
Interpreting viral evolution
Trevor Bedford
Fred Hutchinson Cancer Center / Howard Hughes Medical Institute
11 Jul 2025
SURP Seminar
Fred Hutch
Slides at: bedford.io/talks
We work at the interface of virology, evolution and epidemiology
 Ebola
Ebola
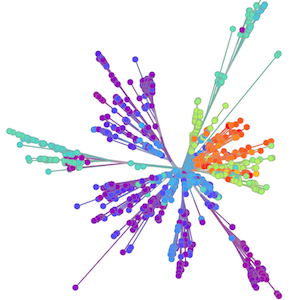
 Zika
Zika

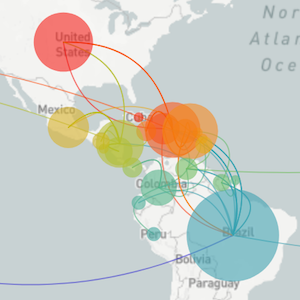
Segue through data vizualization
Data tells a story
Charles Minard and Napoleon's 1812 campaign

If data are looked at in the right way, answers become immediately clear
Edward Tufte




Above all, show the data
A good graphic is honestBarchart of average value
Box-and-whisker plot showing variation
Showing the data
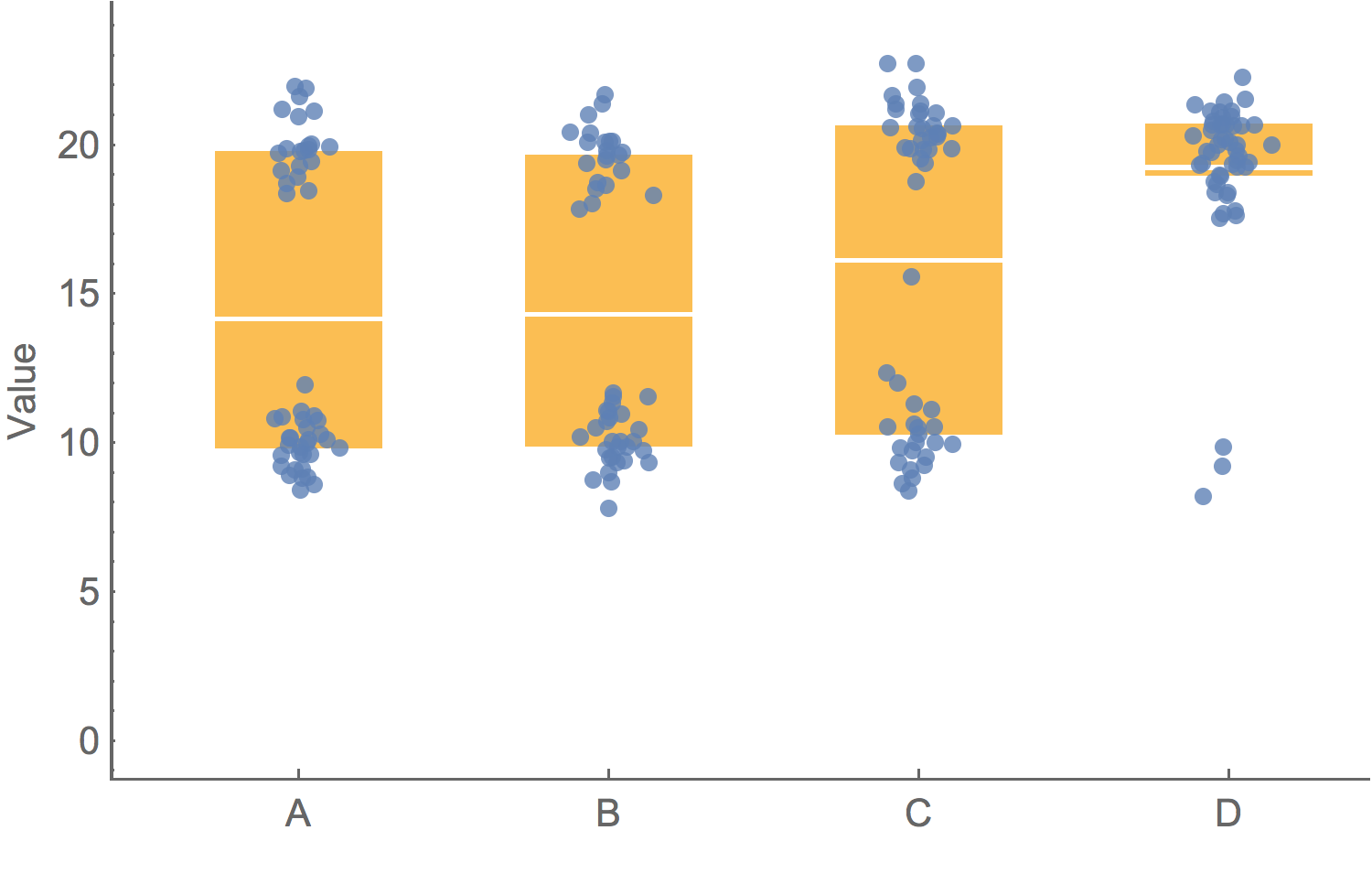
To clarify add detail
"To clarify, add detail... Clutter and overload are not attributes of information, they are failures of design. If the information is in chaos, don’t start throwing out information, instead fix the design."– Tufte
Evolution of Conus shells
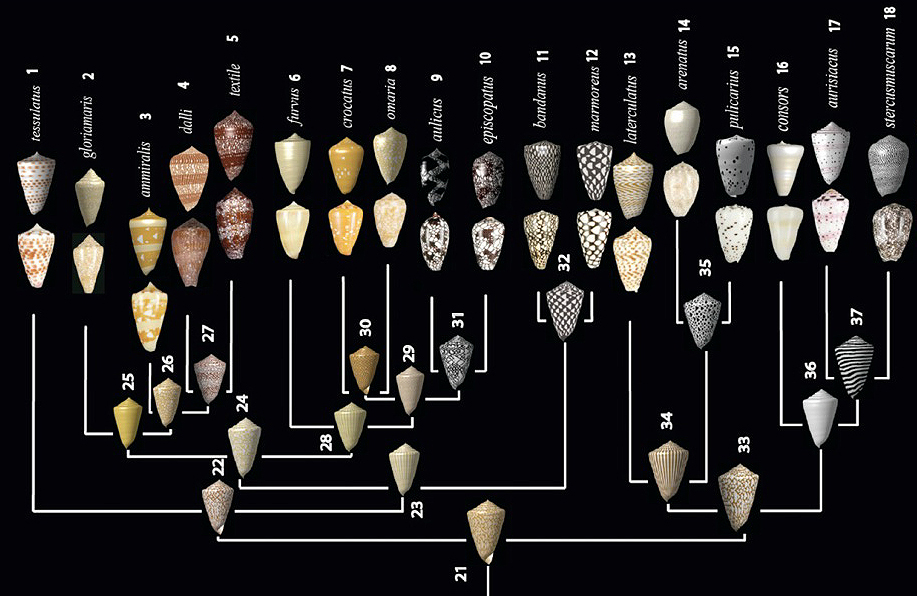 Gong et al. 2012. PNAS.
