Forecasting strain turnover in influenza and dengue viruses
Trevor Bedford and Sidney Bell
6 Nov 2018
SMBE Satellite Workshop on Genome Evolution in Pathogen Transmission and Disease
Kyoto, Japan
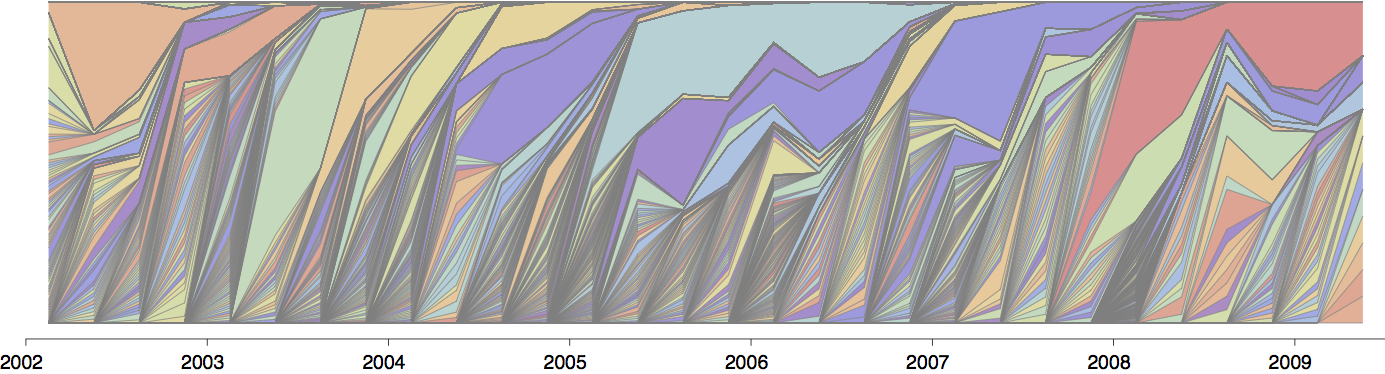
Influenza and dengue viruses

Influenza virus
Population turnover is extremely rapid
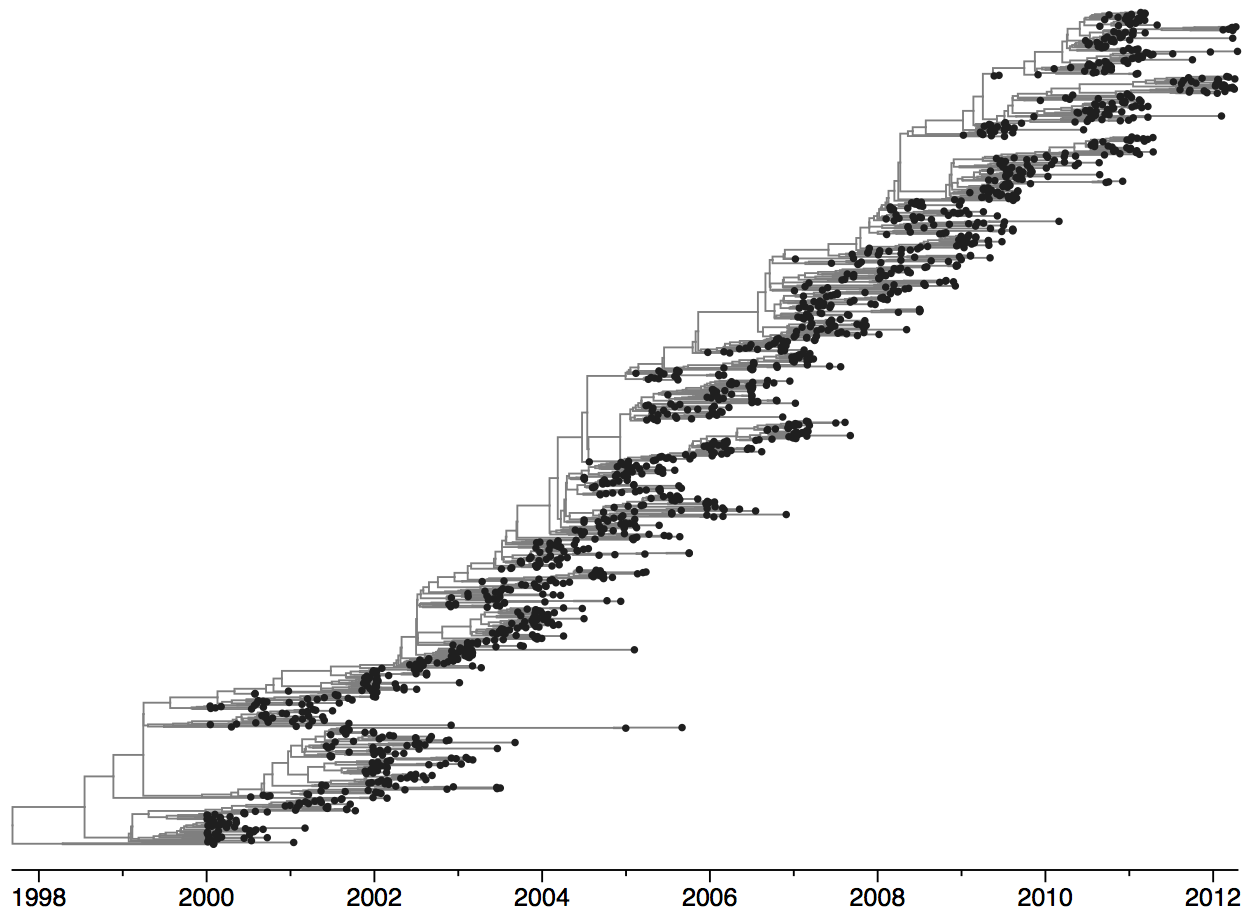
Dynamics driven by antigenic drift
Necessitates vaccine updates every ~2 years
Vaccine strain selection by WHO
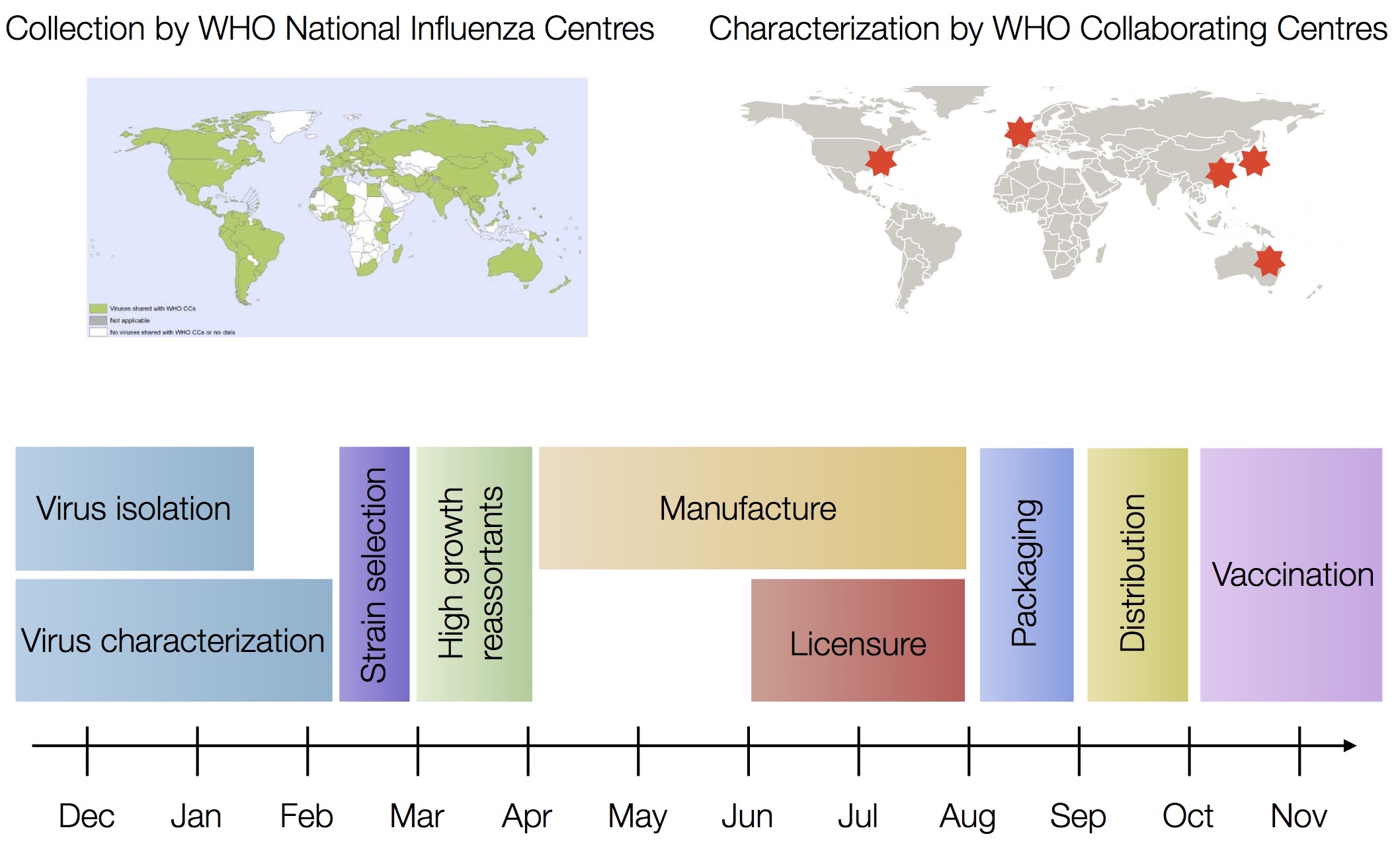
nextflu
