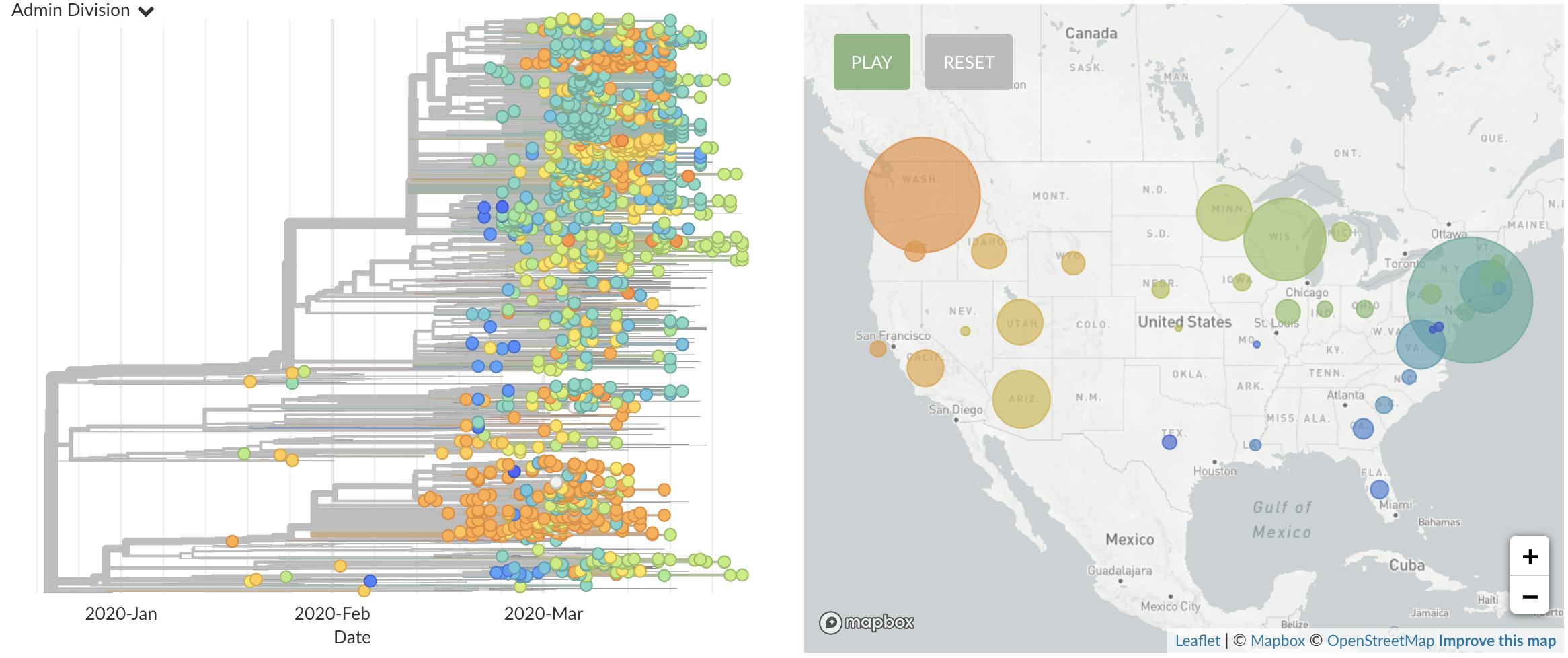
Once in the US, virus spread rapidly
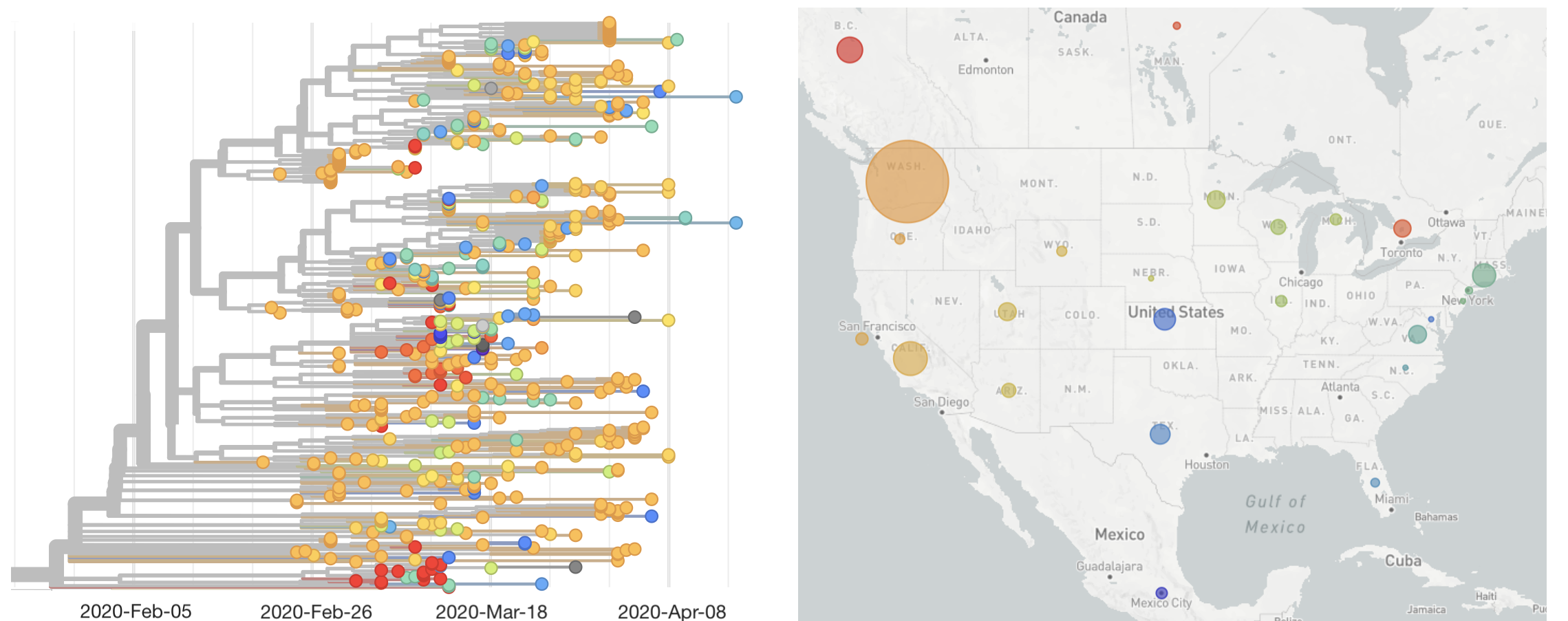
Single introduction ~Feb 1 quickly shows up throughout the country