Gong et al. 2012. PNAS.
Greenland rising
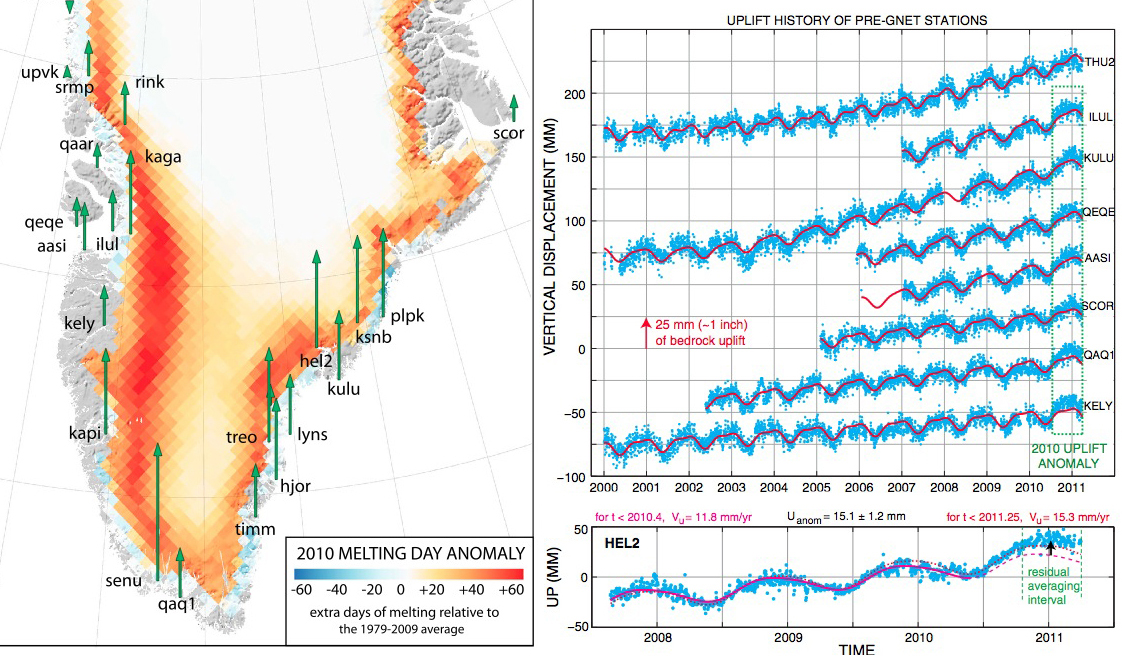 Bevis et al. 2012. PNAS.
Bevis et al. 2012. PNAS.
Small multiples and dimensionality
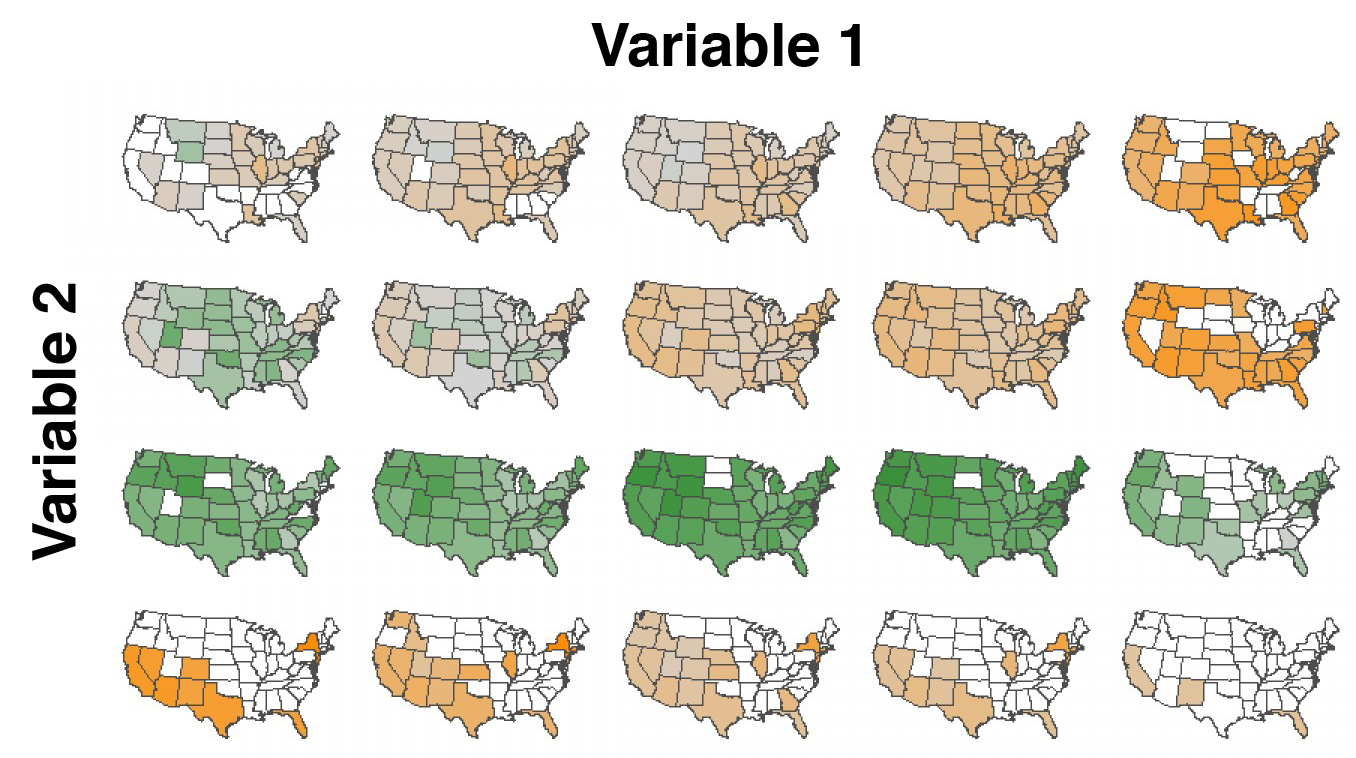
Mapping infection spread
John Snow and the founding of epidemiologyMy research
Methods focus on sequencing to reconstruct pathogen spread
Epidemic process
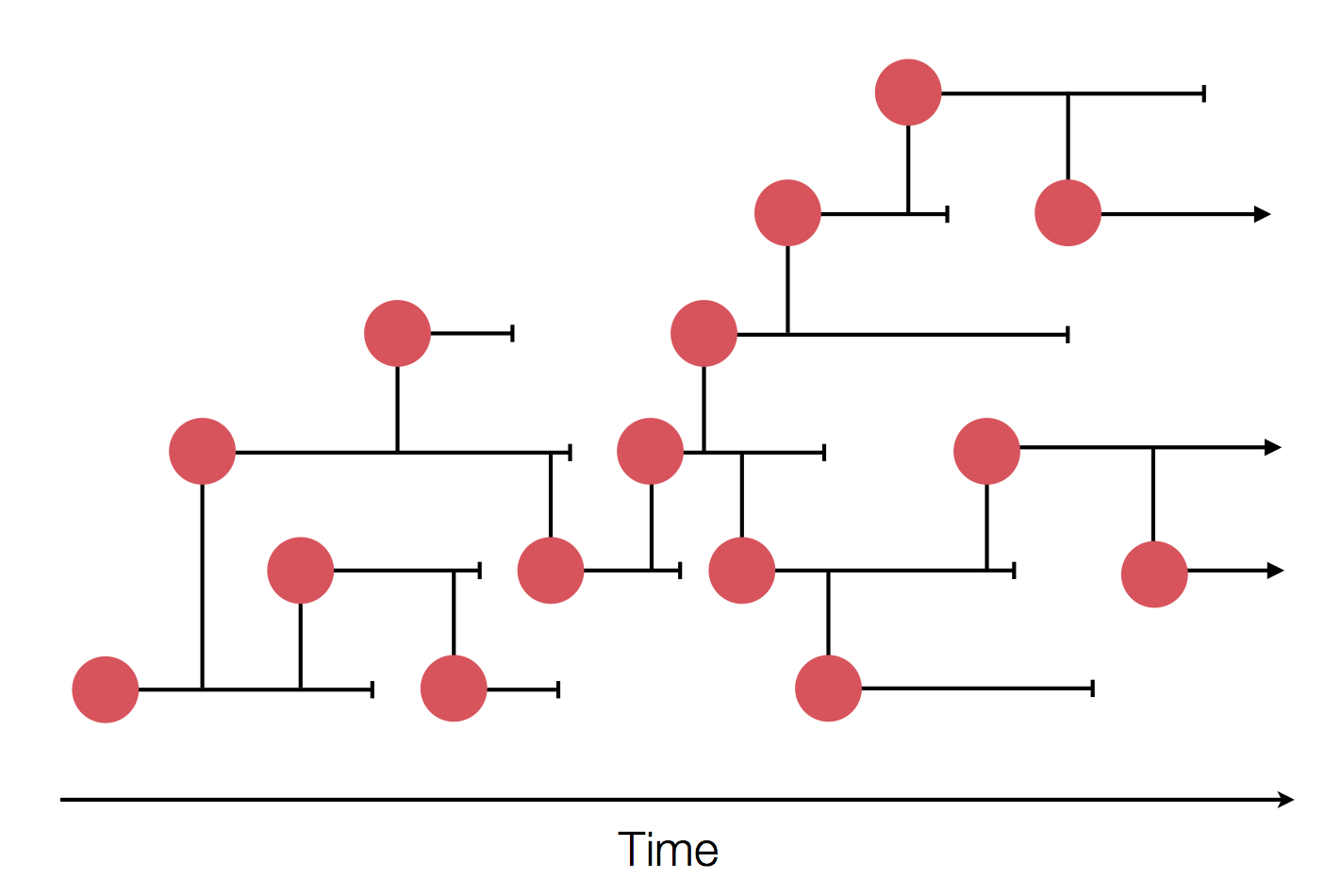
Sample some individuals
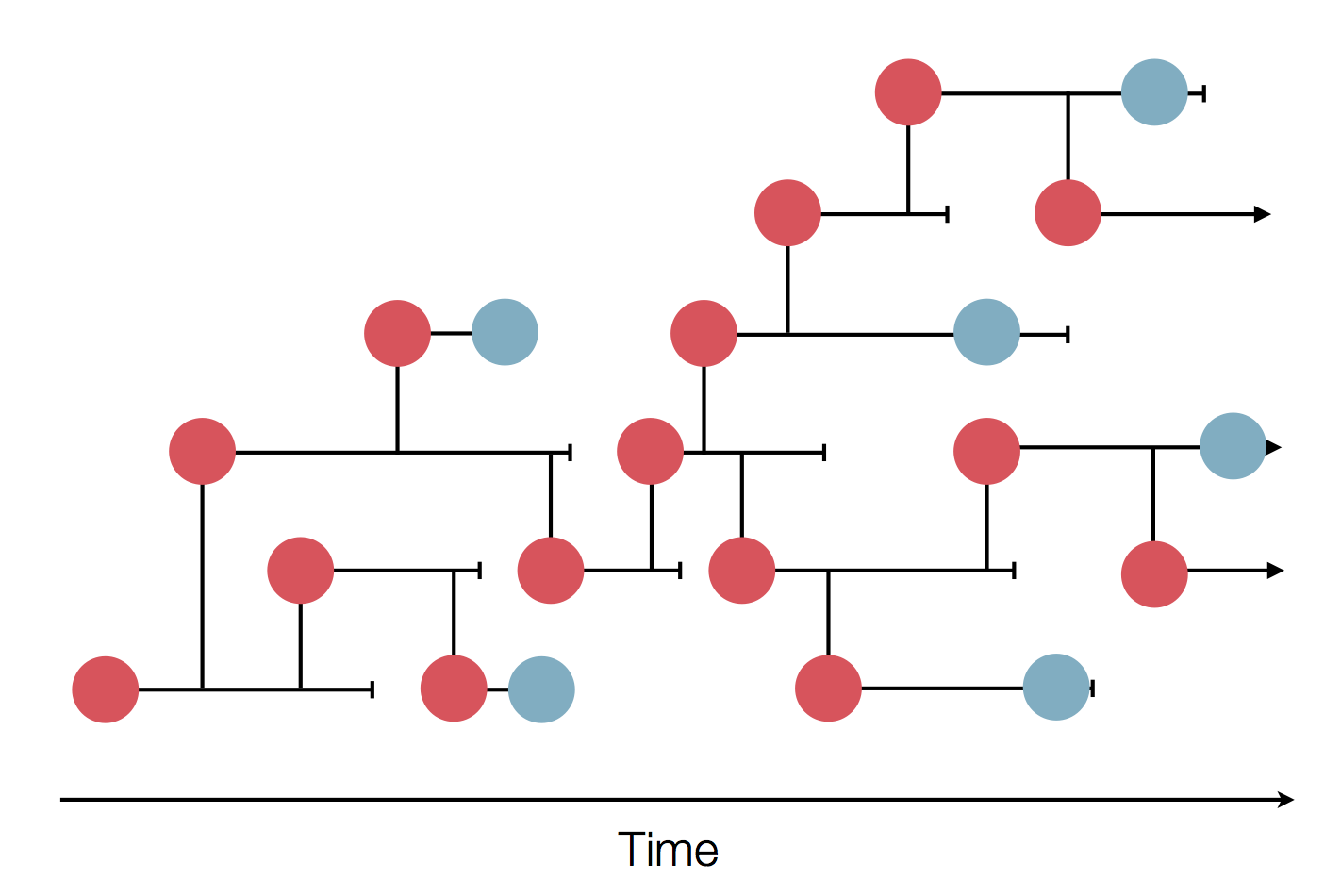
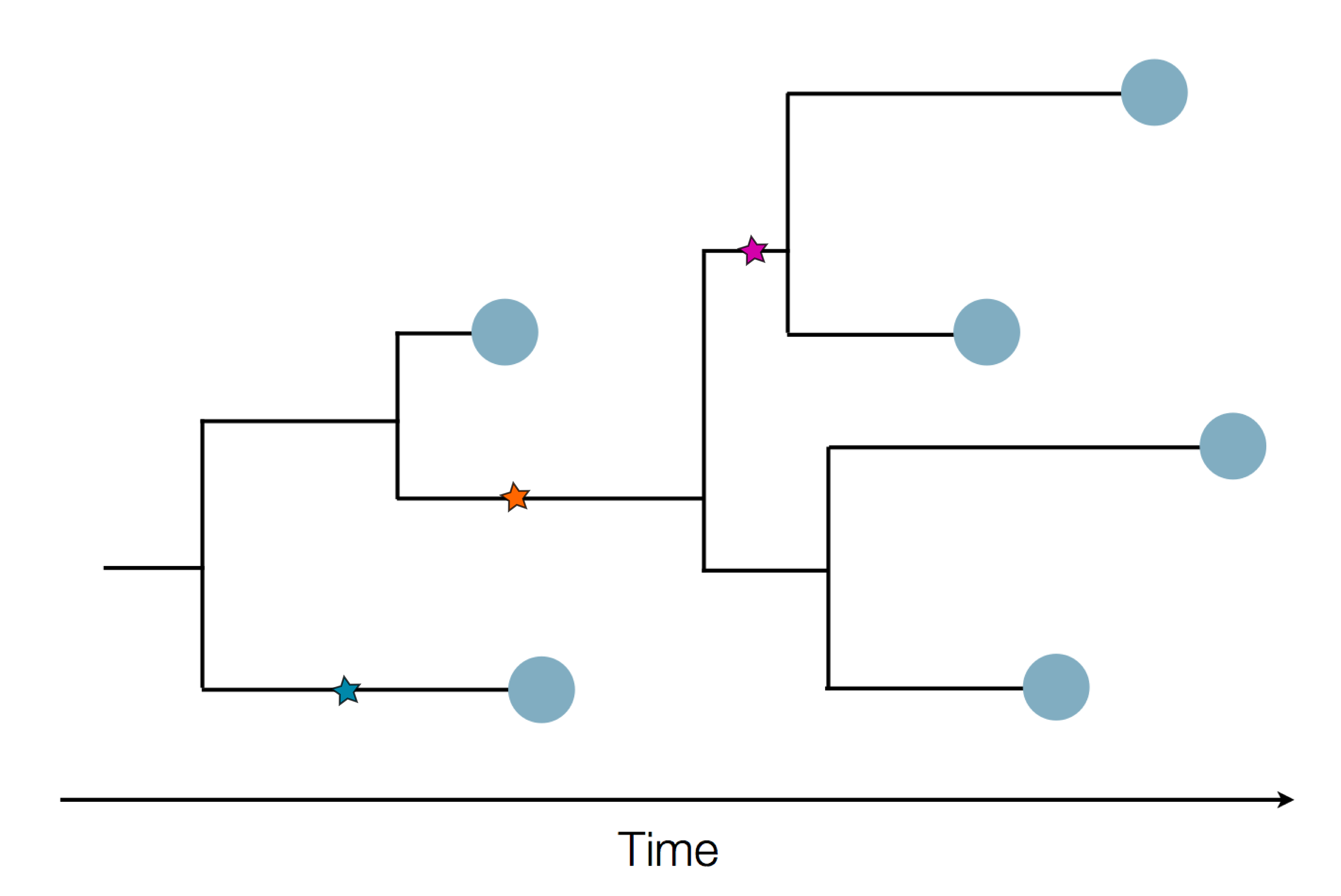
Sequence and determine phylogeny
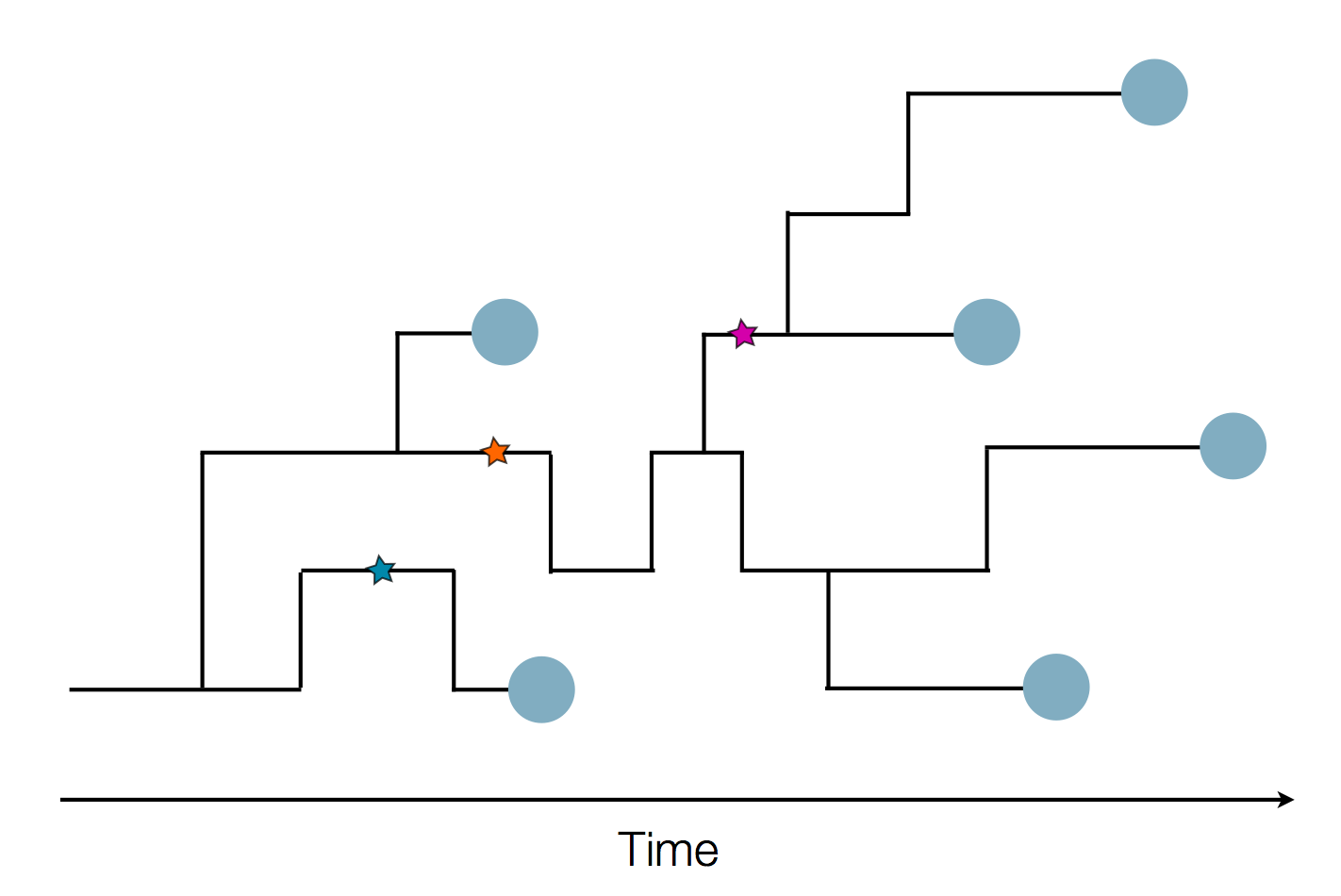
Sequence and determine phylogeny
Pathogen genomes may reveal
- Evolution of new adaptive variants
- Epidemic origins
- Patterns of geographic spread
- Animal-to-human spillover
- Transmission chains
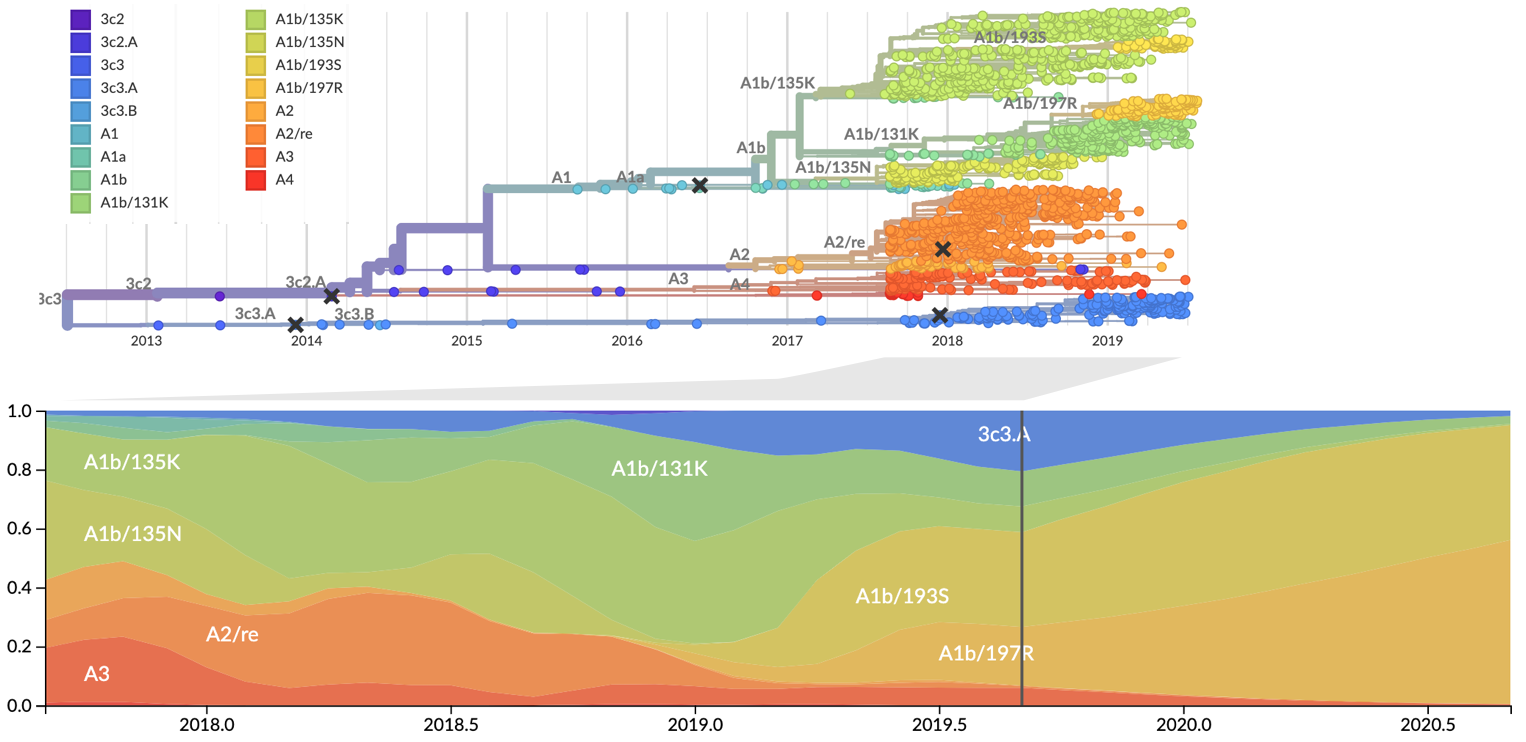
Influenza: Forecasting spread of new variants for vaccine strain selection
Zika: Uncovering origins of the epidemic in the Americas