Project to provide a real-time view of the evolving influenza population
in collaboration with Richard Neher
nextflu pipeline
- Download all recent HA sequences from GISAID
- Filter to remove outliers
- Subsample across time and space
- Align sequences
- Build tree
- Estimate clade frequencies
- Infer antigenic phenotypes
- Export for visualization
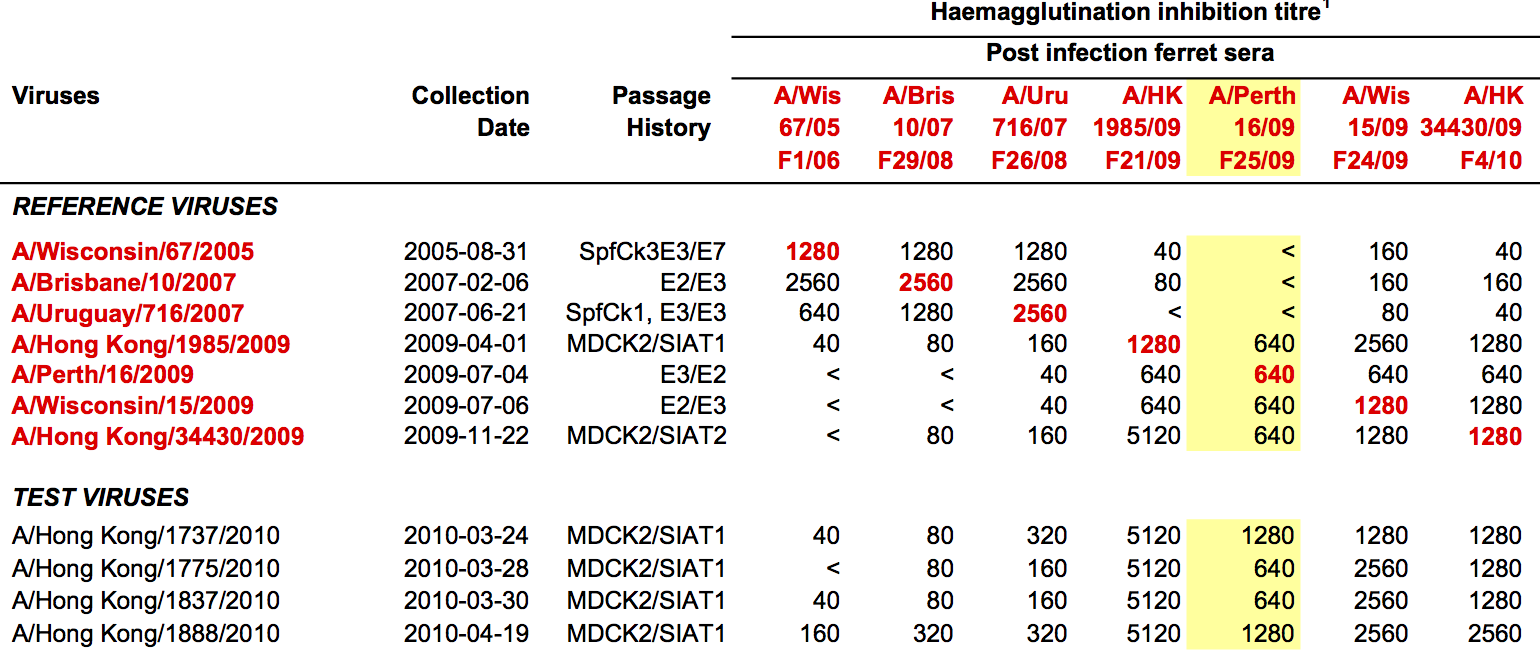
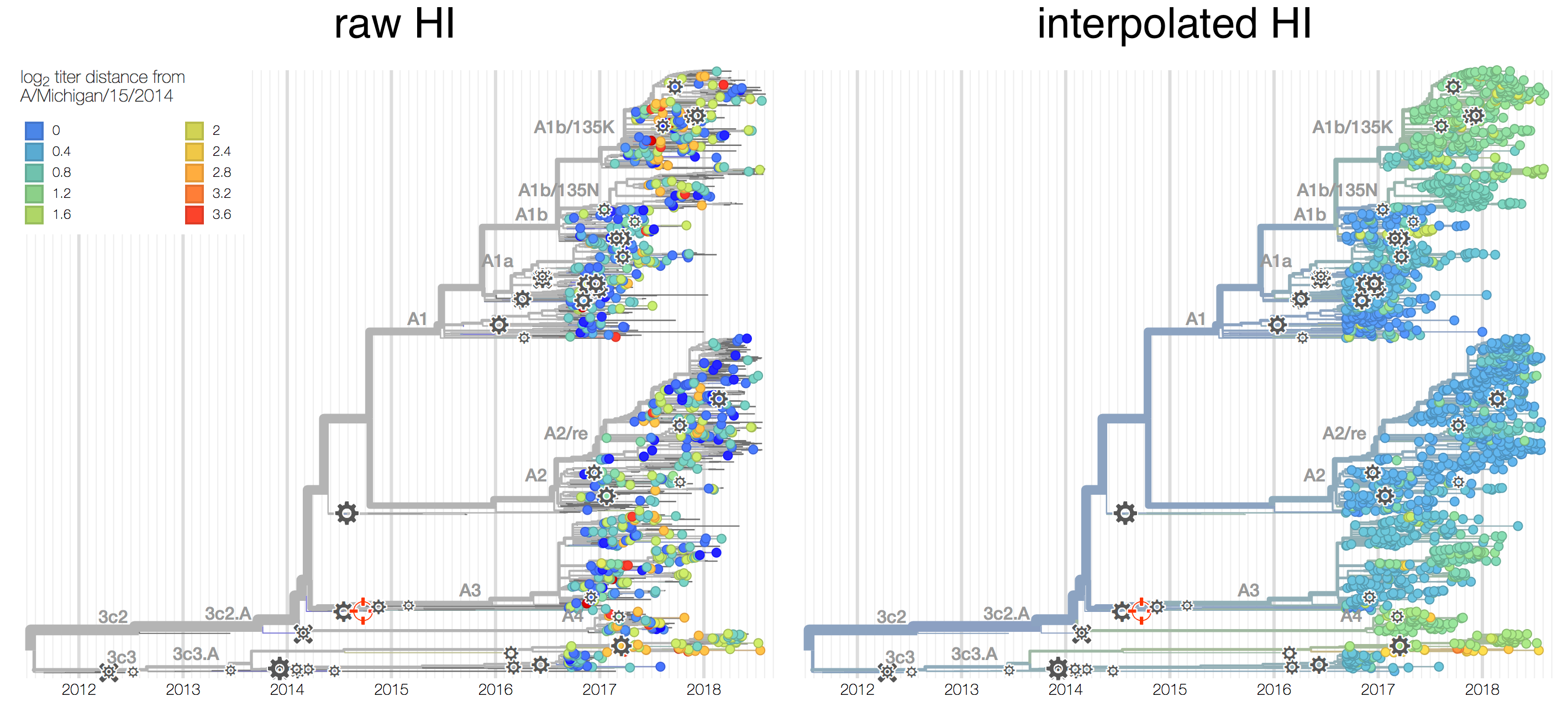
Antigenic phenotype measured by pairwise hemagglutination inhibition (HI) assays
Phylogenetic model that ascribes drops in HI titer data to specific branches