Sequencing immediately useful for epidemiological understanding, but selection and functional impacts should also be studied
Significant interest in spike mutation D614G
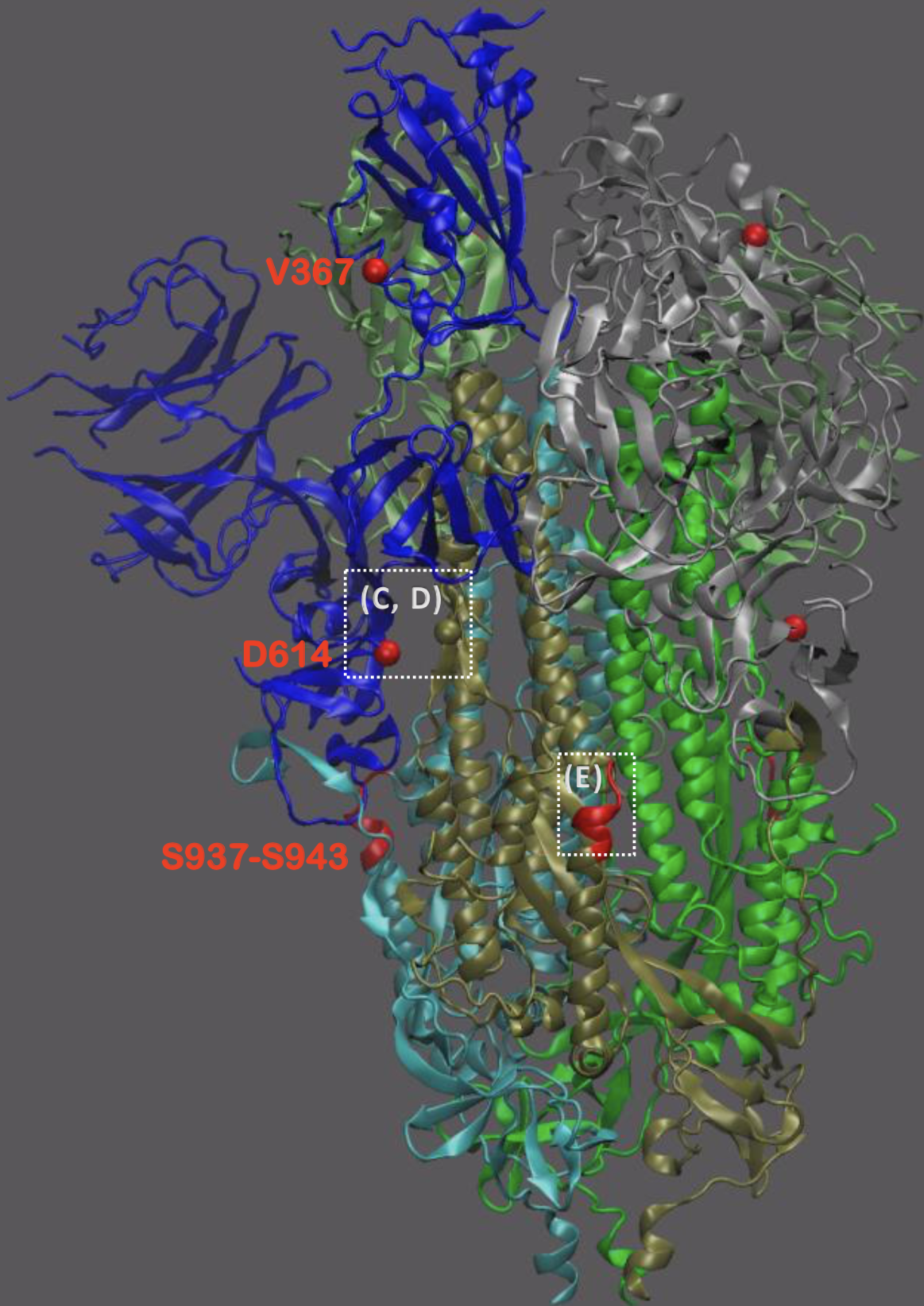
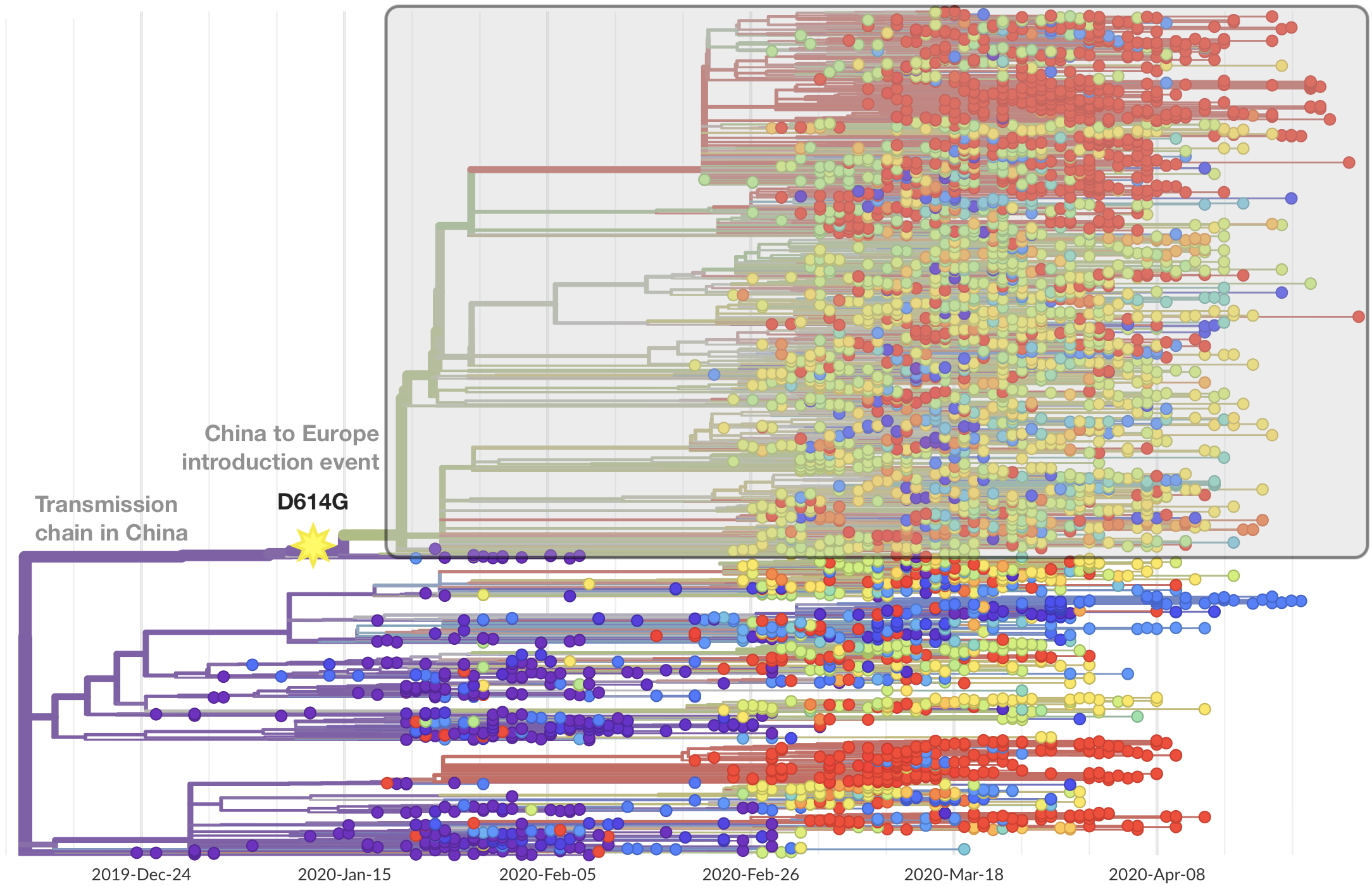
This mutation occurred in the initial European introduction
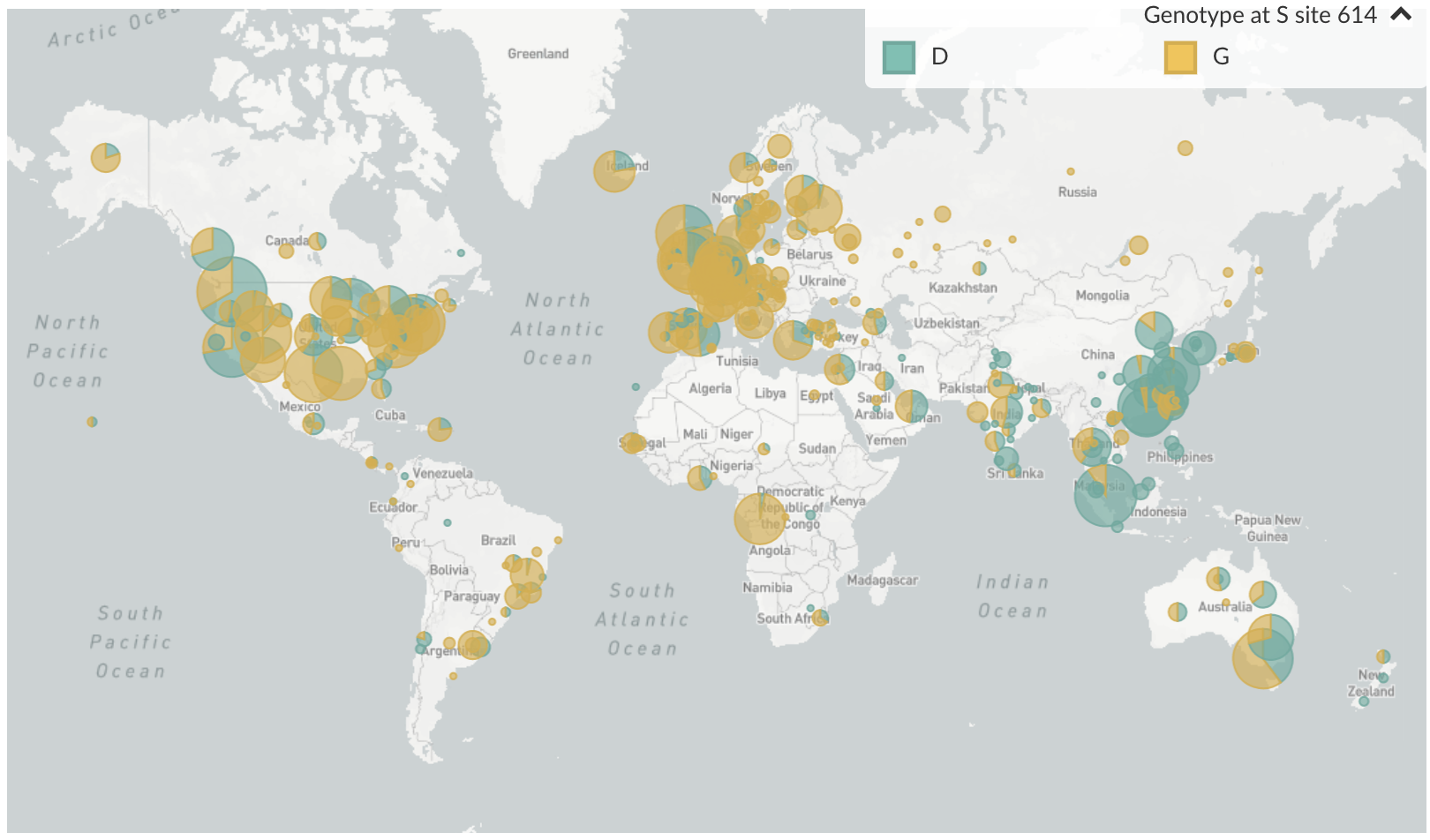