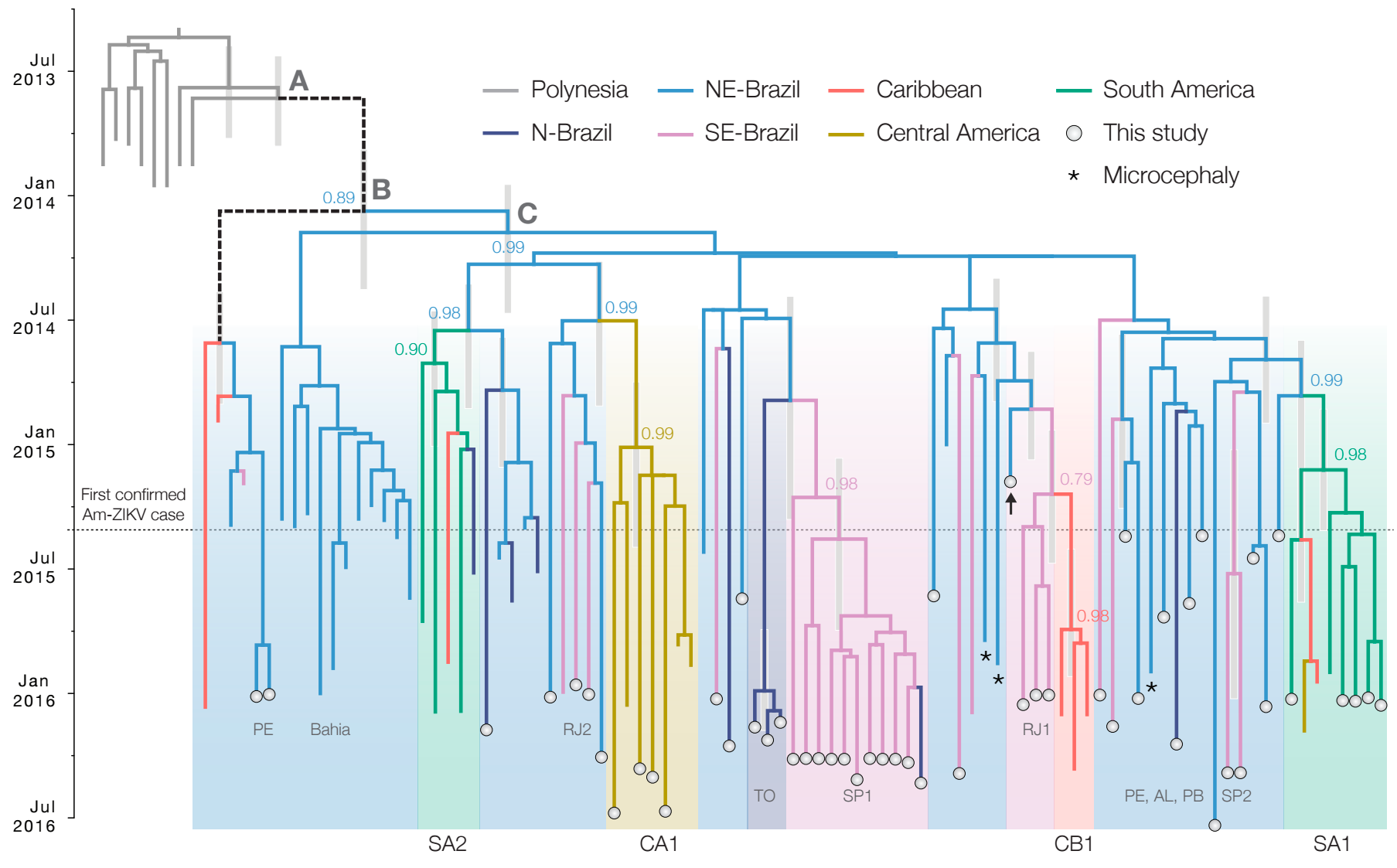 Faria et al. 2017. Nature.
Faria et al. 2017. Nature.
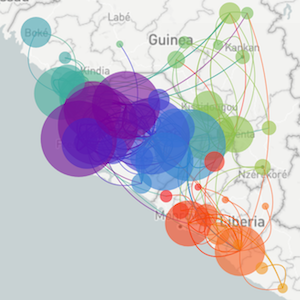
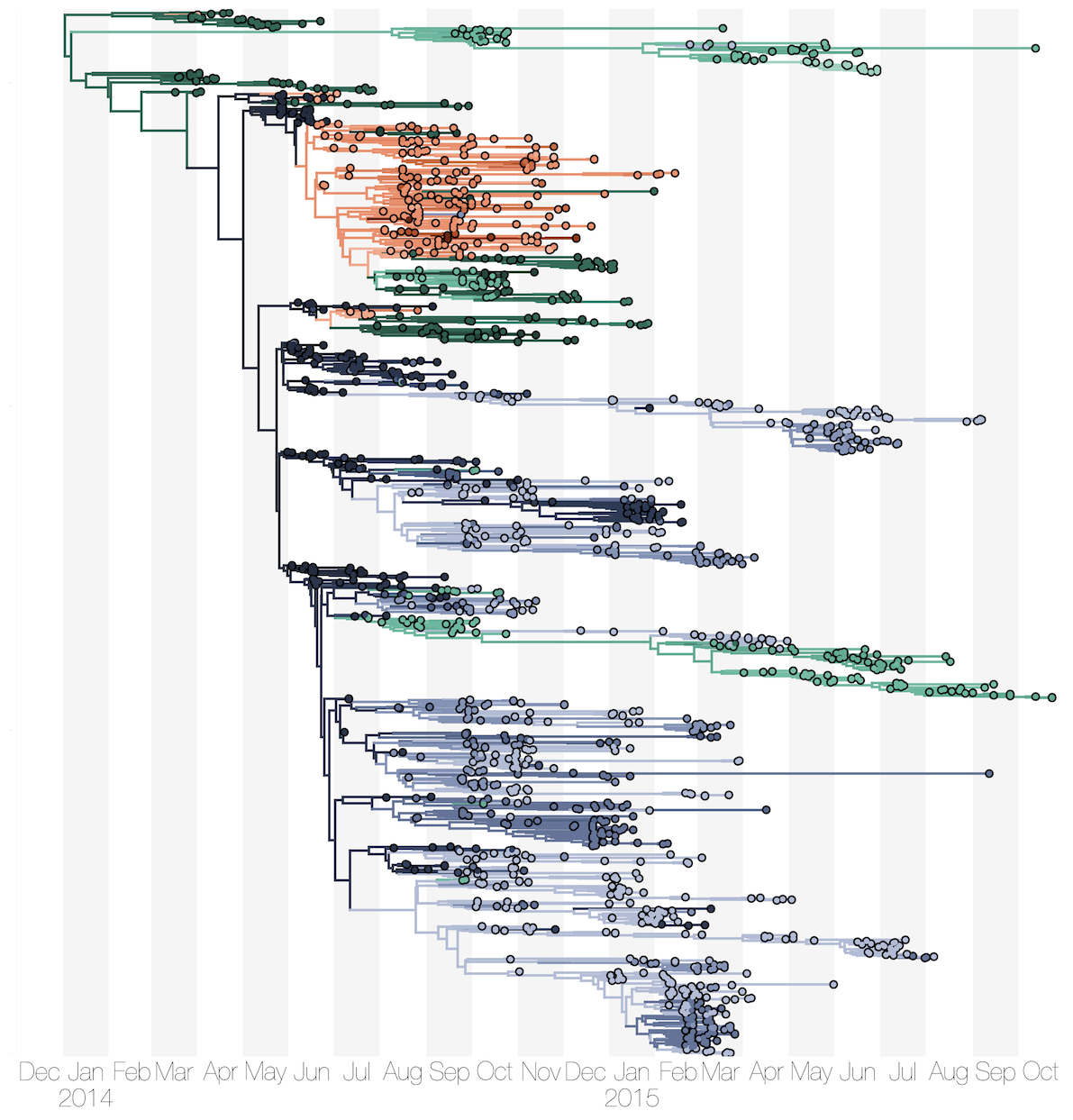
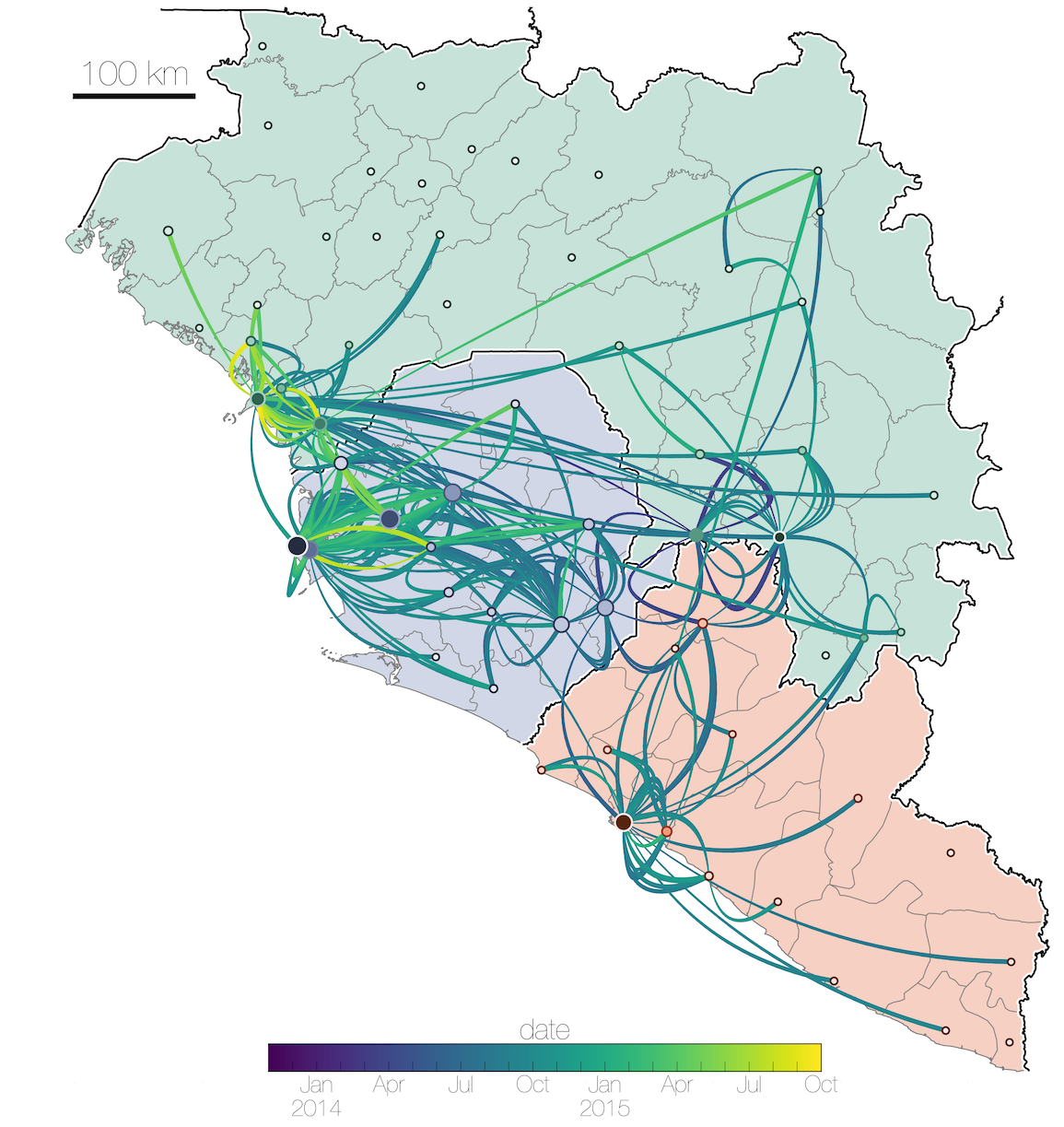
Ebola: Revealing spatial spread and persistence in West Africa
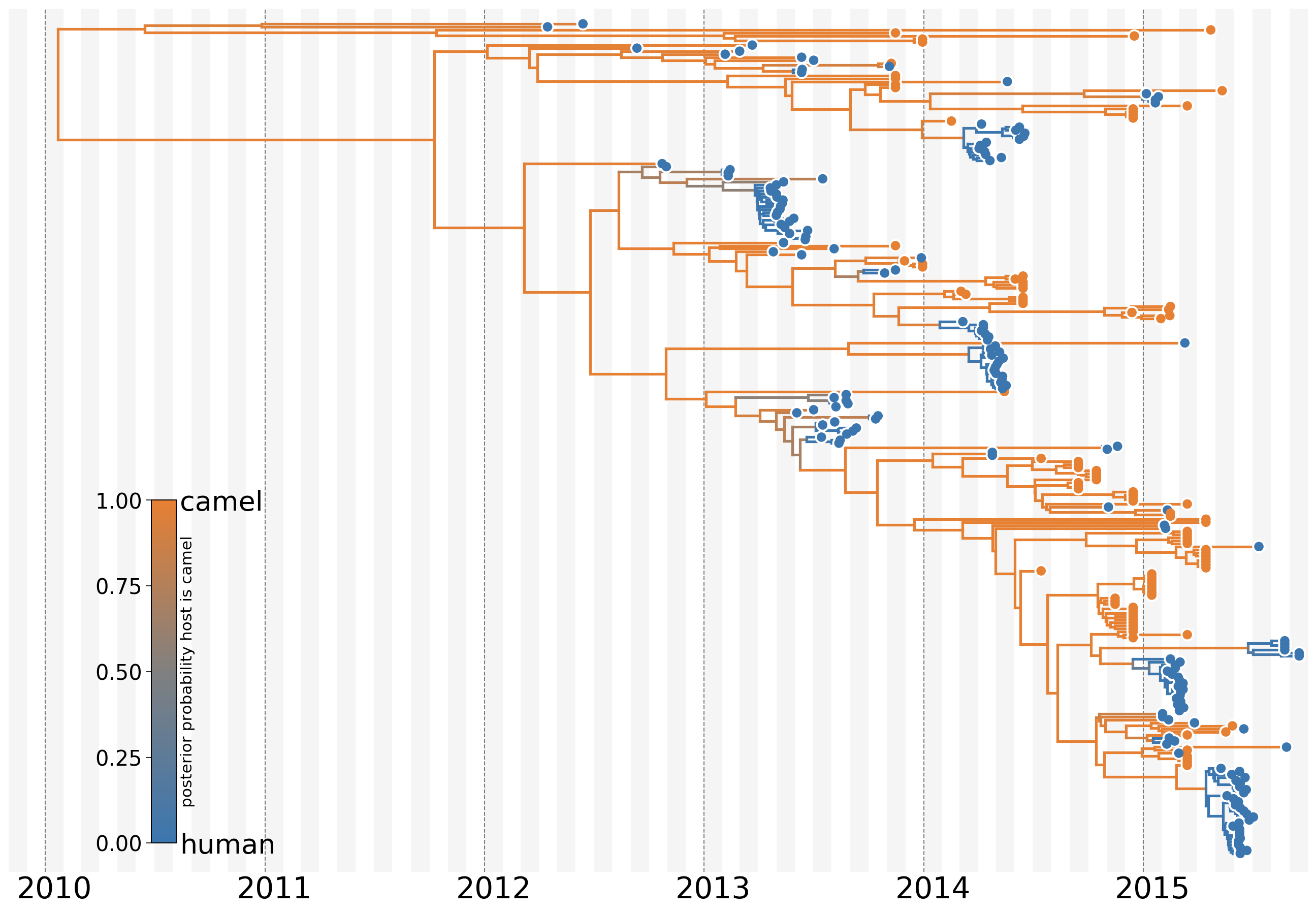
MERS: Repeated spillover into the human population from camel reservoir
Actionable inferences
Nextstrain
Project to conduct real-time genomic epidemiology and evolutionary analysis of emerging epidemics
![]() Richard Neher,
Richard Neher,
![]() Ivan Aksamentov,
Ivan Aksamentov,
![]() John SJ Anderson,
John SJ Anderson,
![]() Kim Andrews,
Kim Andrews,
![]() Jennifer Chang
Jennifer Chang
![]() James Hadfield,
James Hadfield,
![]() Emma Hodcroft,
Emma Hodcroft,
![]() John Huddleston,
John Huddleston,
![]() Jover Lee,
Jover Lee,
![]() Victor Lin,
Victor Lin,
![]() Cornelius Roemer,
Cornelius Roemer,
![]() Thomas Sibley
Thomas Sibley
Nextstrain architecture