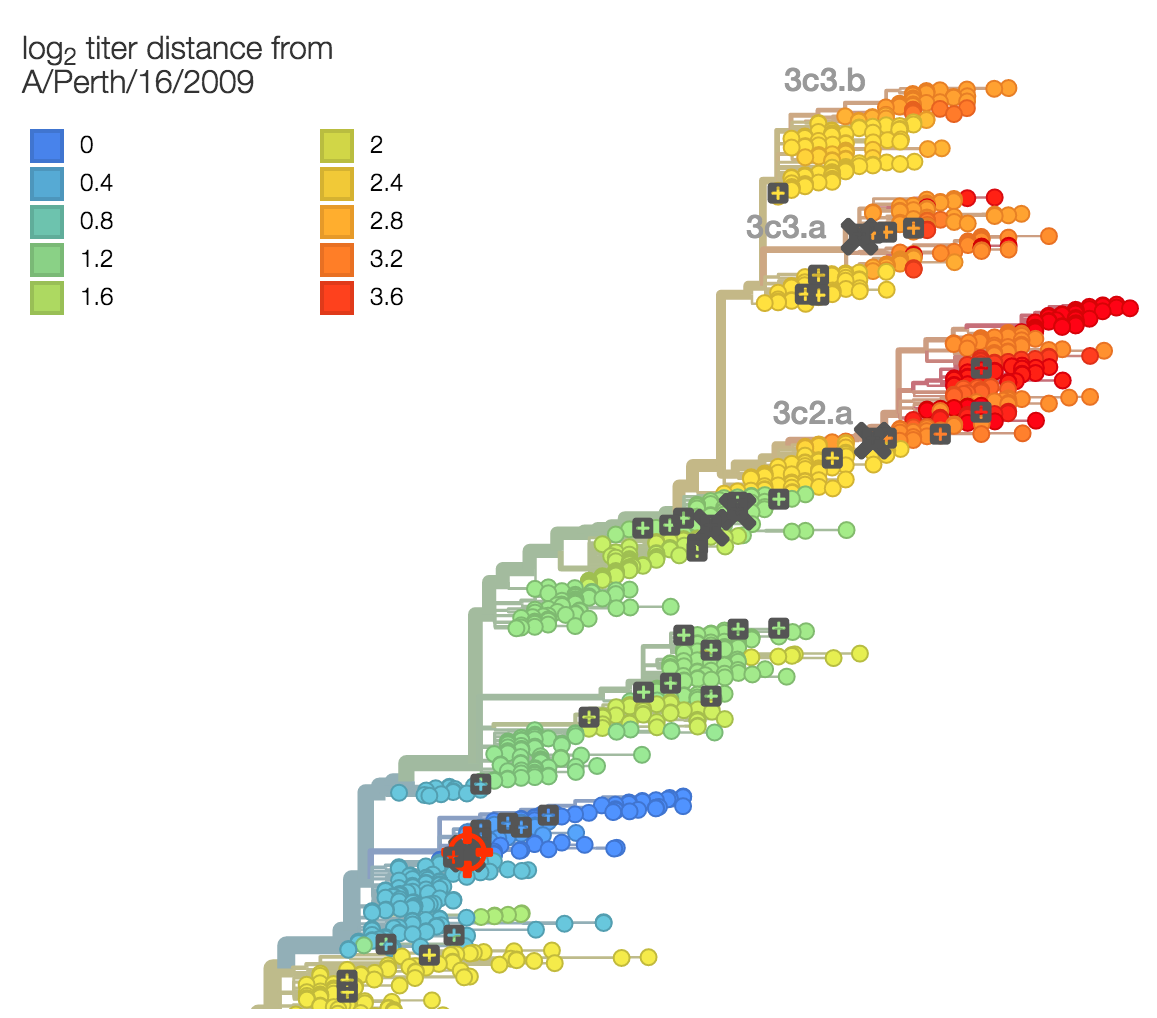
Up-to-date analysis publicly available at:
nextstrain.org/flu
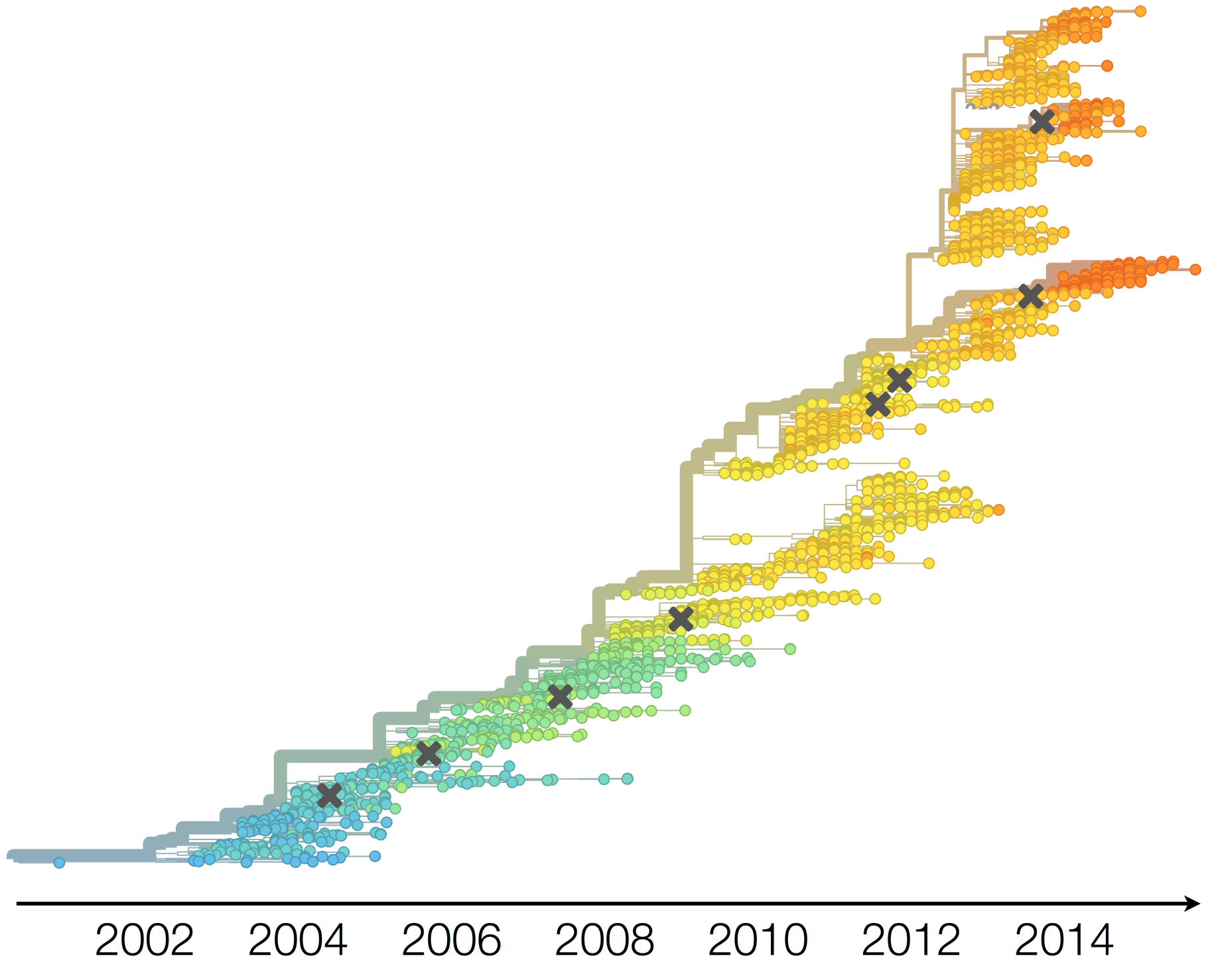
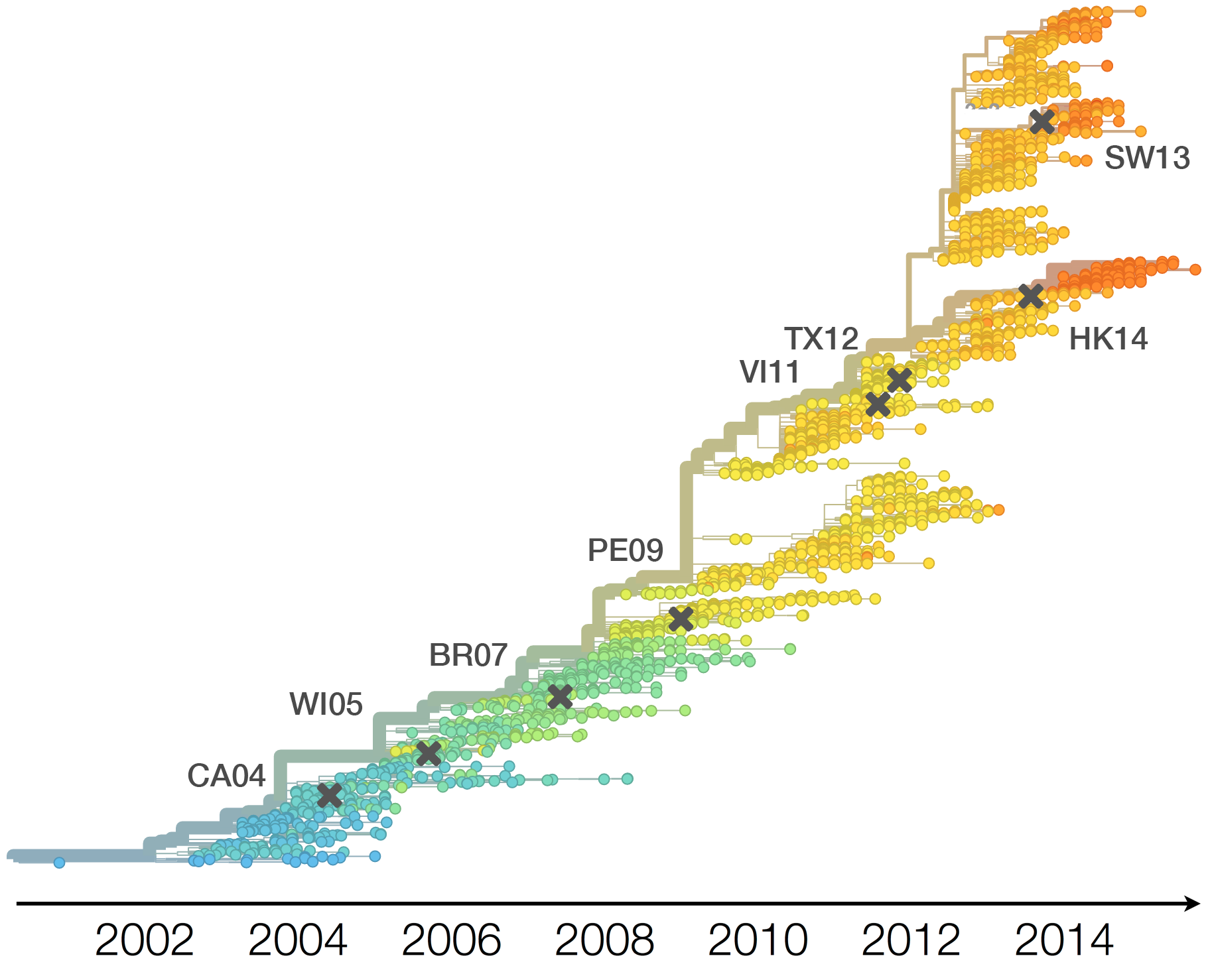
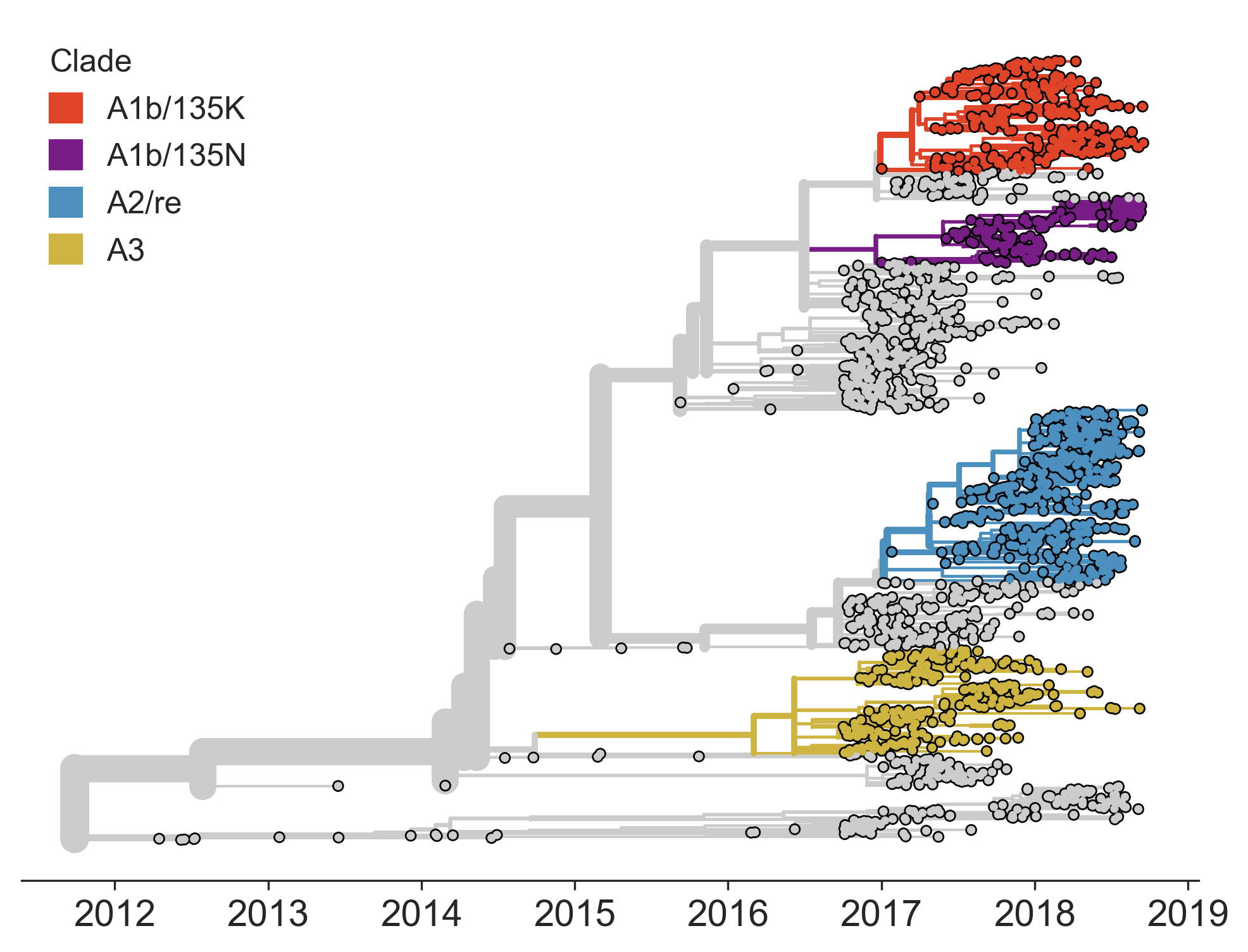
Current diversity
H3N2 diversity as of Sep 2018
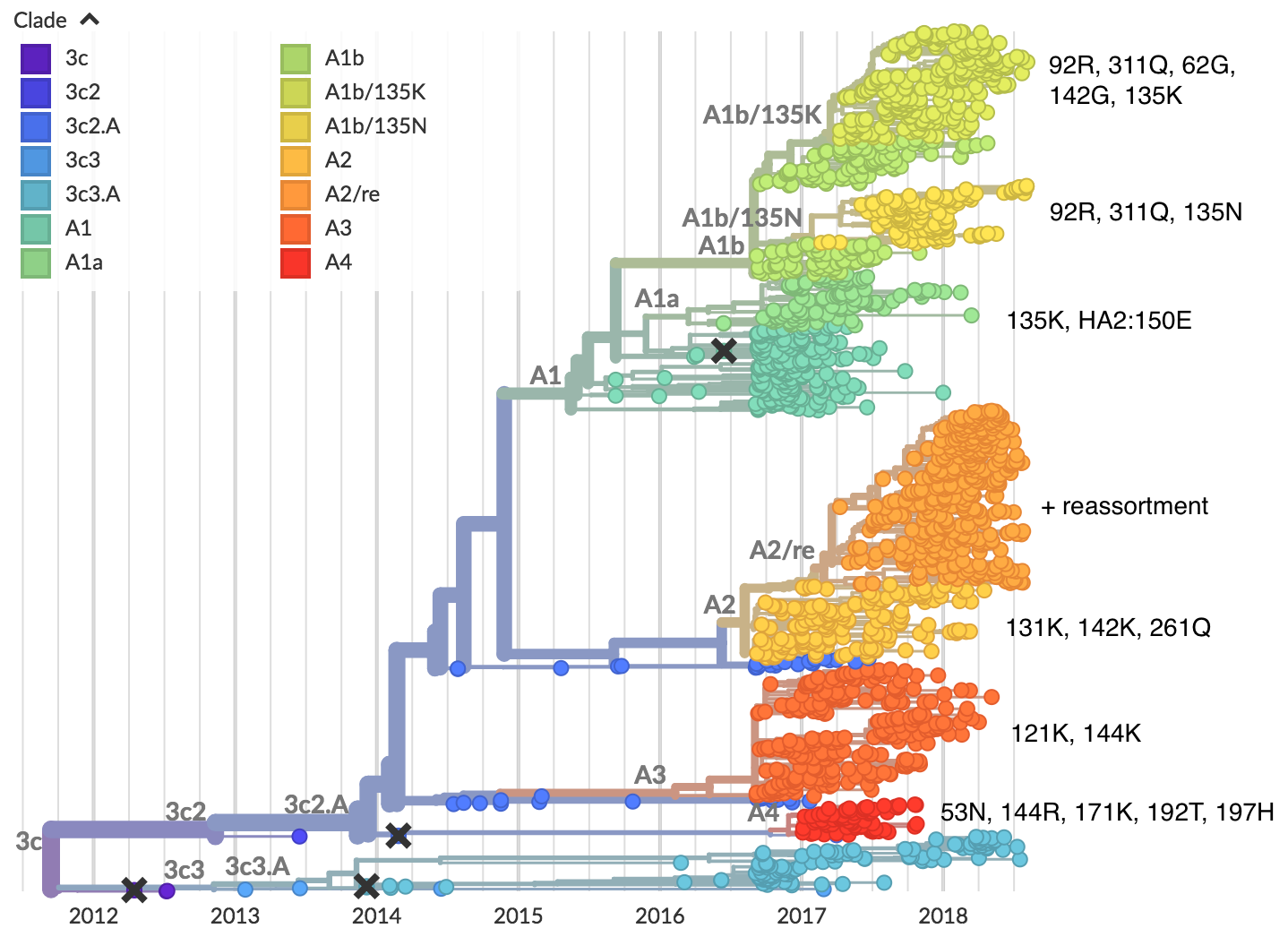
Clade dynamics show recent success of A1b and A2 viruses
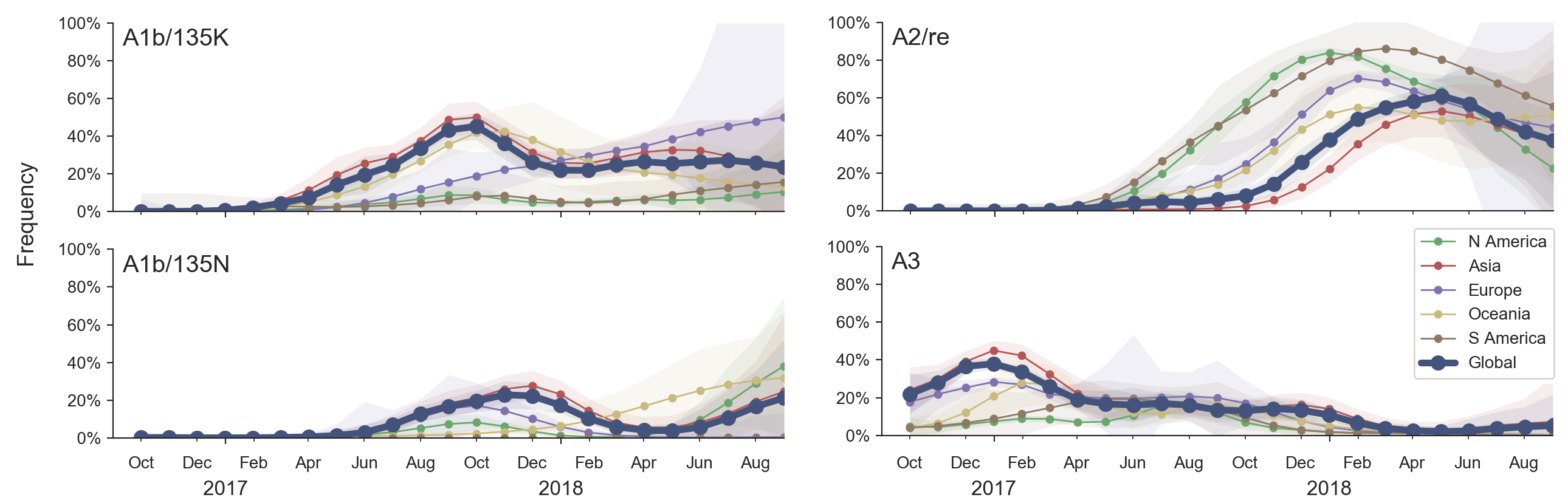
Clade A1b has drifted viruses
Clade A2 appears driven by reassortment event
Clade success generally has a retrospective narrative