D614G is prevalent throughout Europe and mixed in US and Australia
D614G is increasing in frequency across states in US and Australia
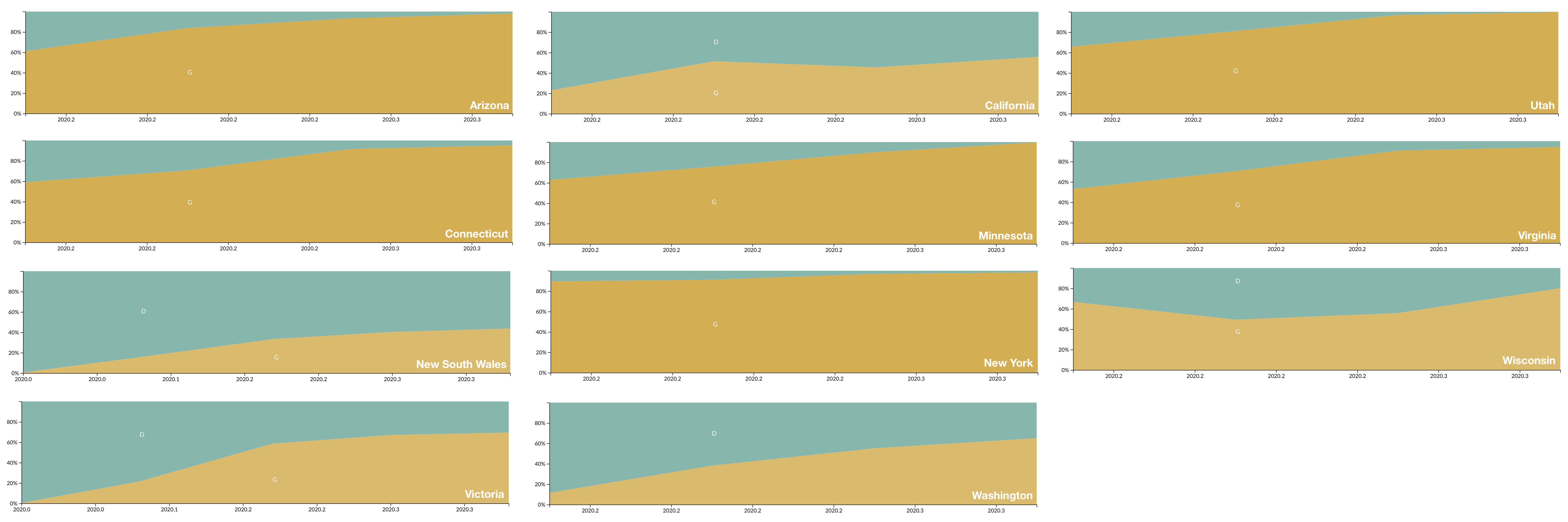