Central aims: (1) rapid and flexible bioinformatic workflows, (2) interactive visualization and (3) always up-to-date analyses at nextstrain.org
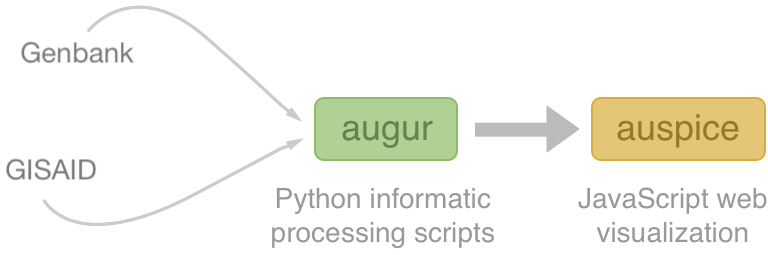
All code open source at github.com/nextstrain
Operationalized during the 2018-2020 outbreak of Ebola in the Democratic Republic of the Congo

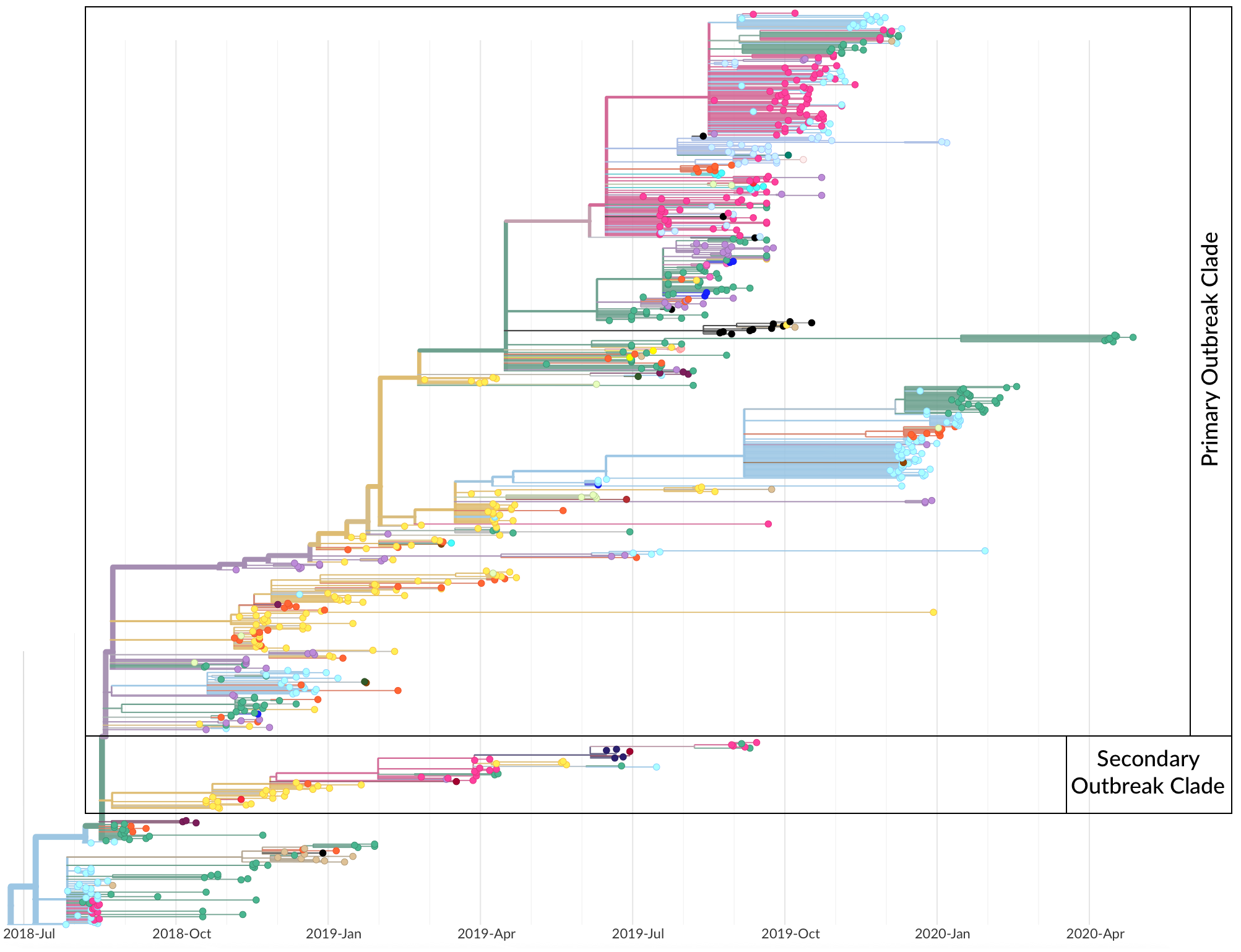 Kinganda-Lusamaki et al. 2021. Nat Med.
Kinganda-Lusamaki et al. 2021. Nat Med.
Genomic epidemiology during the COVID-19 pandemic
To date, over 17M SARS-CoV-2 genomes shared to GISAID and evolution tracked in real-time at nextstrain.org
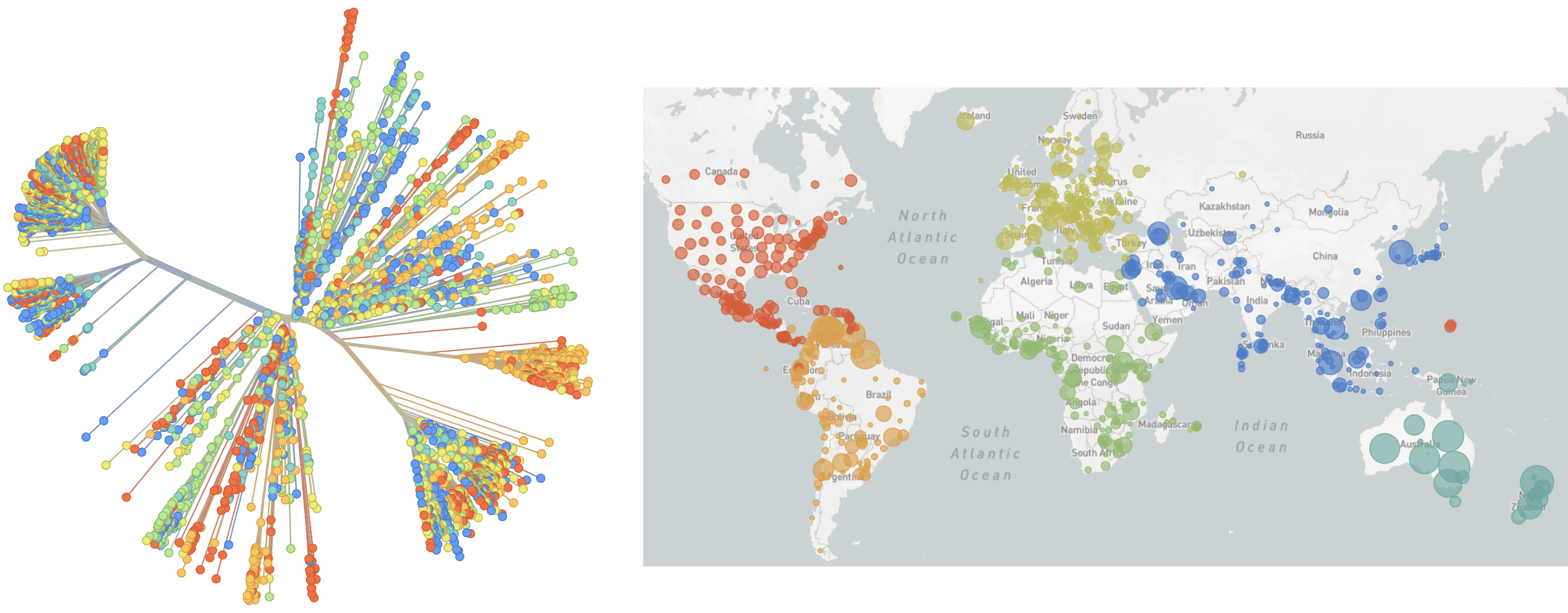
Three key insights that genomic epi provided during pandemic
- Rapid human-to-human spread in Wuhan beyond initial market outbreak
- Extensive local transmission while testing was rare
- Identification of variants of concern and mapping of increased transmission rates
On Jan 19, 2020, the first 13 viral genomes from Wuhan and Bangkok showed little genetic diversity