Forecasting strain turnover
with John Huddleston and Richard Neher
"The future is here, it's just not evenly distributed yet"
— William Gibson

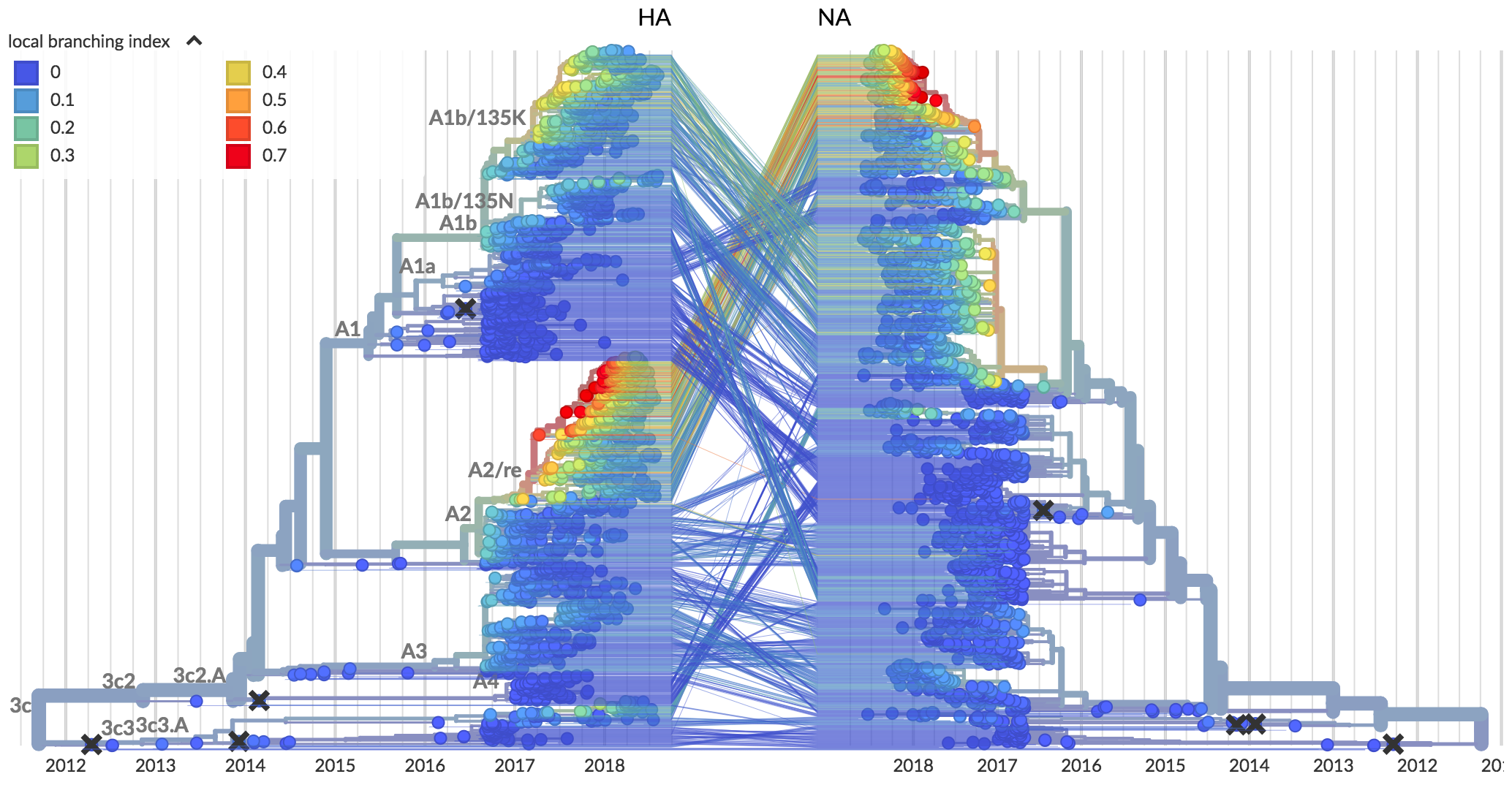
Influenza population turnover
Seek to explain change in clade frequencies over 1 year
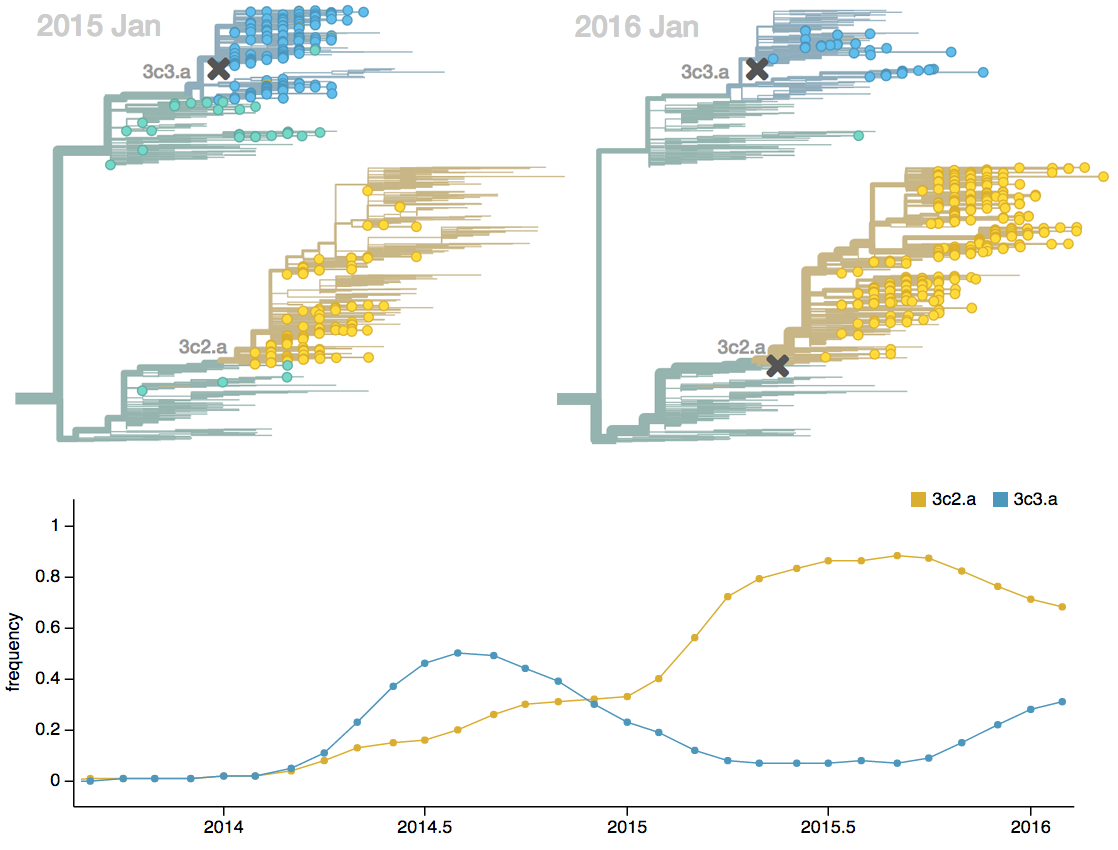
Fitness models can project clade frequencies
Clade frequencies $X$ derive from the fitnesses $f$ and frequencies $x$ of constituent viruses, such that
$$\hat{X}_v(t+\Delta t) = \sum_{i:v} x_i(t) \, \mathrm{exp}(f_i \, \Delta t)$$