D614G is increasing in frequency across states in US and Australia
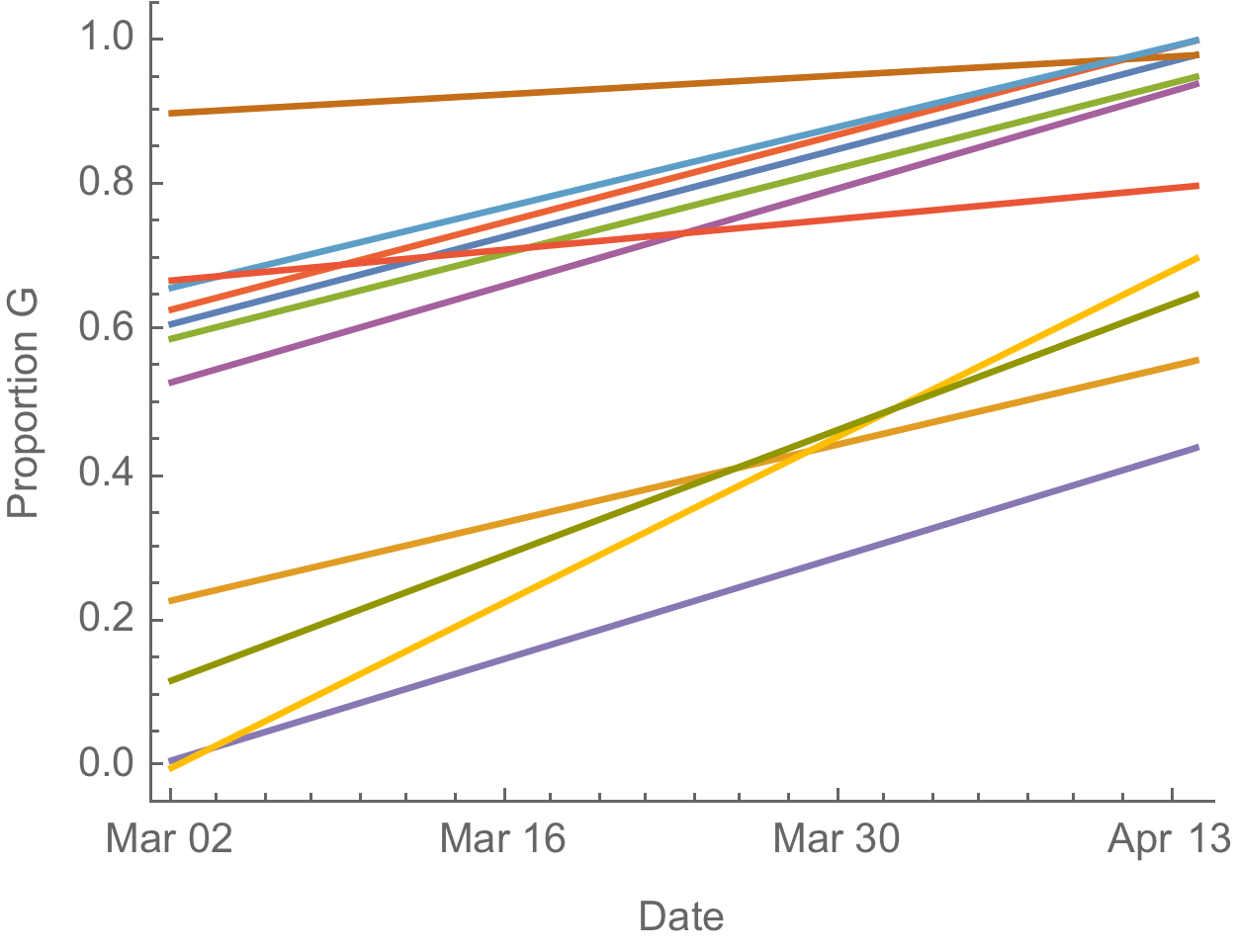
The success of D614G can be explained by either:
- D614G is more transmissible and has higher $R_0$