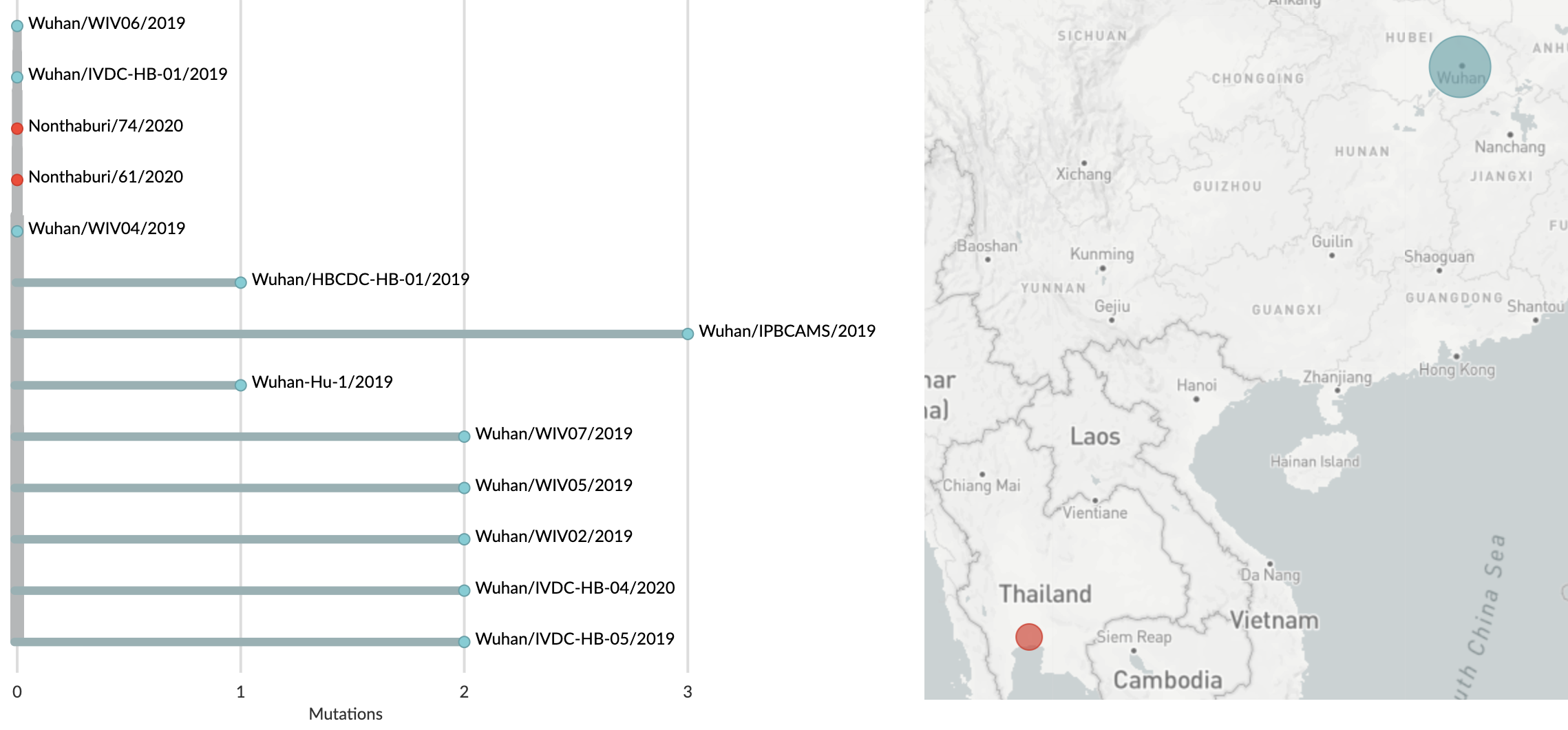 Data from CAMS, China CDC, Fudan University, Hubei CDC, Thai MOPH, WIV;
Figure from nextstrain.org
Data from CAMS, China CDC, Fudan University, Hubei CDC, Thai MOPH, WIV;
Figure from nextstrain.org
Allowed us to infer introduction between Nov 15 and Dec 15 and subsequent rapid human-to-human spread
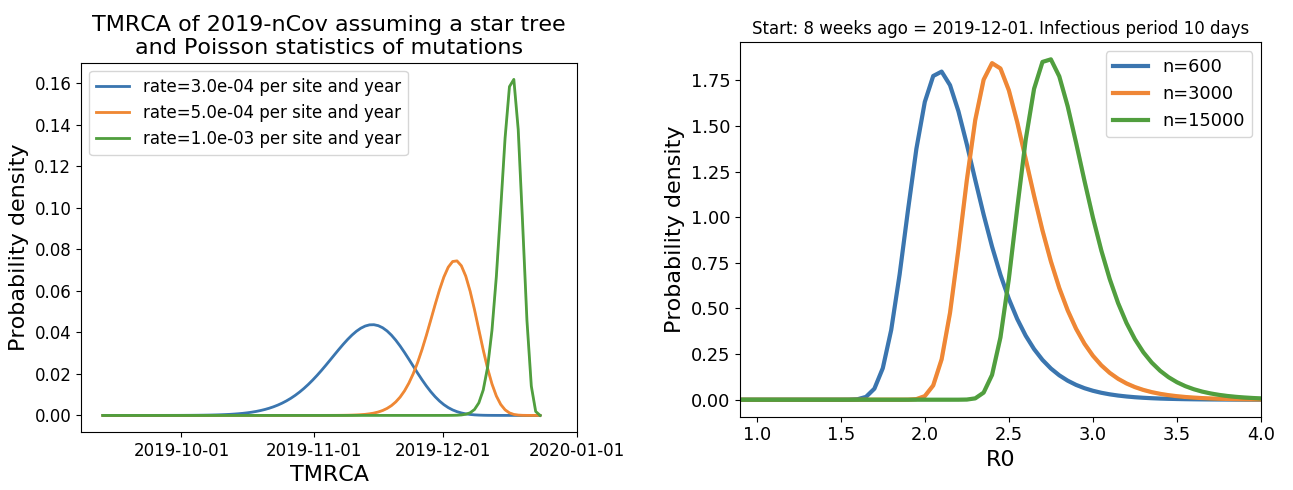 Bedford et al. 2020. Technical Report.
Bedford et al. 2020. Technical Report.
Rapid global epidemic spread from China
Epidemic in the USA was introduced from China in late Jan and from Europe during Feb
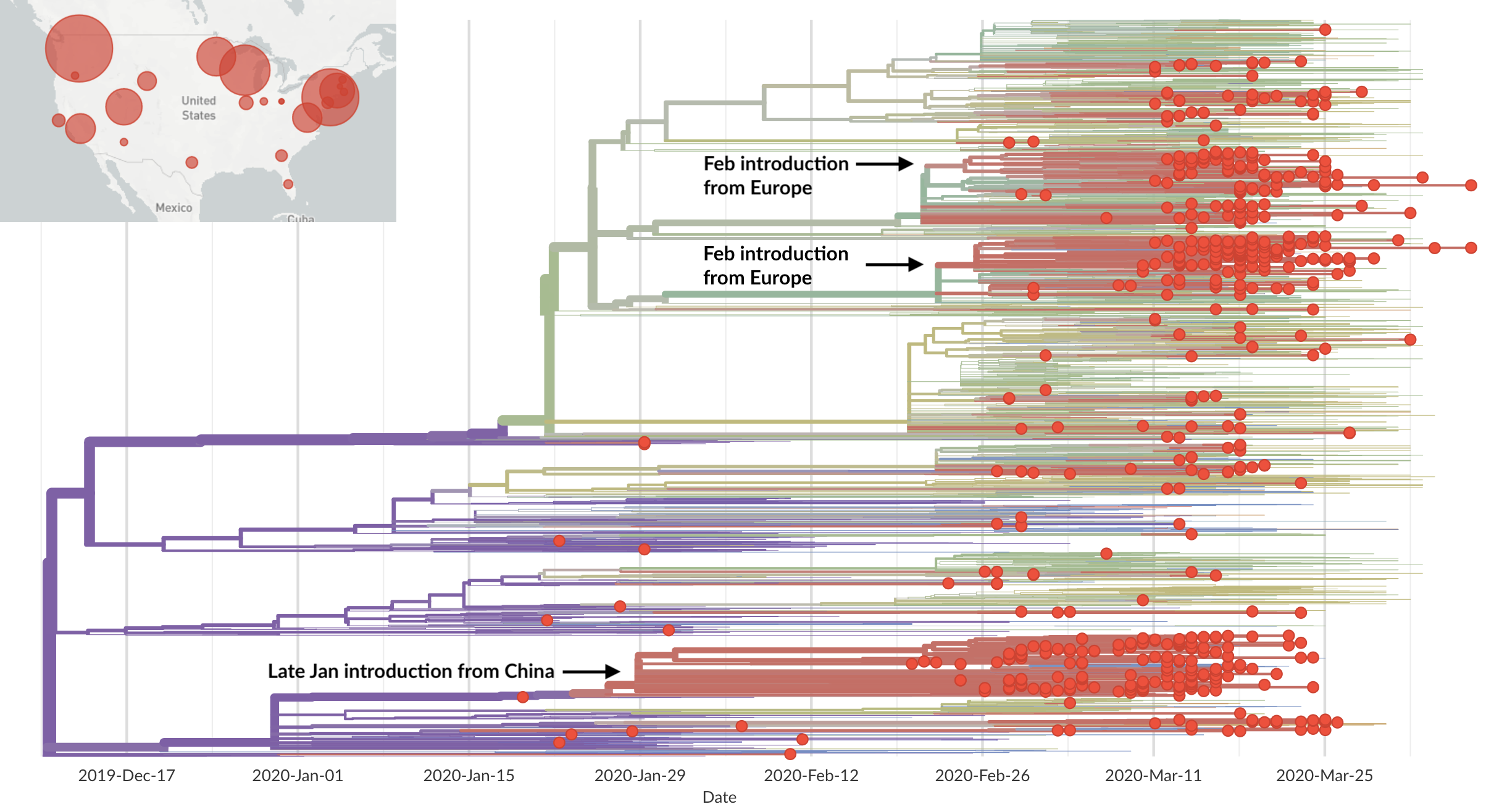 nextstrain.org
nextstrain.org
Early sequencing provided best estimate of extent of local outbreak
After initial wave, with mitigation
efforts and decreased travel,
regional clades emerge
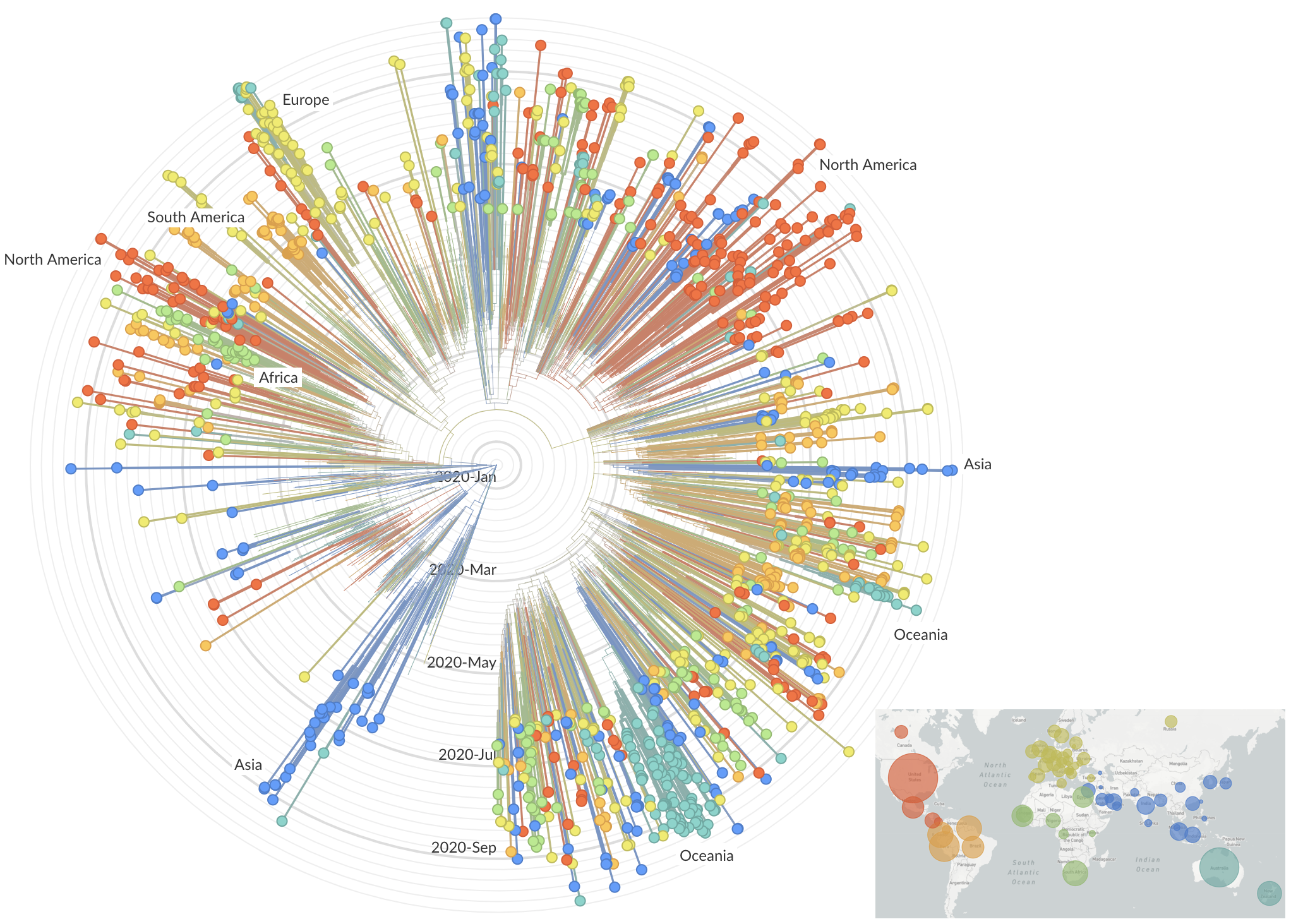 nextstrain.org
nextstrain.org
Emergence of Alpha in the UK with excess spike mutations
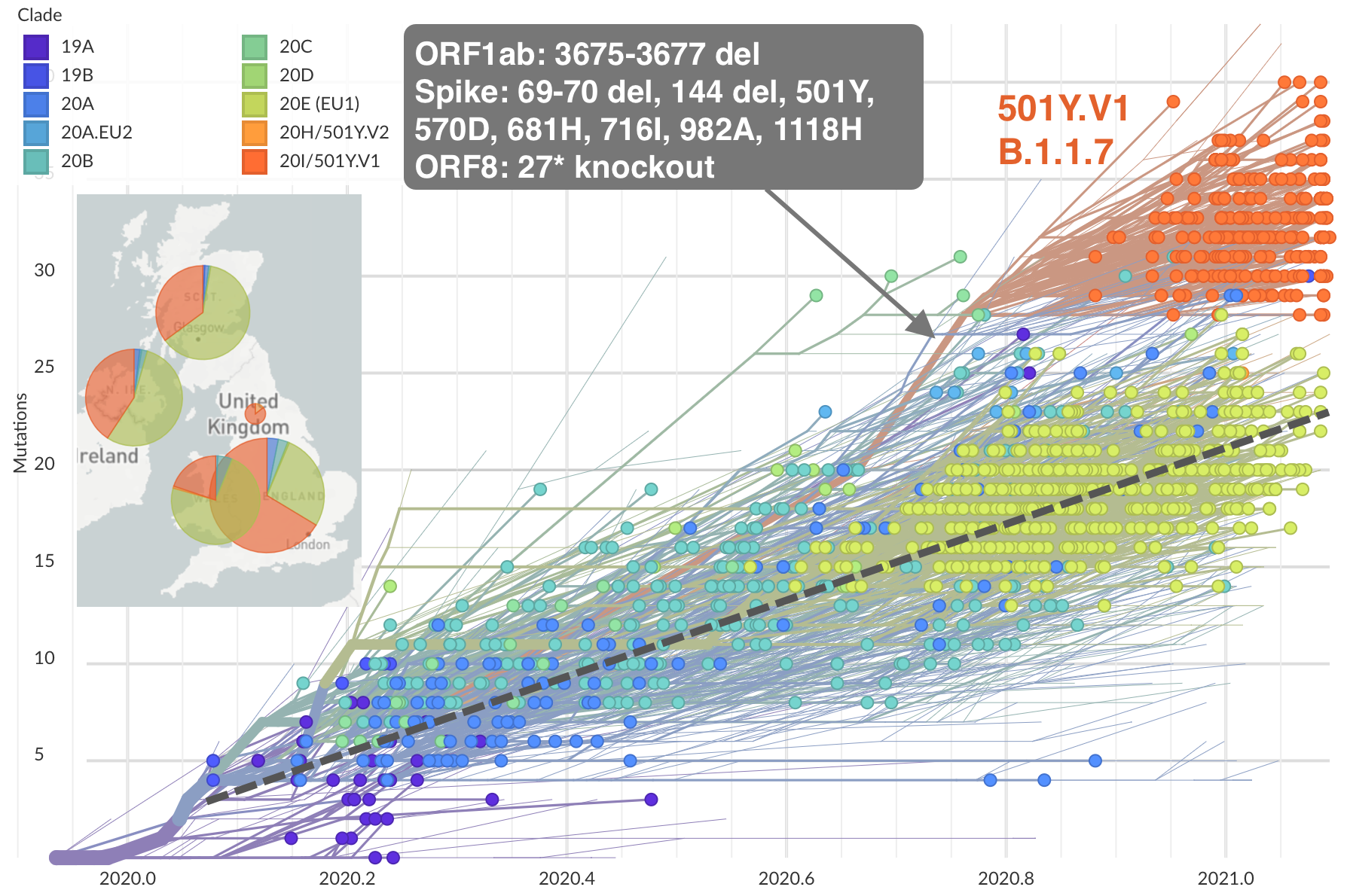 Alpha described in Rambaut et al. 2020. Virological.org.