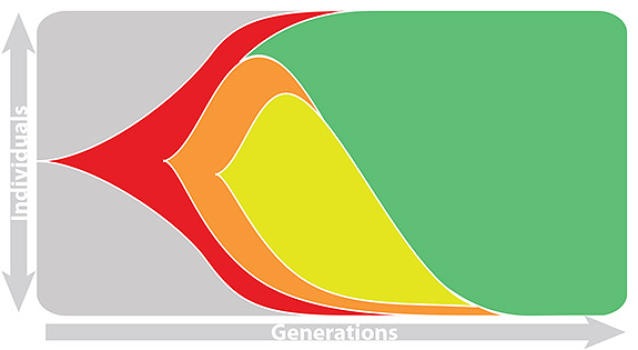
This captures clonal interference between competing lineages
Fitness estimated from viral attributes
The fitness $f$ of virus $i$ is estimated as
$$\hat{f}_i = \beta^\mathrm{A} \, f_i^\mathrm{A} + \beta^\mathrm{B} \, f_i^\mathrm{B} + \ldots$$
where A, B, etc... are different standardized viral attributes
We learn $\beta$ coefficients from most recent 12 years of H3N2 evolution
Optimal $\beta$ coefficients minimize sum of squared errors between observed clade frequencies and frequency estimated in a 1-year look ahead