- founder effects and epidemiological confounding
Additional evidence from Ct values of clinical specimens
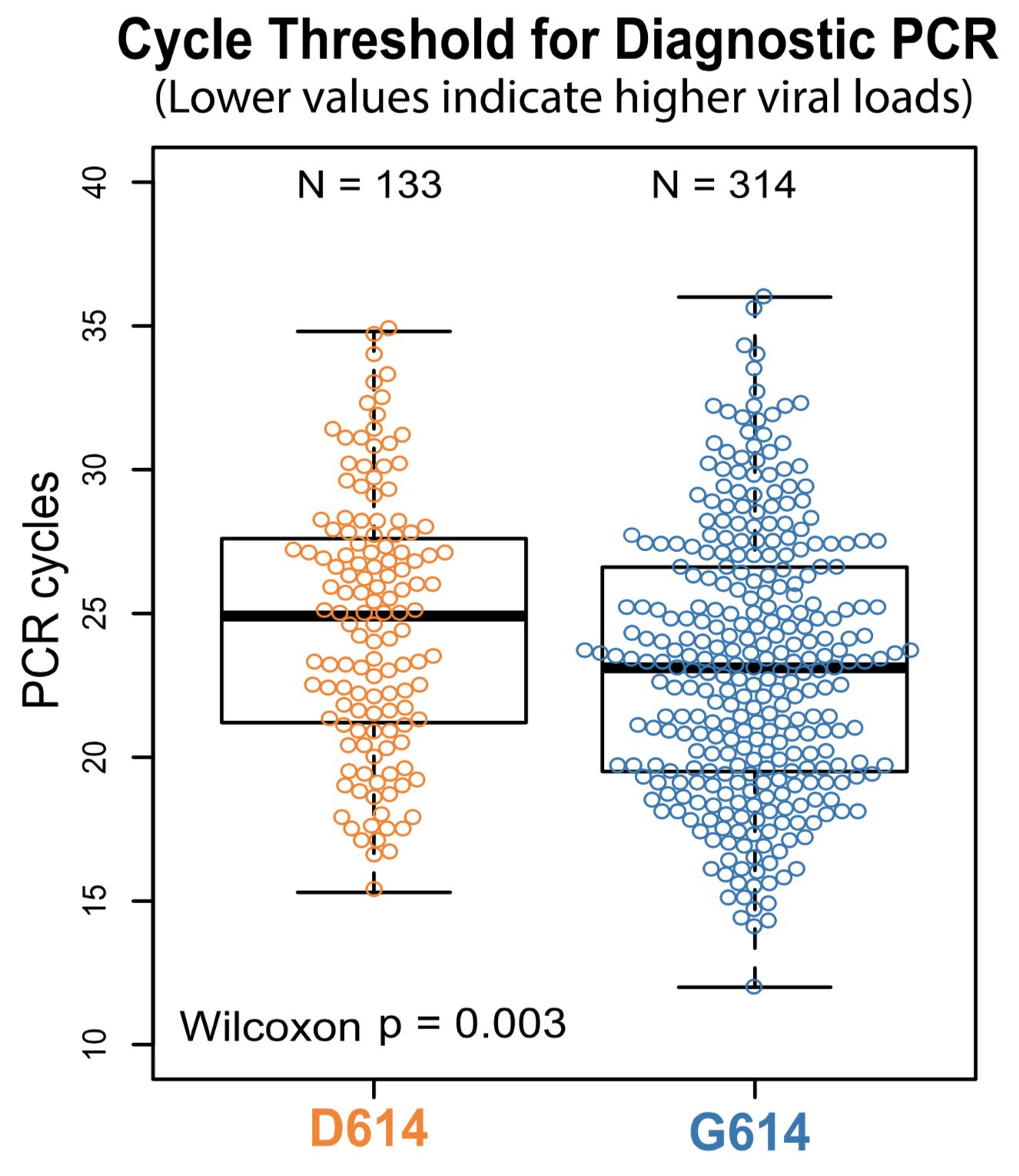
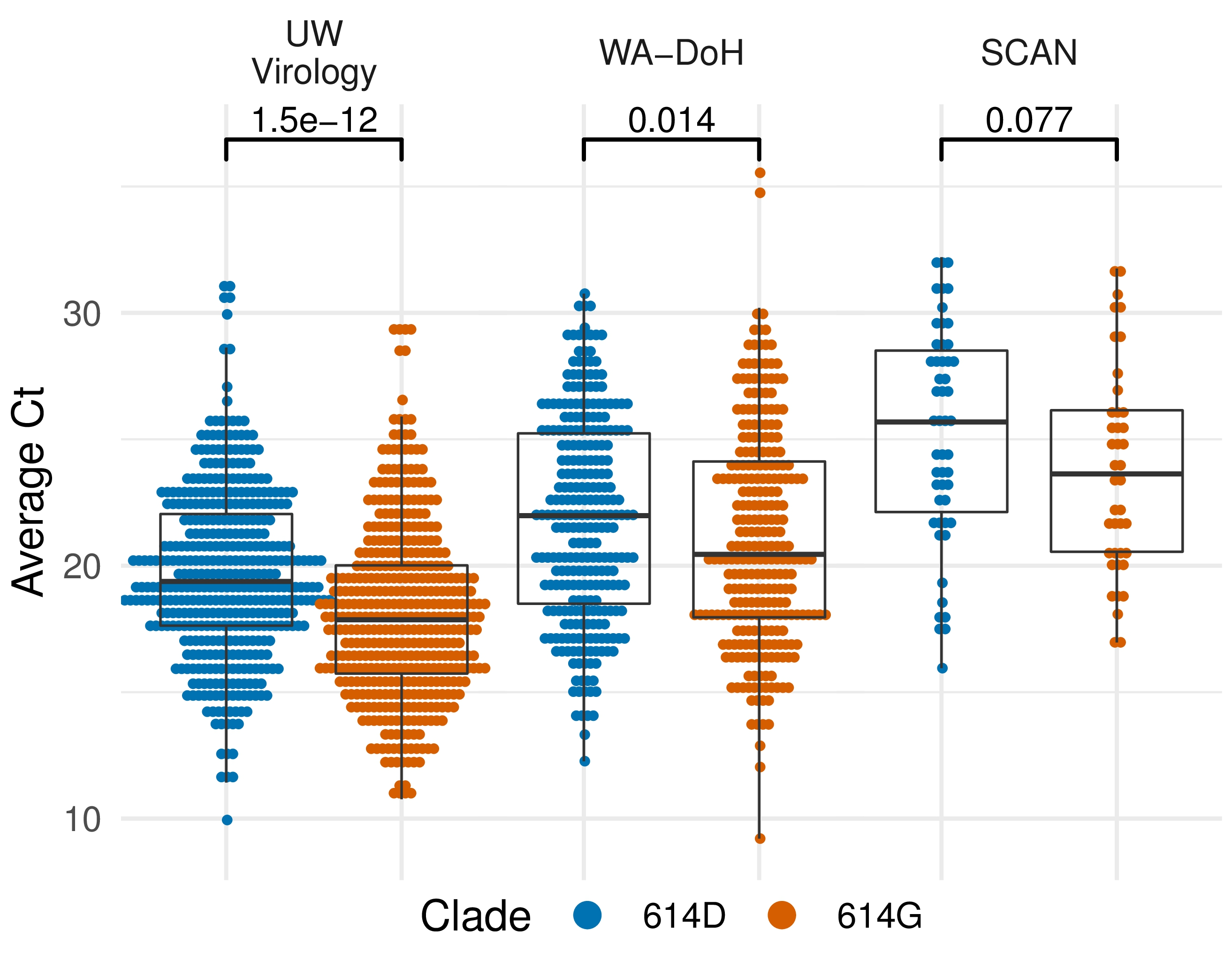
Moving forward
- Genomic approaches immediately useful to surveillance, particularly to distinguish endogenous spread from importations
- Longer term tracking of antigenic drift for vaccine strain updating
- Our best way out of this mess is with test-trace-isolate
Acknowledgements
Genomic epi: Data producers from all over the world, GISAID and the Nextstrain team