Figure from nextstrain.org
Alpha described in Rambaut et al. 2020. Virological.org.
Figure from nextstrain.org
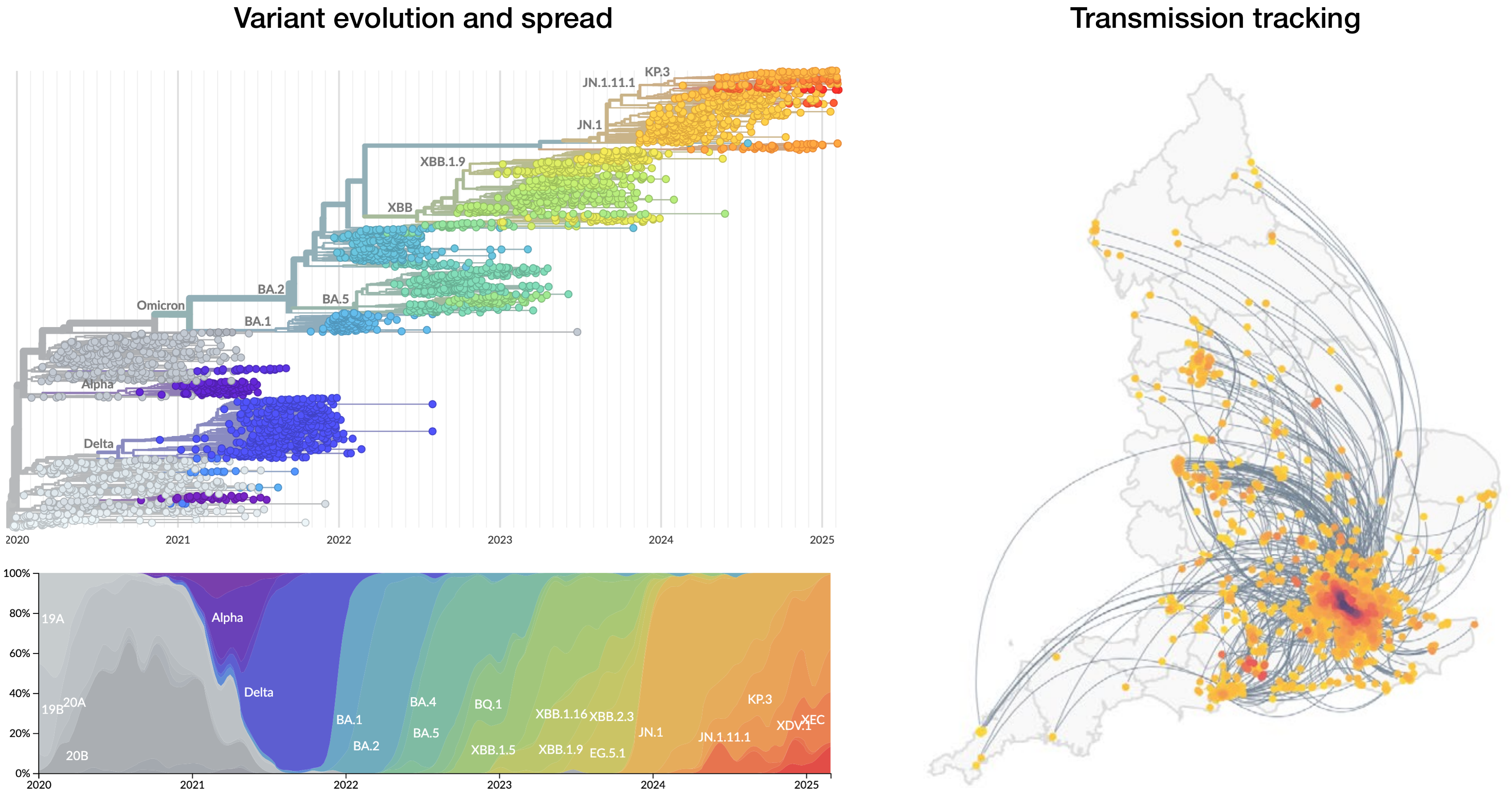
Further emergence of variants with increased transmissibility
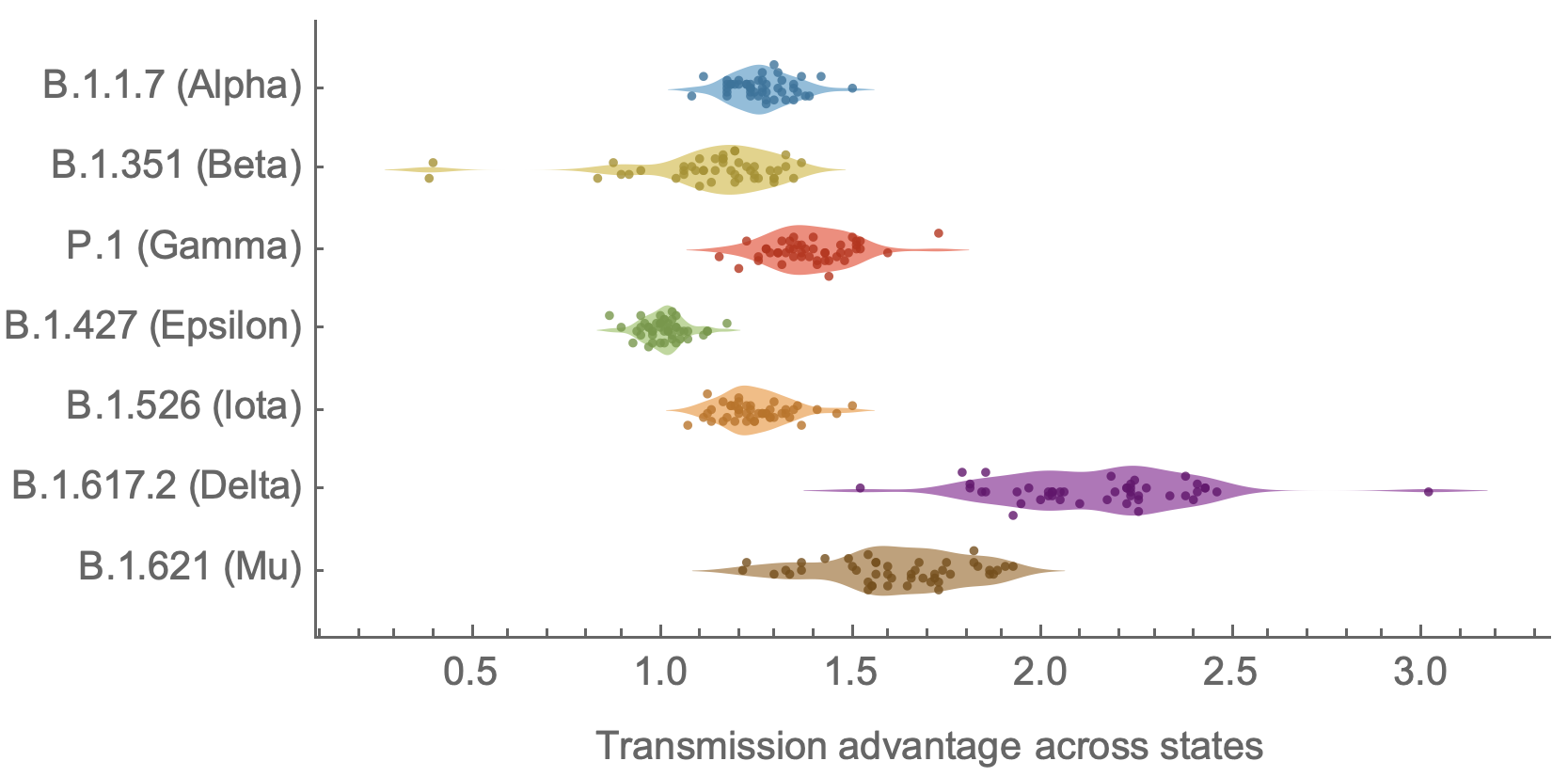
Variant emergence and spread has continued
 nextstrain.org
nextstrain.org
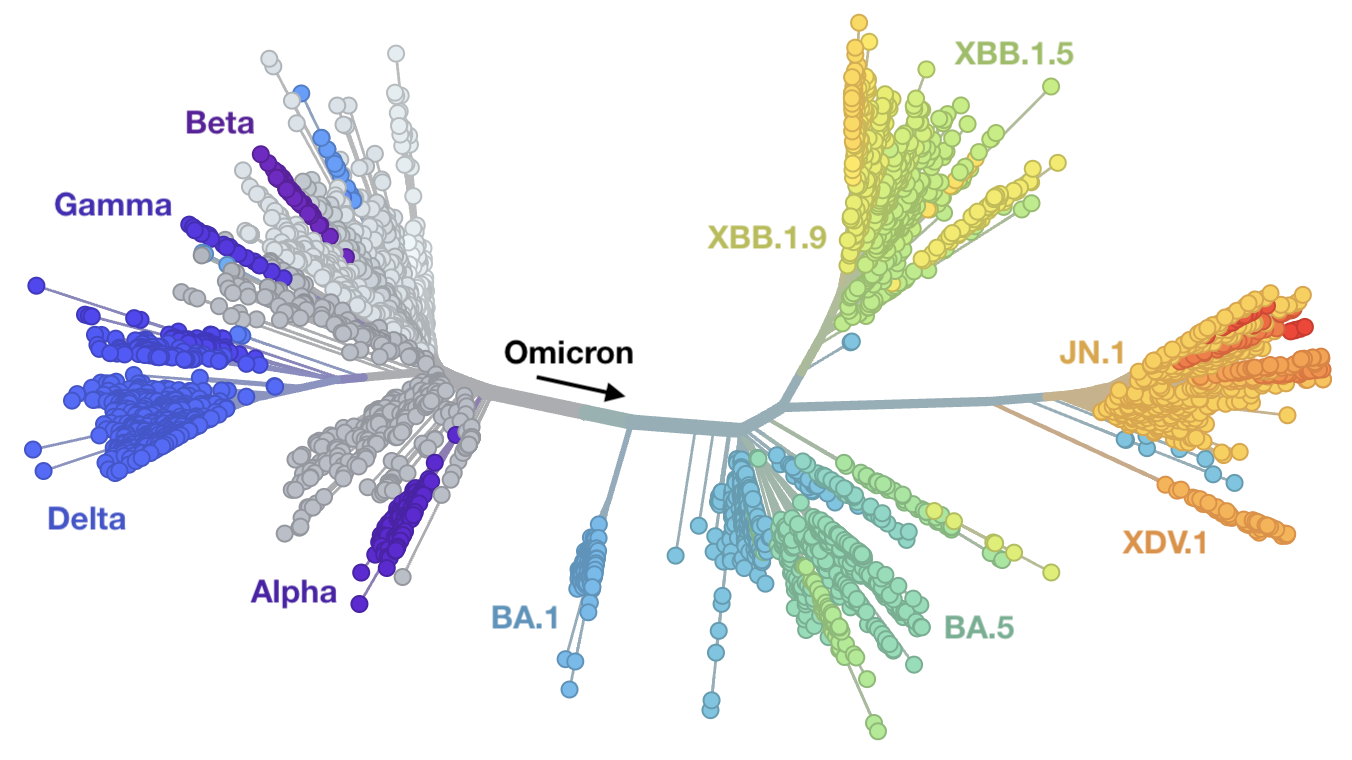
Rapid displacement of existing diversity by emerging variants that escape from existing population immunity