Fit to a subset of non-nested clades
Fitness predictors used alone show moderate success
| Model | $\beta$ coefficient | Growth correlation | Growth accuracy |
|---|---|---|---|
| Antigenic drift based on HI model (cTiterSub) | 0.33 | 0.29 | 69% |
| Cross-immunity based on epitope mutations (ep_x) | 0.50 | 0.18 | 60% |
| Protein function based on deep mutational scanning (dms) | 0.20 | 0.01 | 57% |
| Clade growth based on local branching index (lbi) | 0.39 | 0.30 | 61% |
Combine predictors into a single model
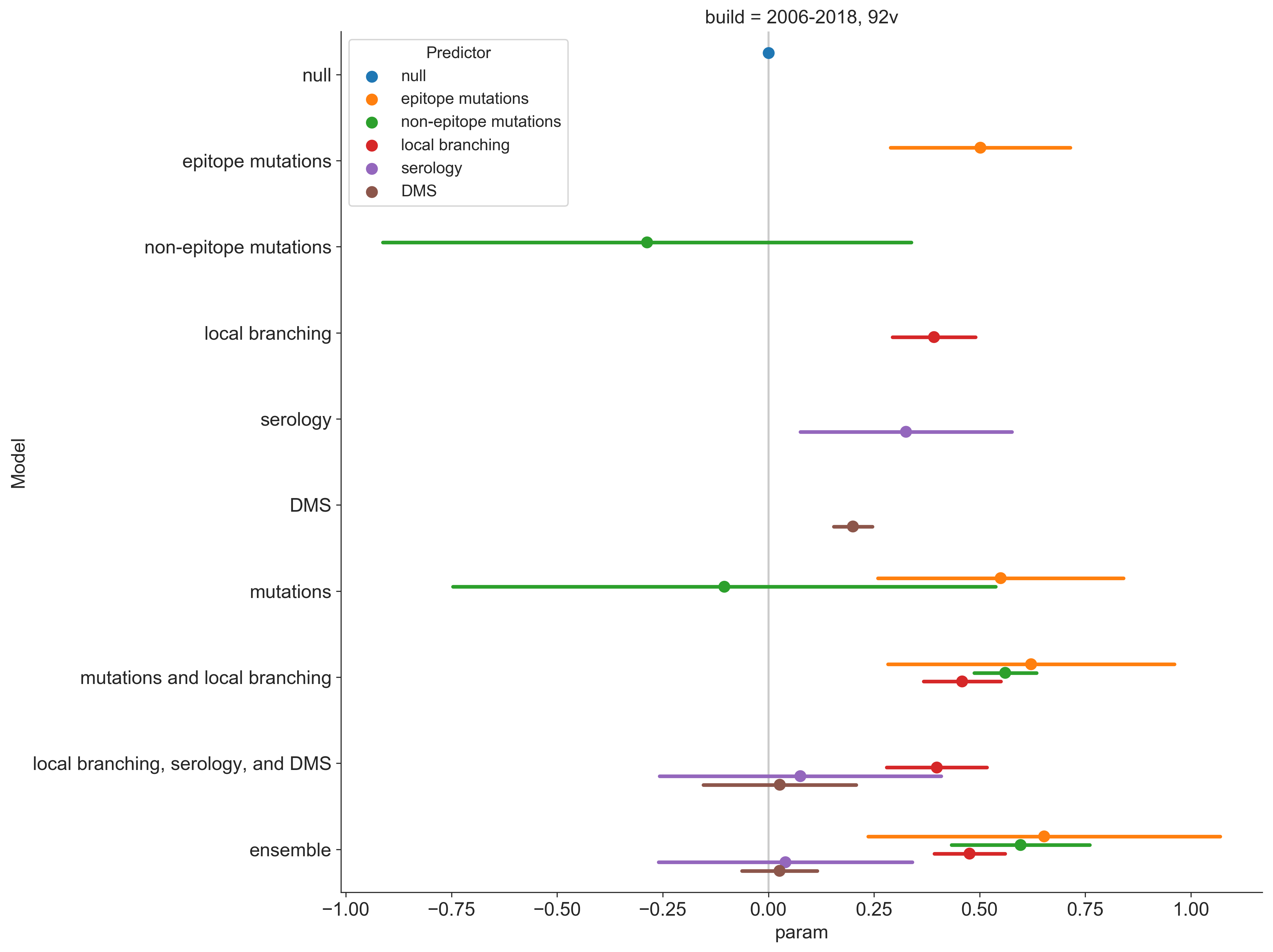
Prospective trajectories from ensemble model
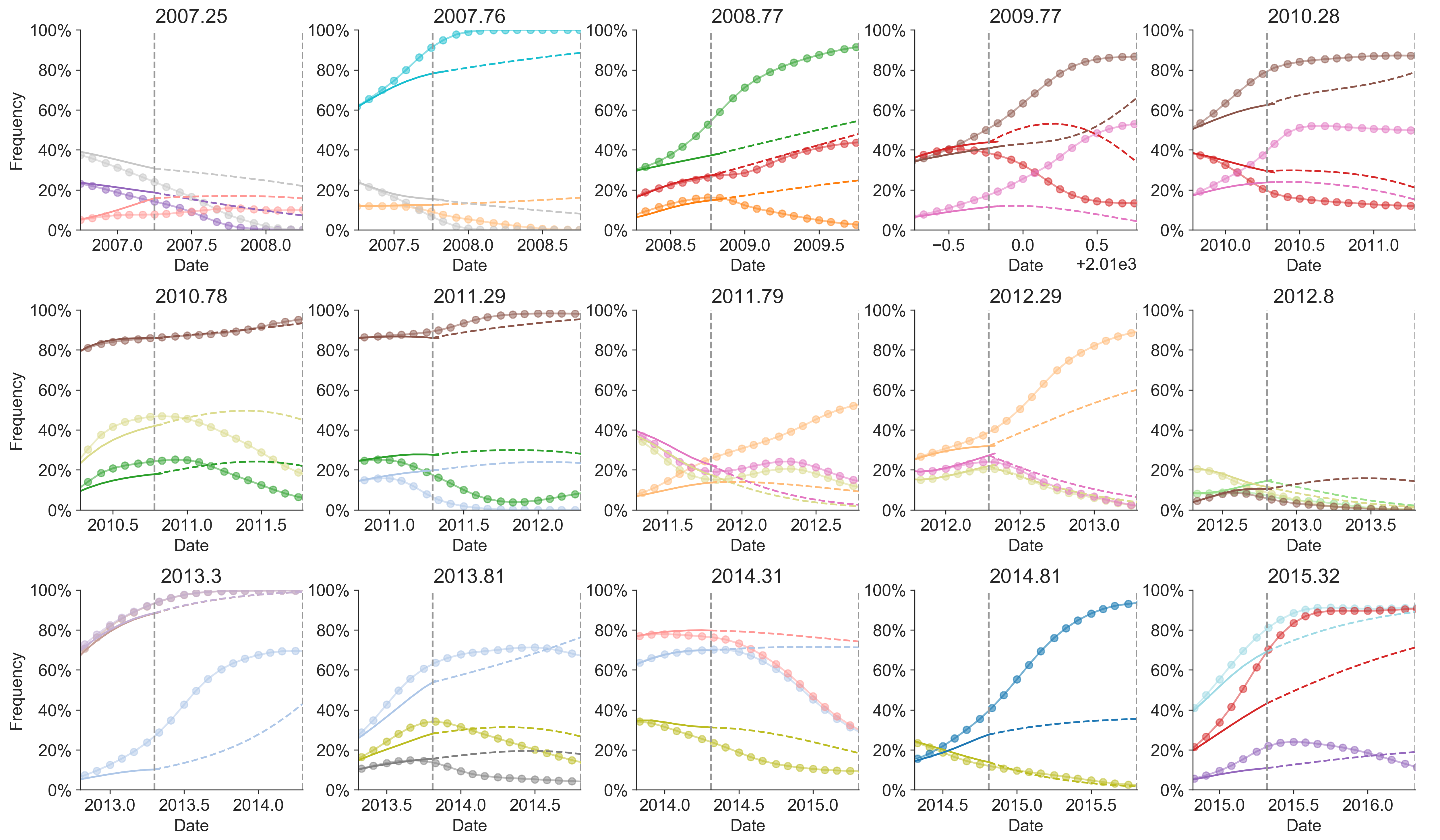
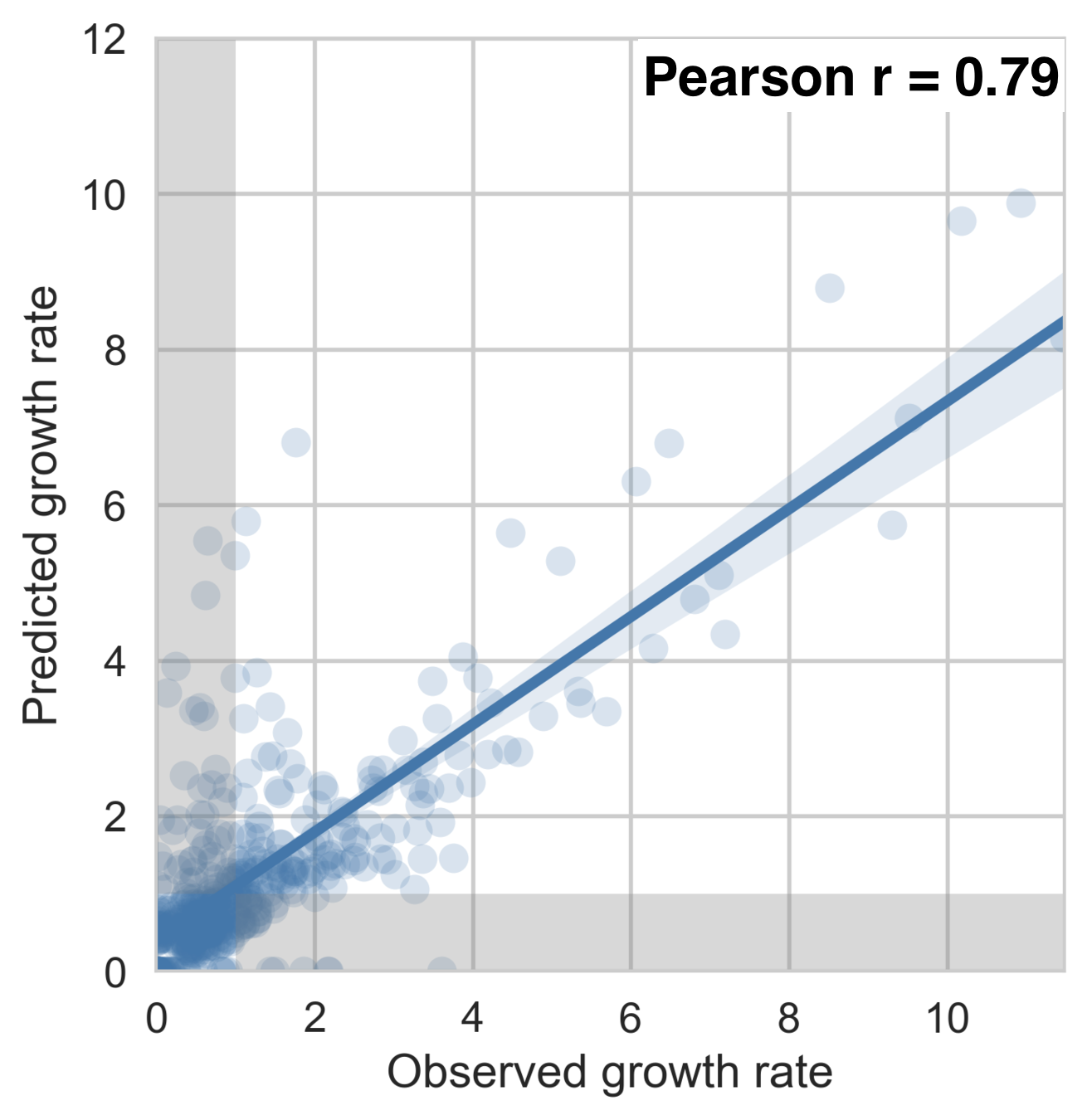
Growth rate of clades is well predicted
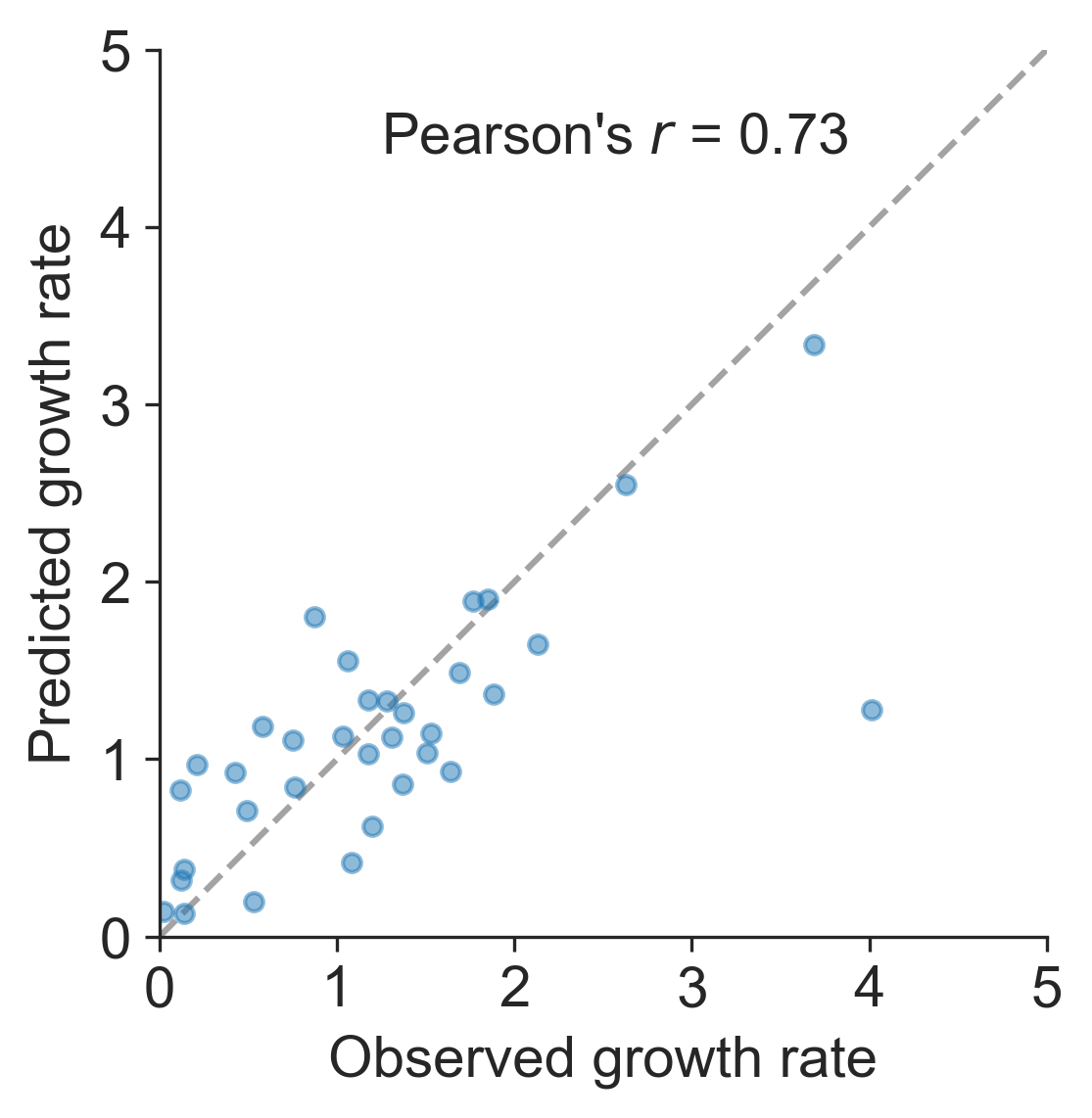
Prediction of growth vs decline also well predicted
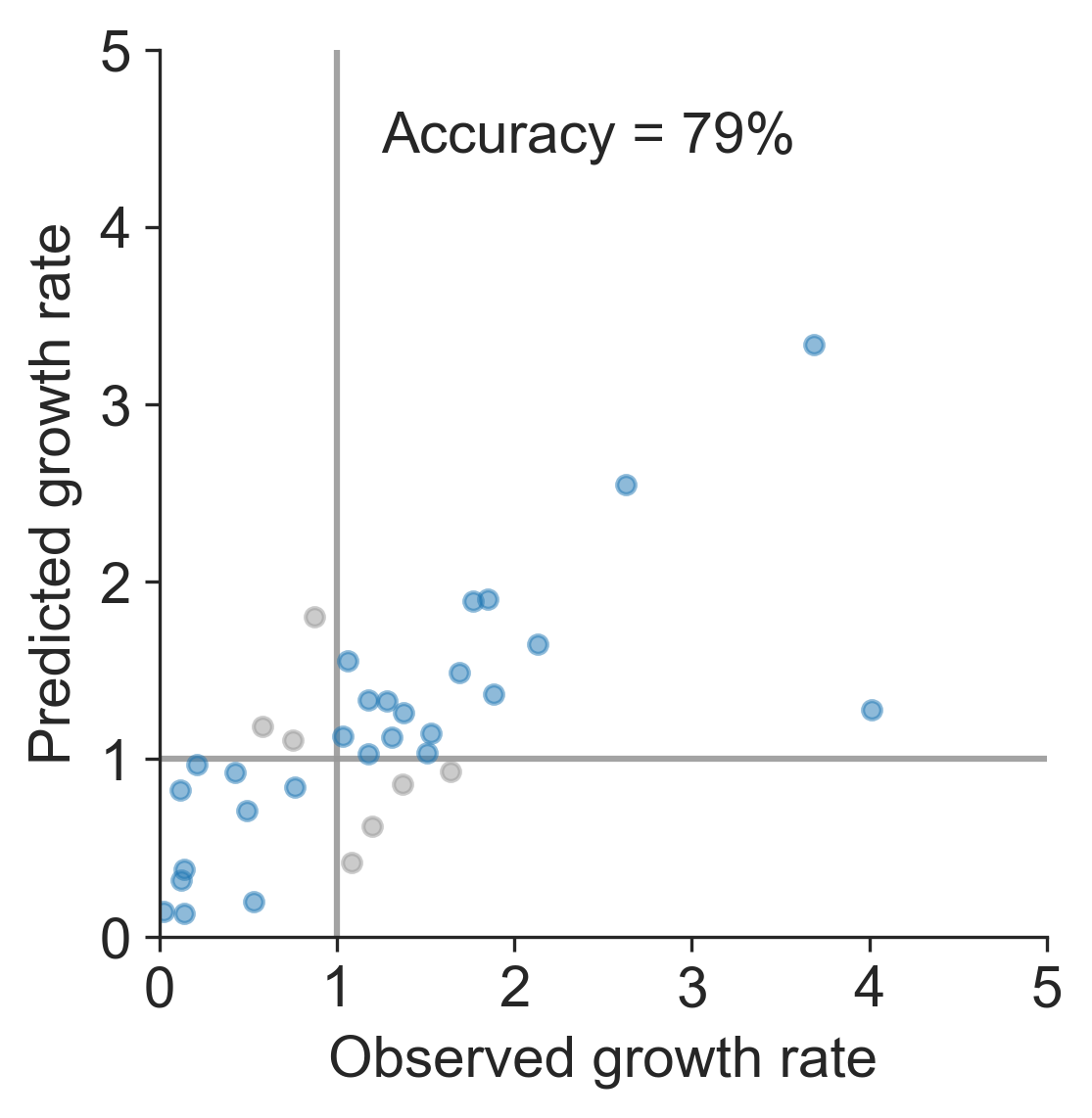
Currently circulating virus clades
Projections of these clades
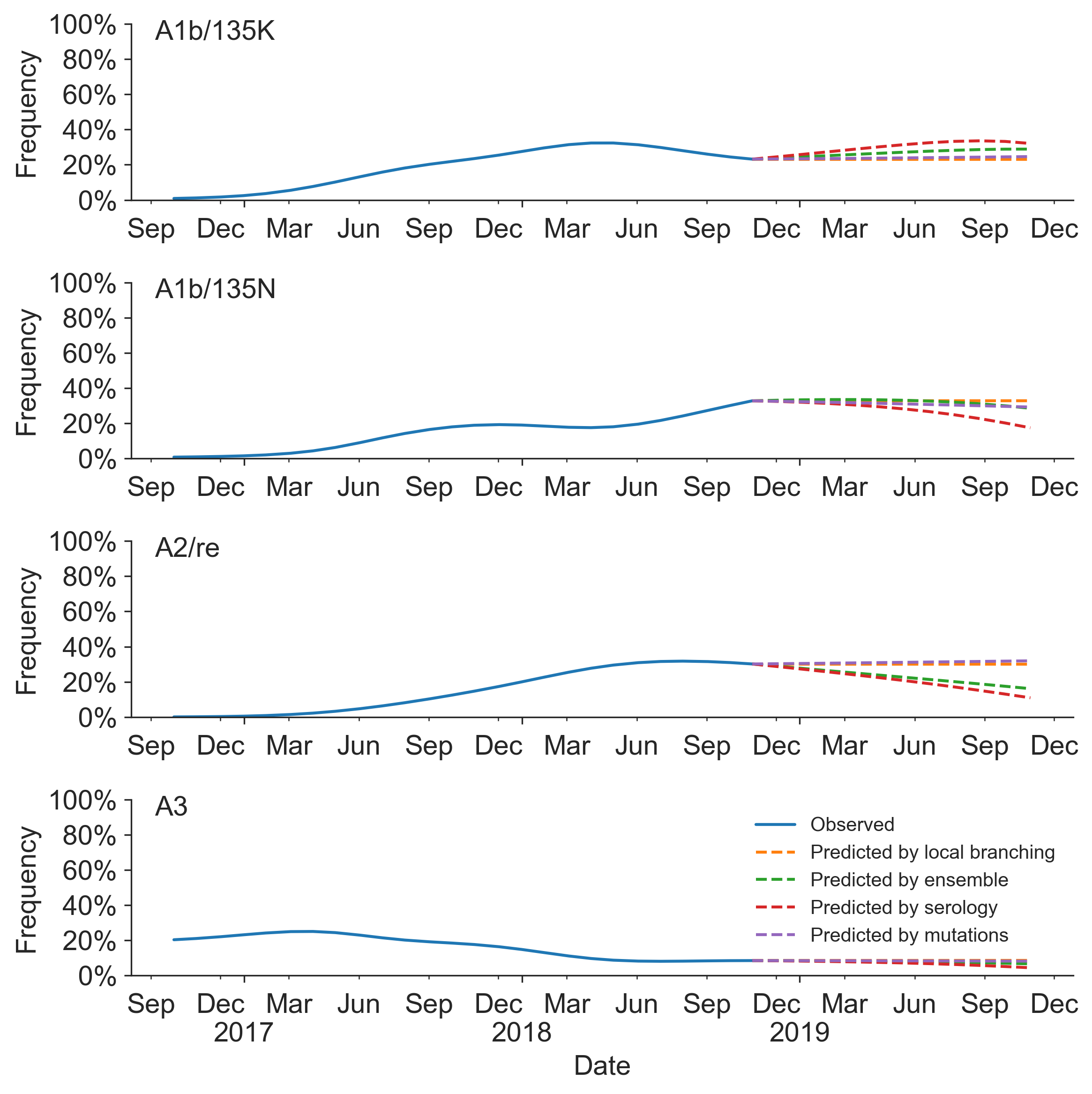
Unlikely to supplant expert predictions, but useful for vaccine candidate selection
Dengue virus
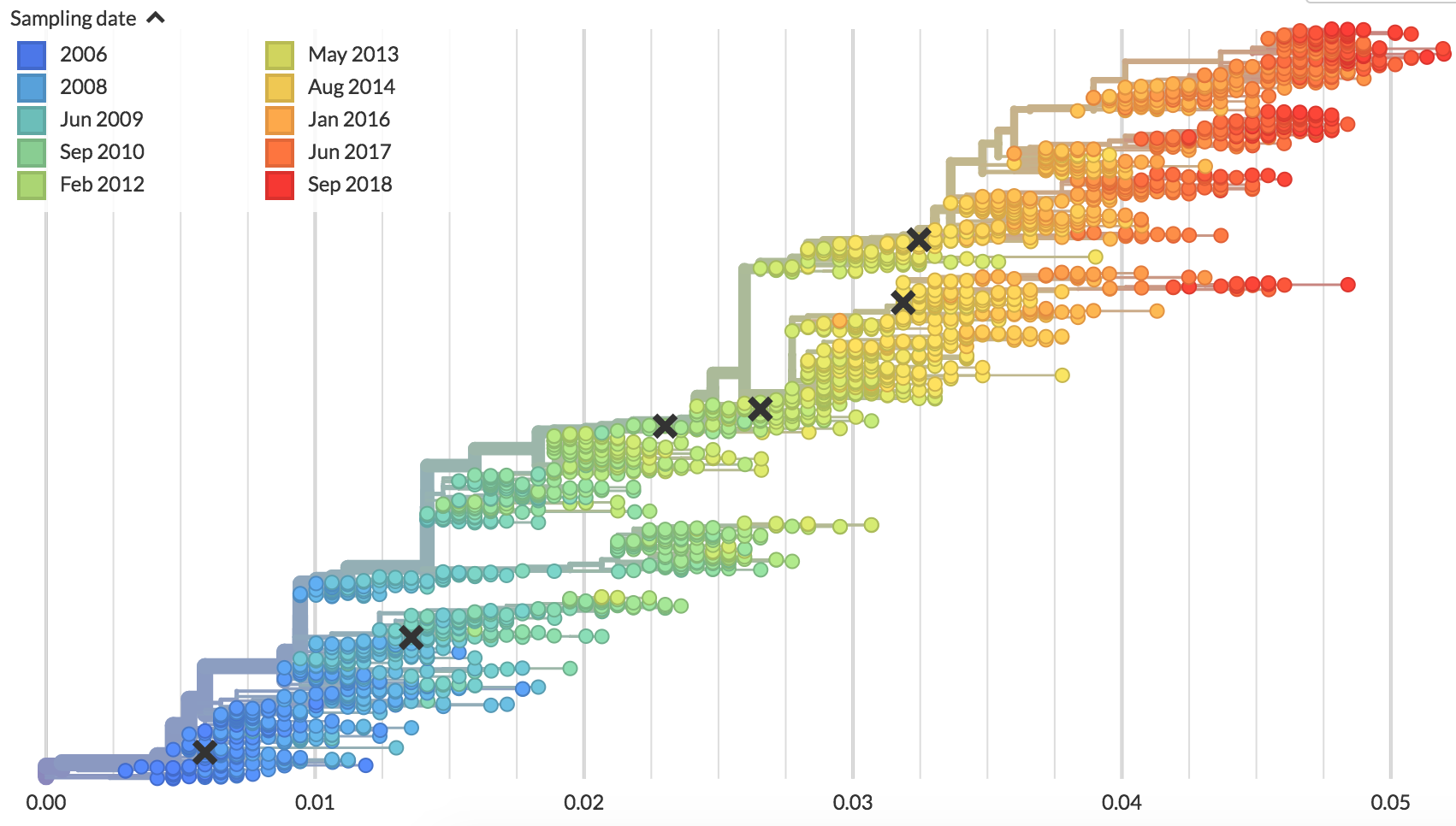