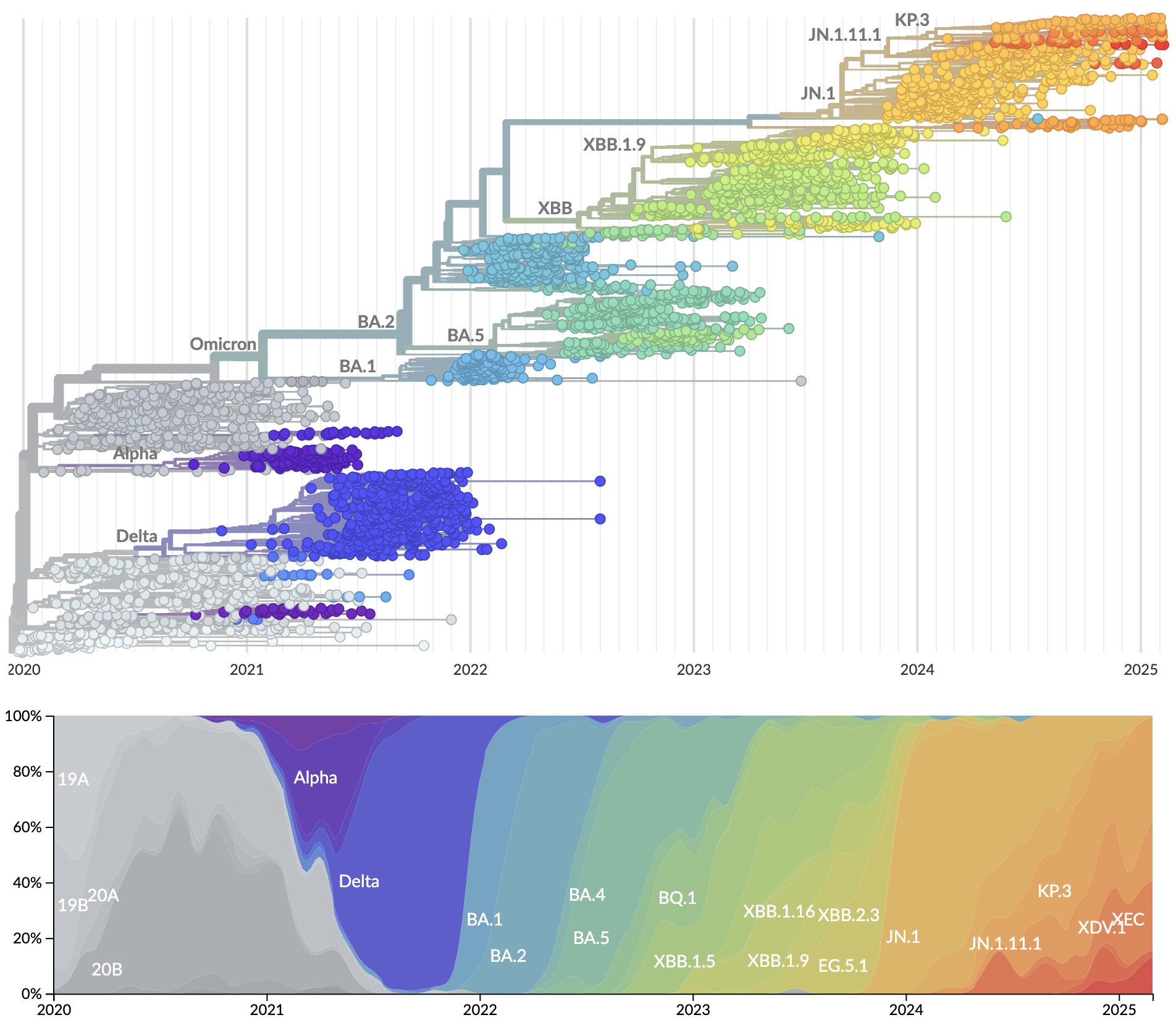
Mutations in S1 domain of spike protein driving displacement
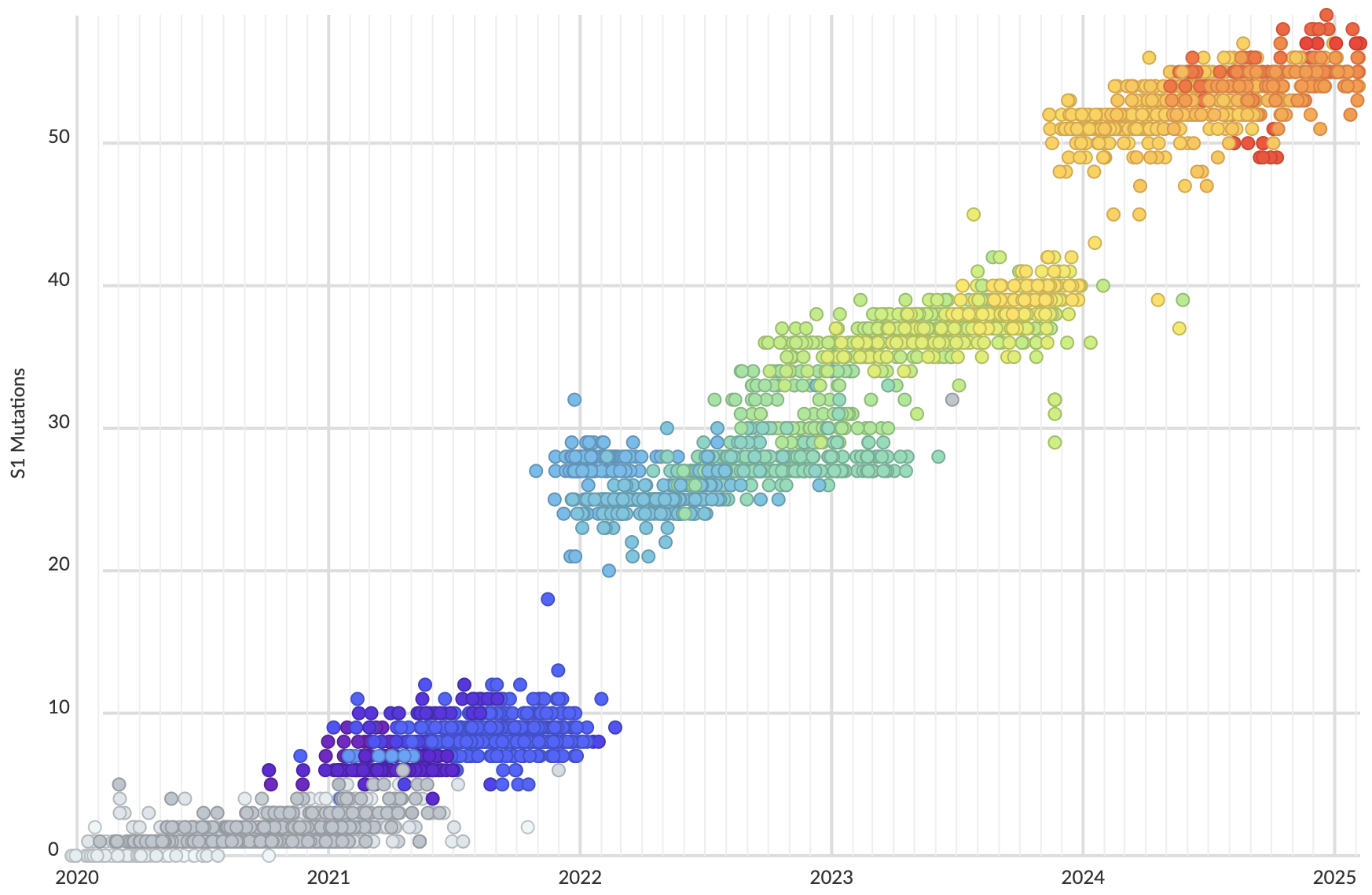 nextstrain.org
nextstrain.org
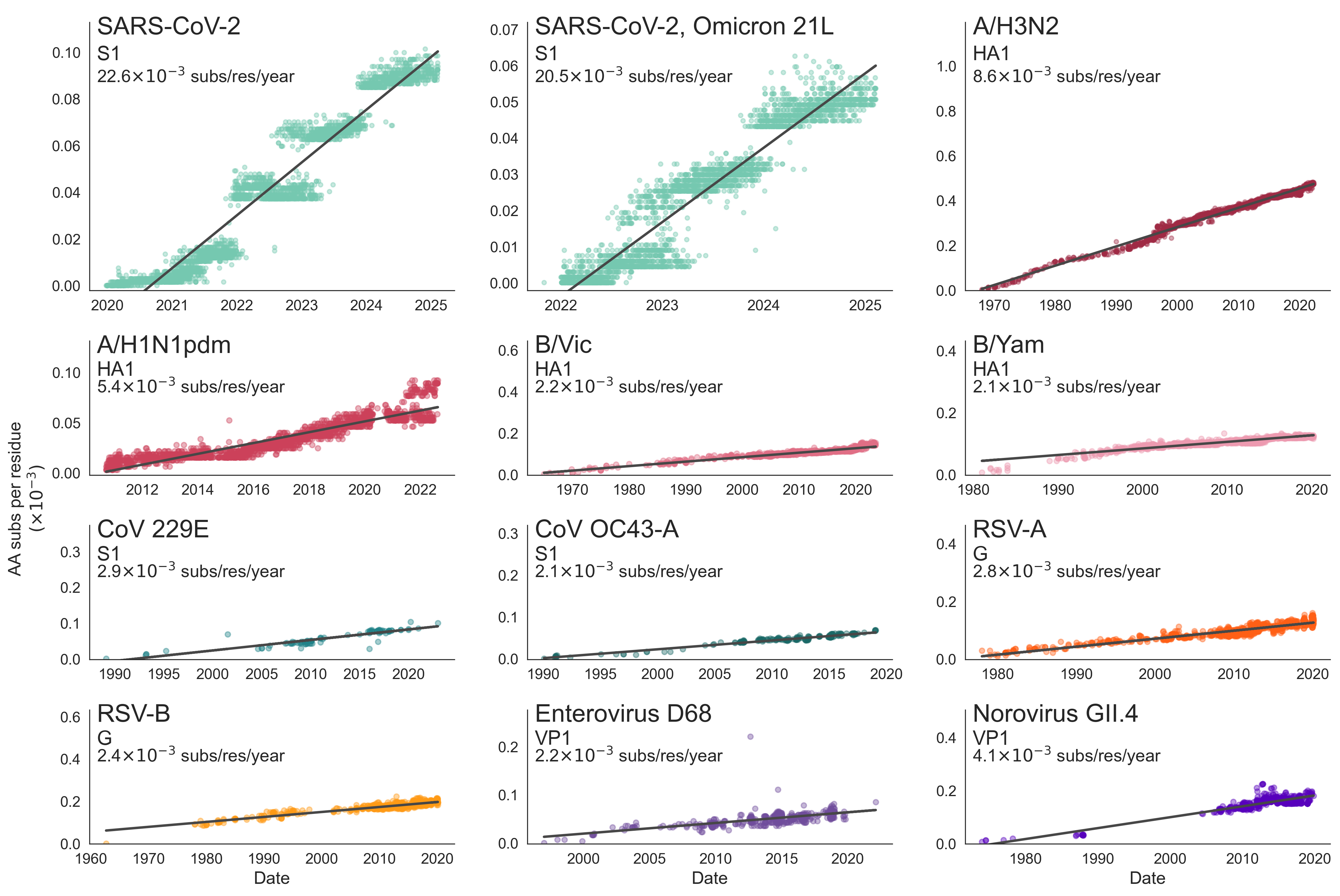
SARS-CoV-2 evolution fast relative to previous endemic viruses
Current directions
Wealth of genome data represents a foundational resource for tracking viral evolution and epidemiology
 McCrone et al. 2022. Nature.
McCrone et al. 2022. Nature.
Though we're still only scratching the surface of this data, in large part due to methodological limitations
AI approaches
- New approaches follow recent human language AI models and treat DNA nucleotides or protein amino acids as a text corpus
- These models follow the familiar generative logic of being trained to predict the next token (nucleotide / amino acid)
- The resulting models have a rich biological understanding encoded into the resulting internal model representations
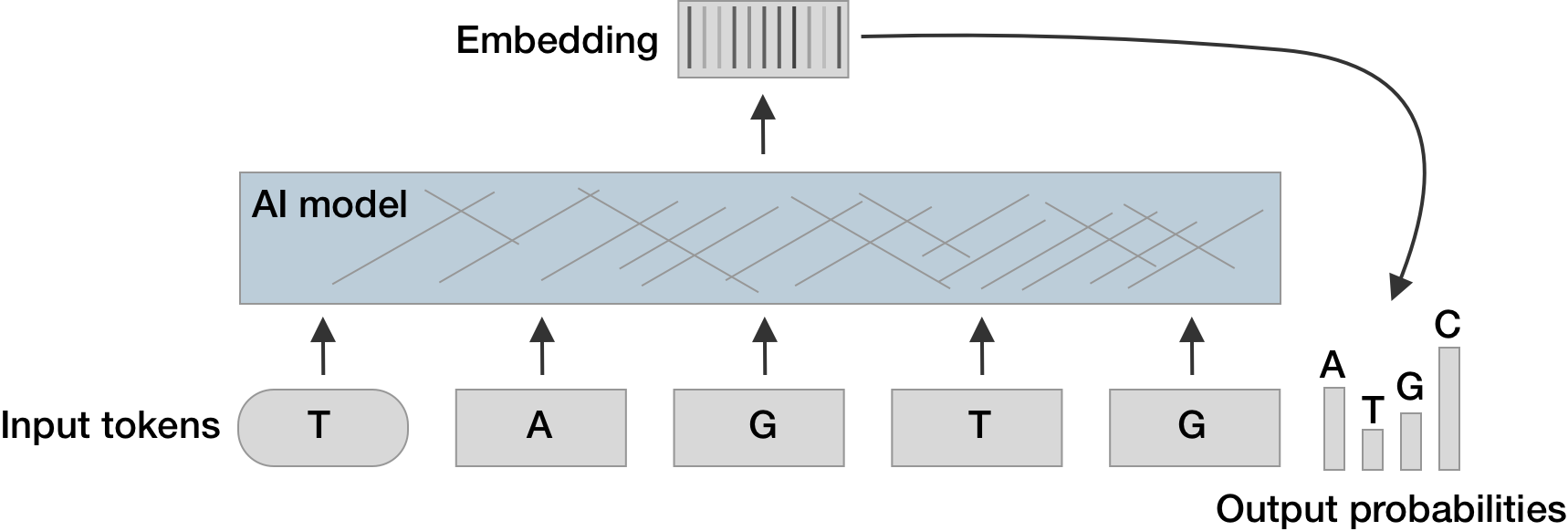
Large language models are based on a transformer architecture
- Models are trained to predict the next token from the preceeding tokens
- Words (or parts of words) are converted into input embedding vectors and these vectors are multiplied through a large stack of matrices
- The resulting output embedding vectors determine predicted token probabilities
Sequence language models have amazing potential
Evo 2 is trained on 9.3 trillion DNA bases and predicts mutation effects, learns biological features and can generate quasi-life-like sequences
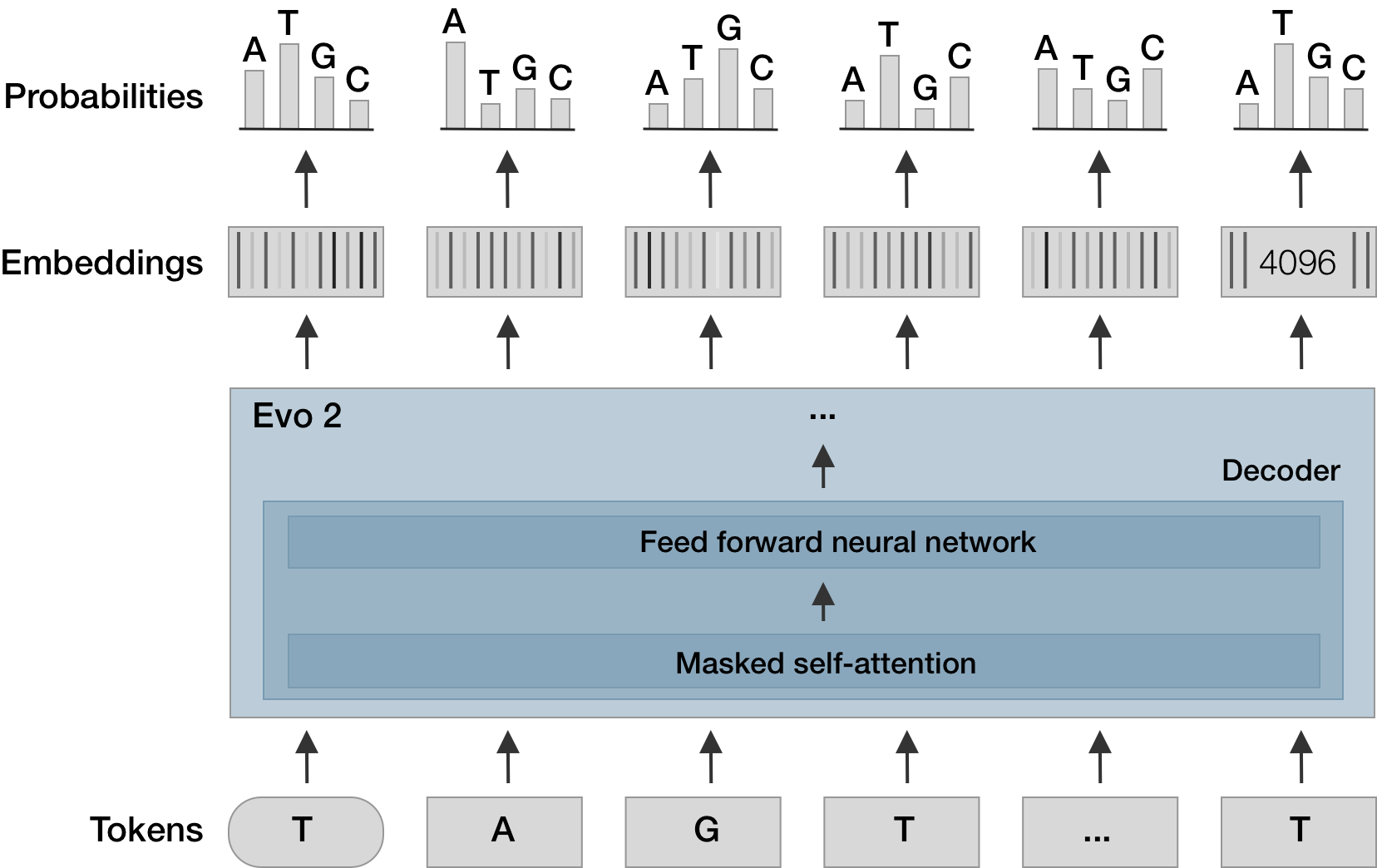
Evo 2 predicts fitness effects of genomic changes
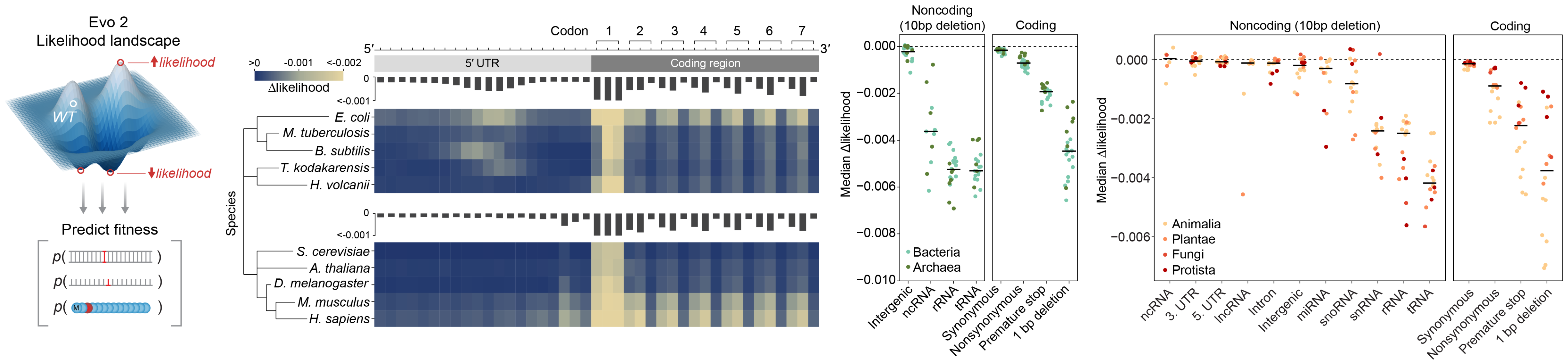 Brixi et al. 2025. bioRxiv.
Brixi et al. 2025. bioRxiv.
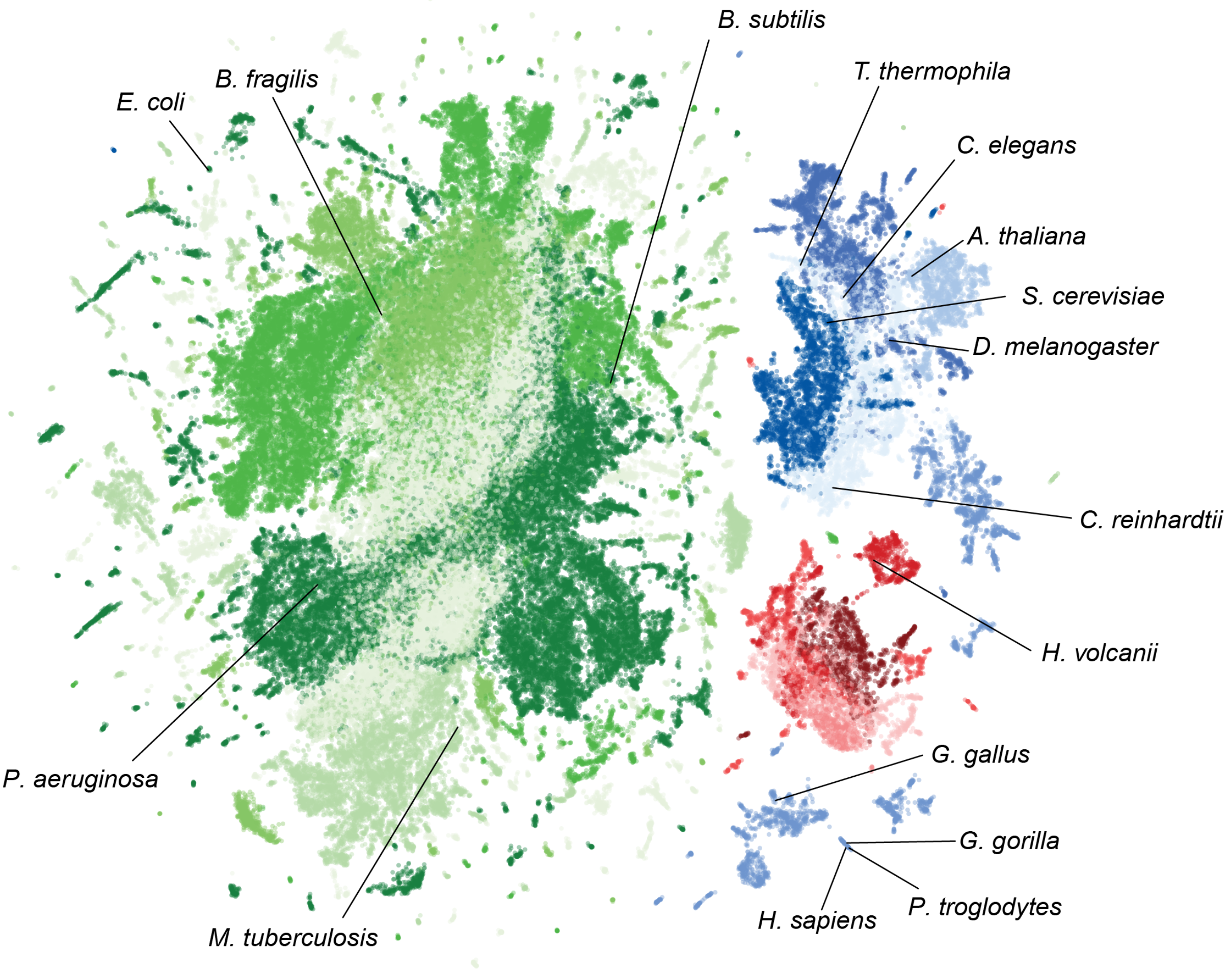
Under the hood, biological features inform Evo 2 embeddings
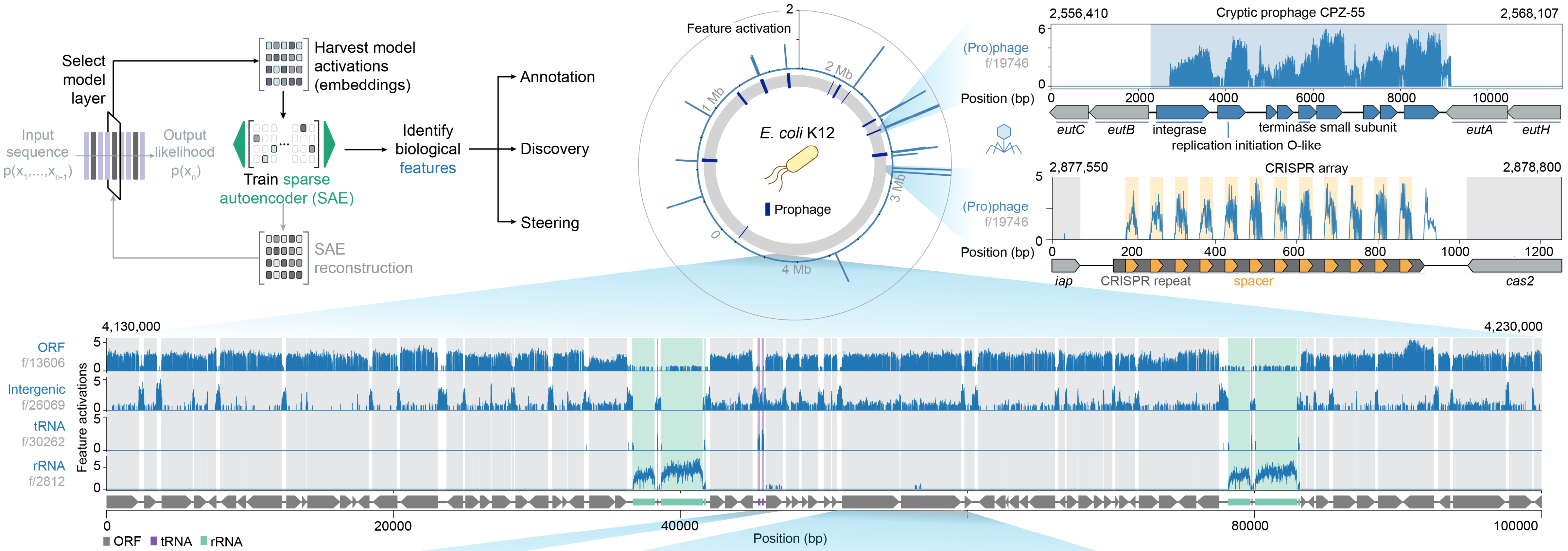 Brixi et al. 2025. bioRxiv.
Brixi et al. 2025. bioRxiv.
Acknowledgements
Influenza: WHO Global Influenza Surveillance Network, GISAID, Richard Neher, John Huddleston. Zika: Nick Loman, Nuno Faria, Oli Pybus, Josh Quick, Ingra Claro, Julien Thézé, Jaquilene de Jesus, Marta Giovanetti, Moritz Kraemer, Sarah Hill, Ester Sabino, Luiz Alcantara, Allison Black. Ebola: Gytis Dudas, Andrew Rambaut, Luiz Carvalho, Philippe Lemey, Marc Suchard, Andrew Tatem, Eddy Kinganda-Lusamaki, Placide Mbala-Kingebeni, Catherine Pratt, Jean-Jacques Muyembe Tamfum. MERS: Gytis Dudas, Andrew Rambaut, Luiz Carvalho. SARS-CoV-2: Data producers from all over the world, GISAID, the Nextstrain team, Marlin Figgins, Katie Kistler. Nextstrain: Richard Neher, Ivan Aksamentov, John SJ Anderson, Kim Andrews, Jennifer Chang, James Hadfield, Emma Hodcroft, John Huddleston, Jover Lee, Victor Lin, Colin Megill, Cornelius Roemer, Thomas Sibley.
Bedford Lab:
![]() John Huddleston,
John Huddleston,
![]() James Hadfield,
James Hadfield,
![]() Katie Kistler,
Katie Kistler,
![]() Thomas Sibley,
Thomas Sibley,
![]() Jover Lee,
Jover Lee,
![]() Marlin Figgins,
Marlin Figgins,
![]() Victor Lin,
Victor Lin,
![]() Jennifer Chang,
Jennifer Chang,
![]() Nashwa Ahmed,
Nashwa Ahmed,
![]() Cécile Tran Kiem,
Cécile Tran Kiem,
![]() Kim Andrews,
Kim Andrews,
![]() Philippa Steinberg,
Philippa Steinberg,
![]() Jacob Dodds,
Jacob Dodds,
![]() John SJ Anderson,
John SJ Anderson,
![]() Nobuaki Masaki,
Nobuaki Masaki,
![]() Amin Bemanian,
Amin Bemanian,
![]() Carlos Avendano,
Carlos Avendano,
![]() Aayush Verma,
Aayush Verma,
![]() Lucy O'Brien
Lucy O'Brien