Different tempos of evolution
Dengue (serotype 2)
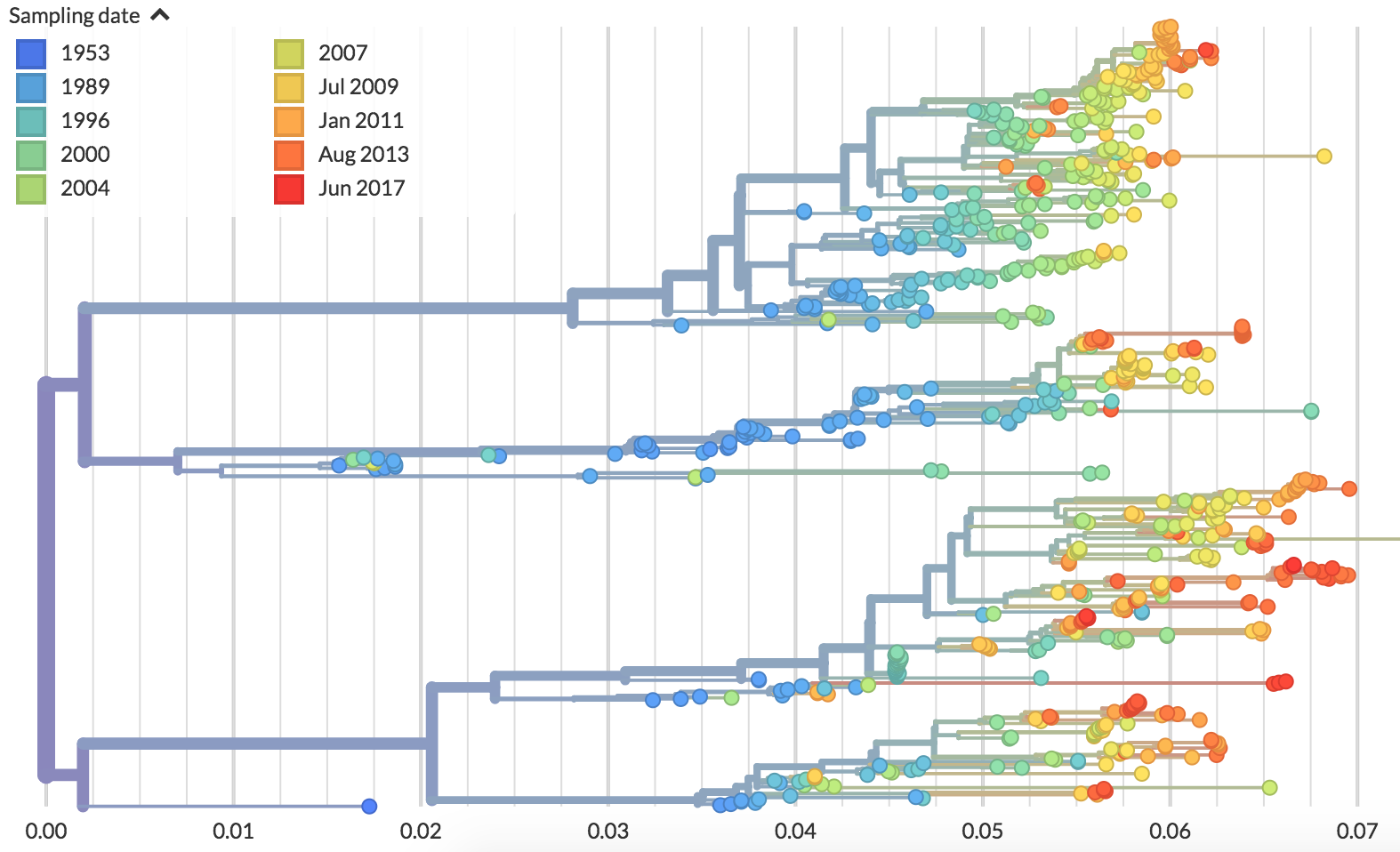
Flu (H3N2)
Four (uniform?) serotypes of dengue
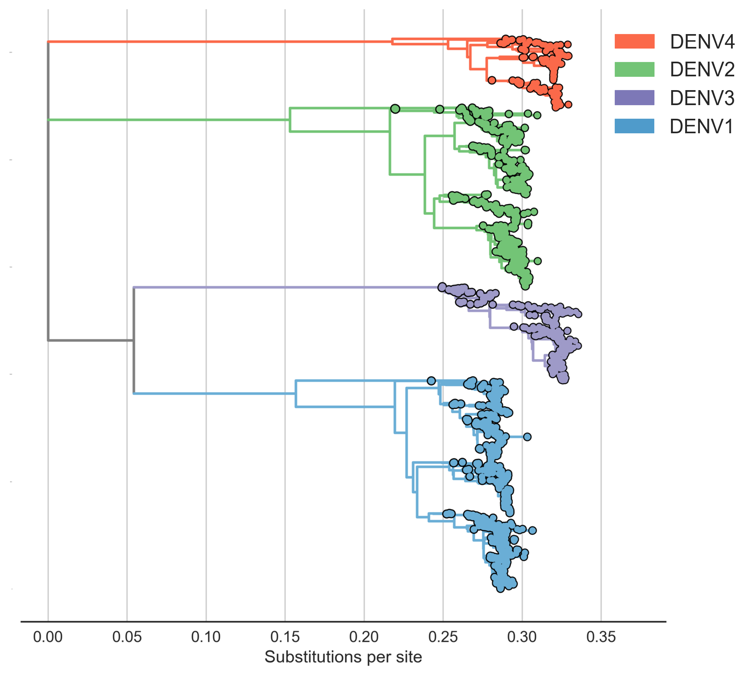
Each serotype is genetically diverse
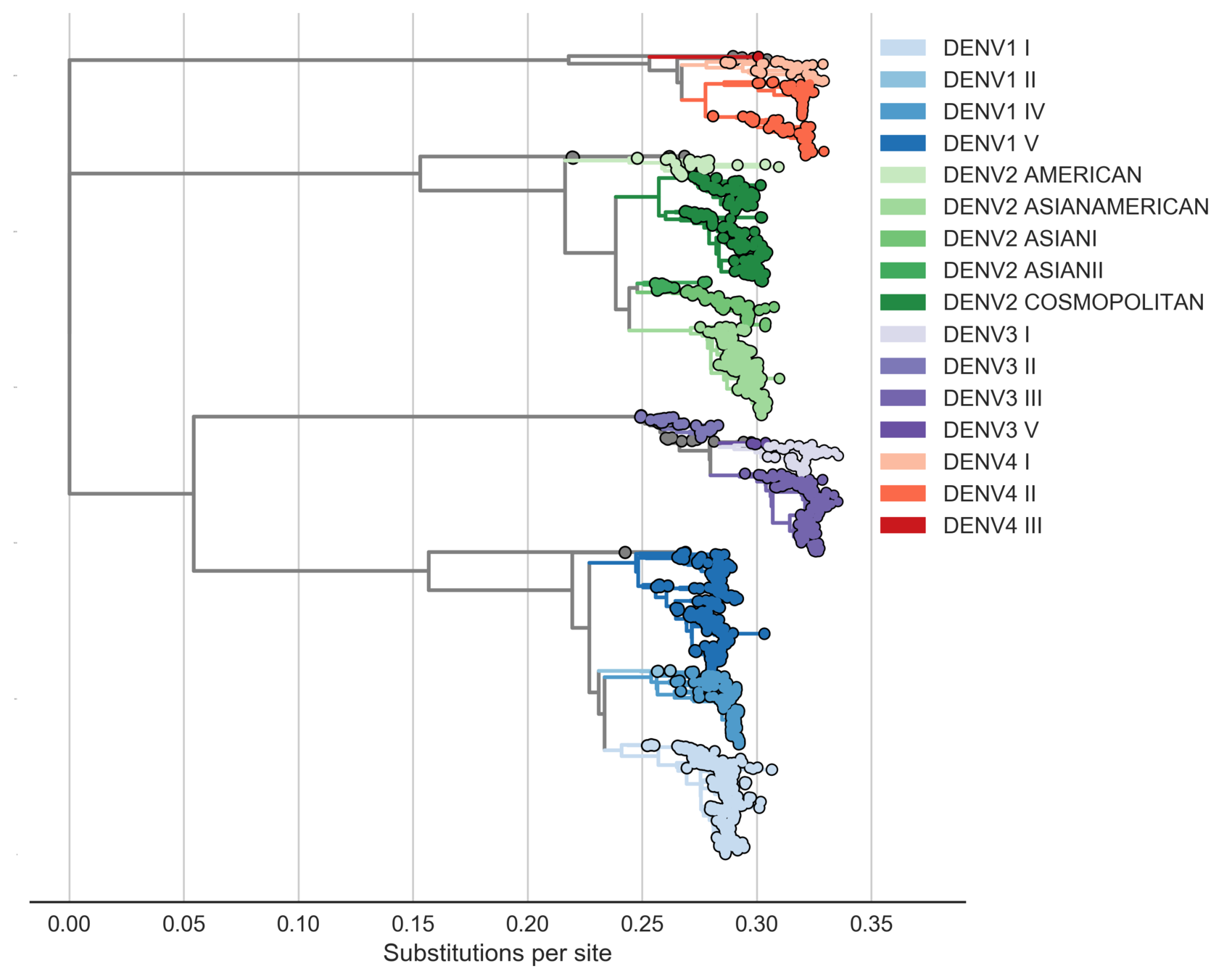
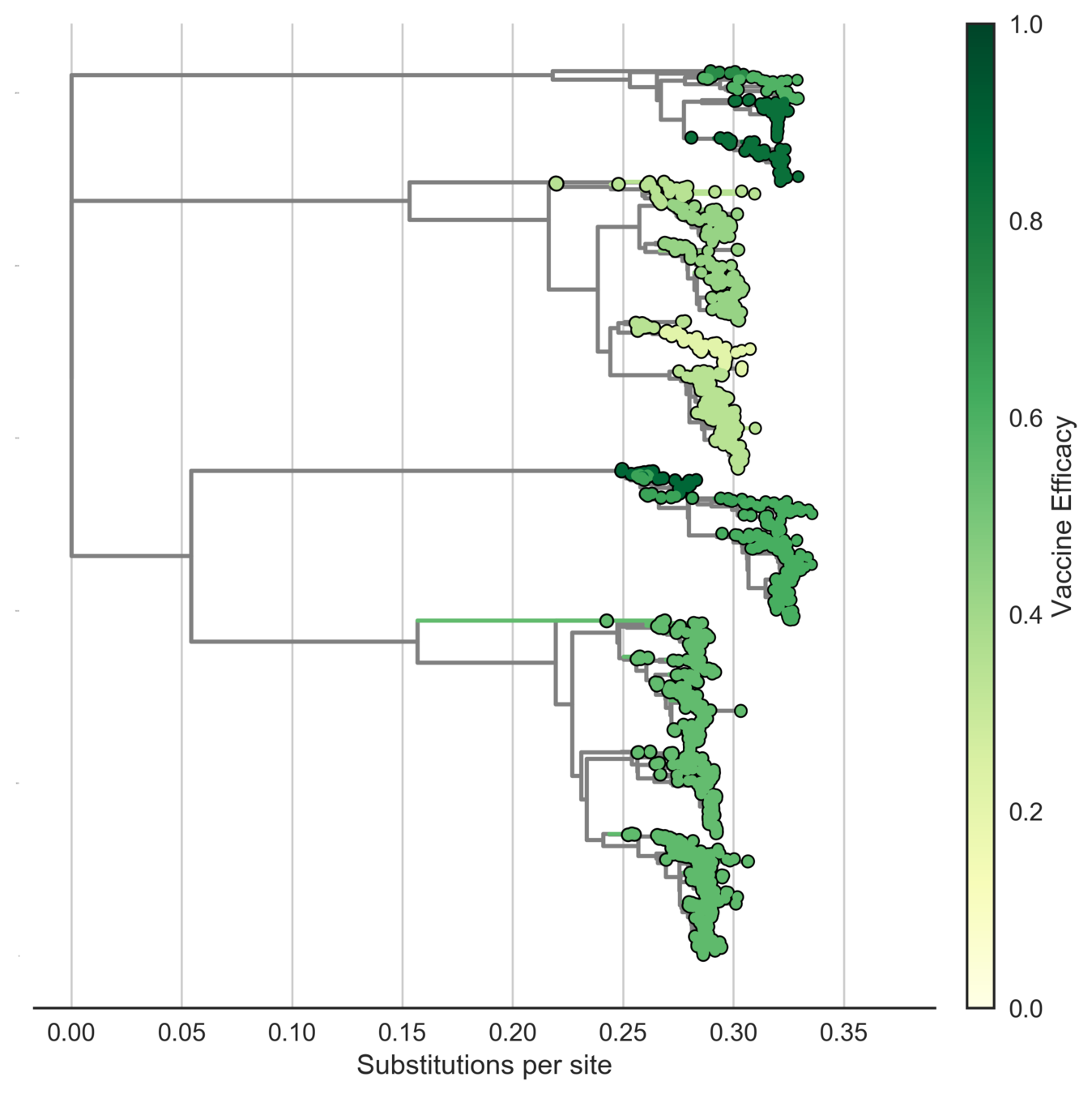
Sanofi vaccine efficacy varies by genotype
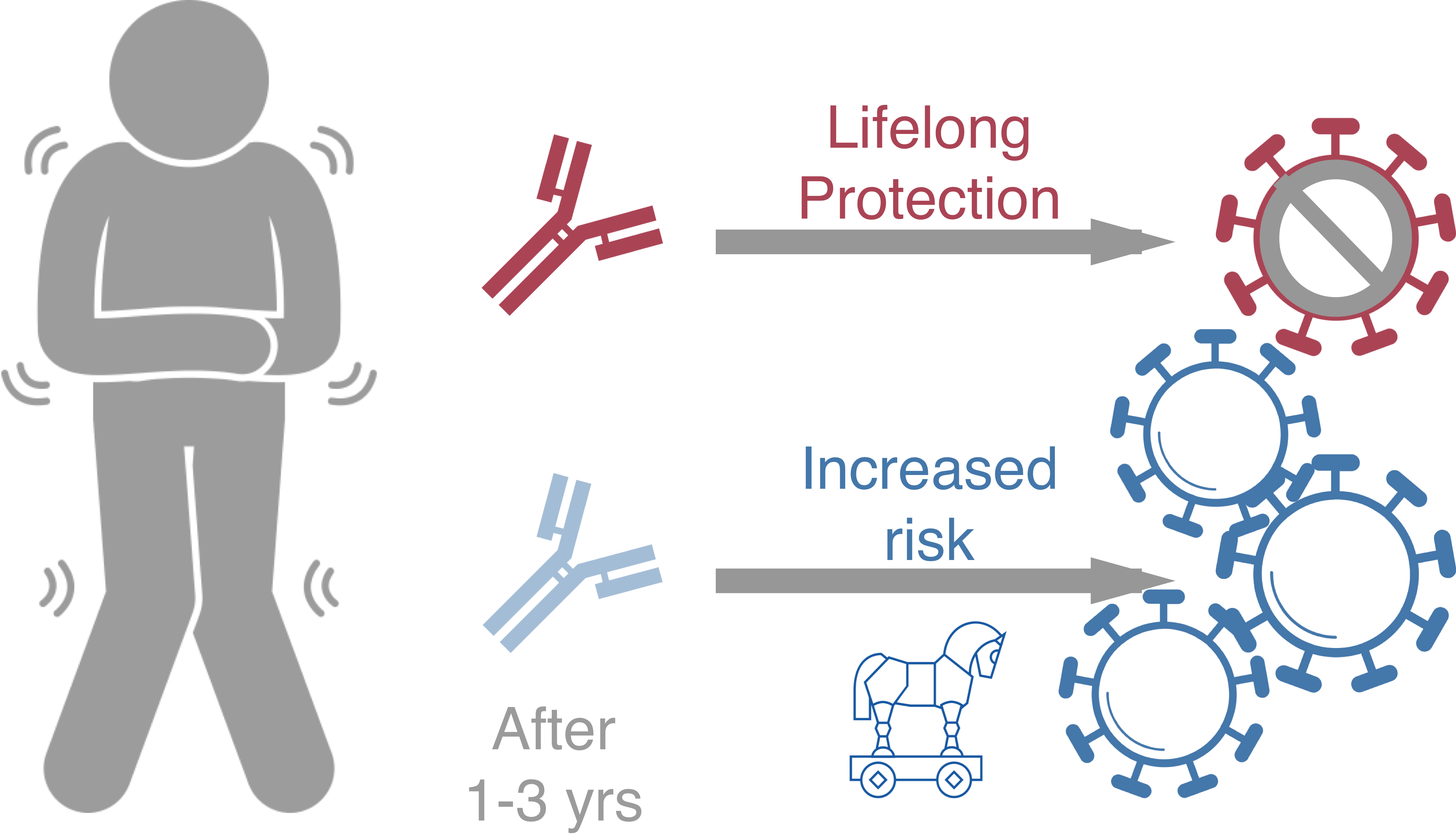
DENV4-I associated with adverse events
Original antigenic sin drives
dengue case outcomes
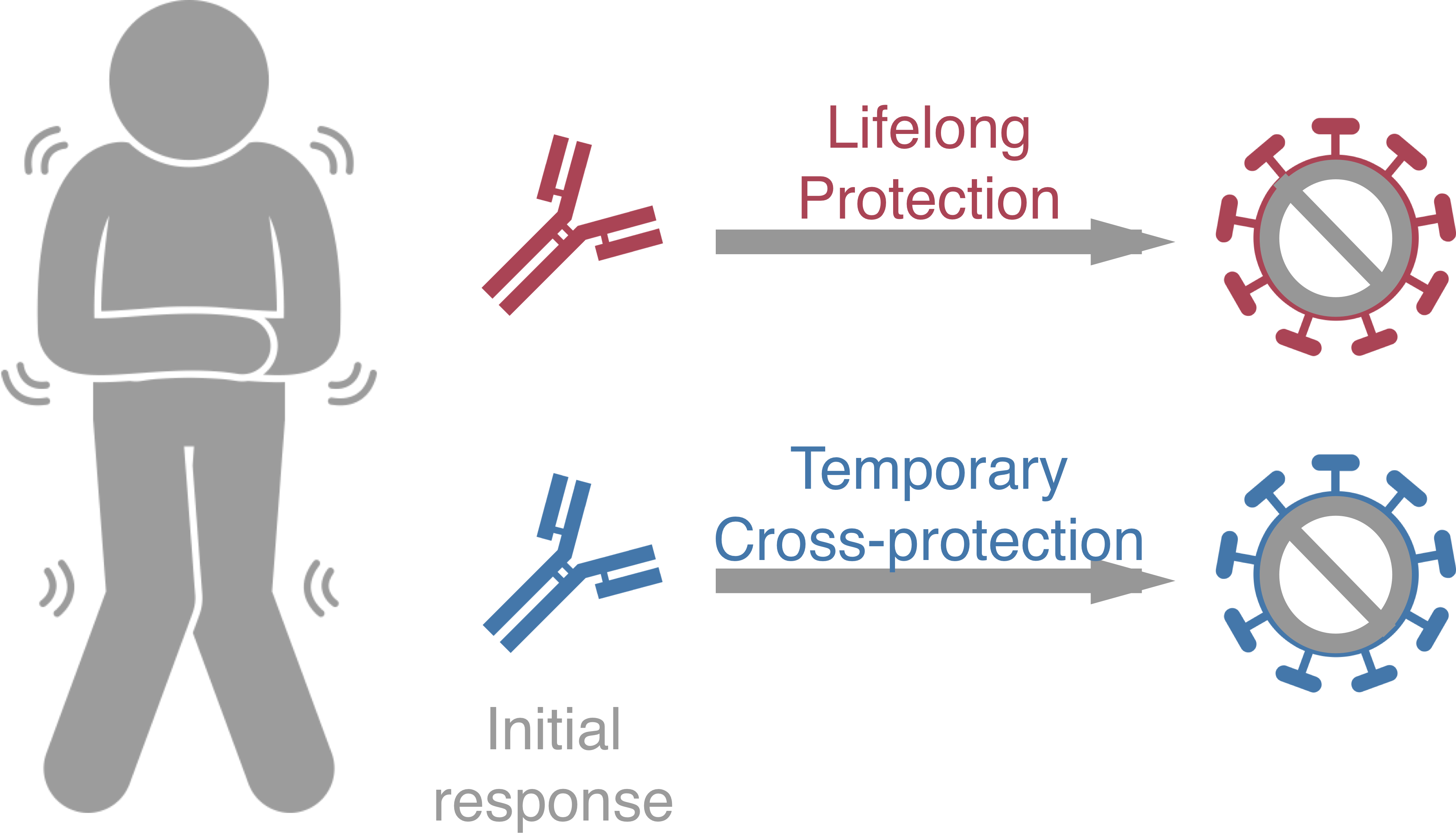
Original antigenic sin drives
dengue case outcomes
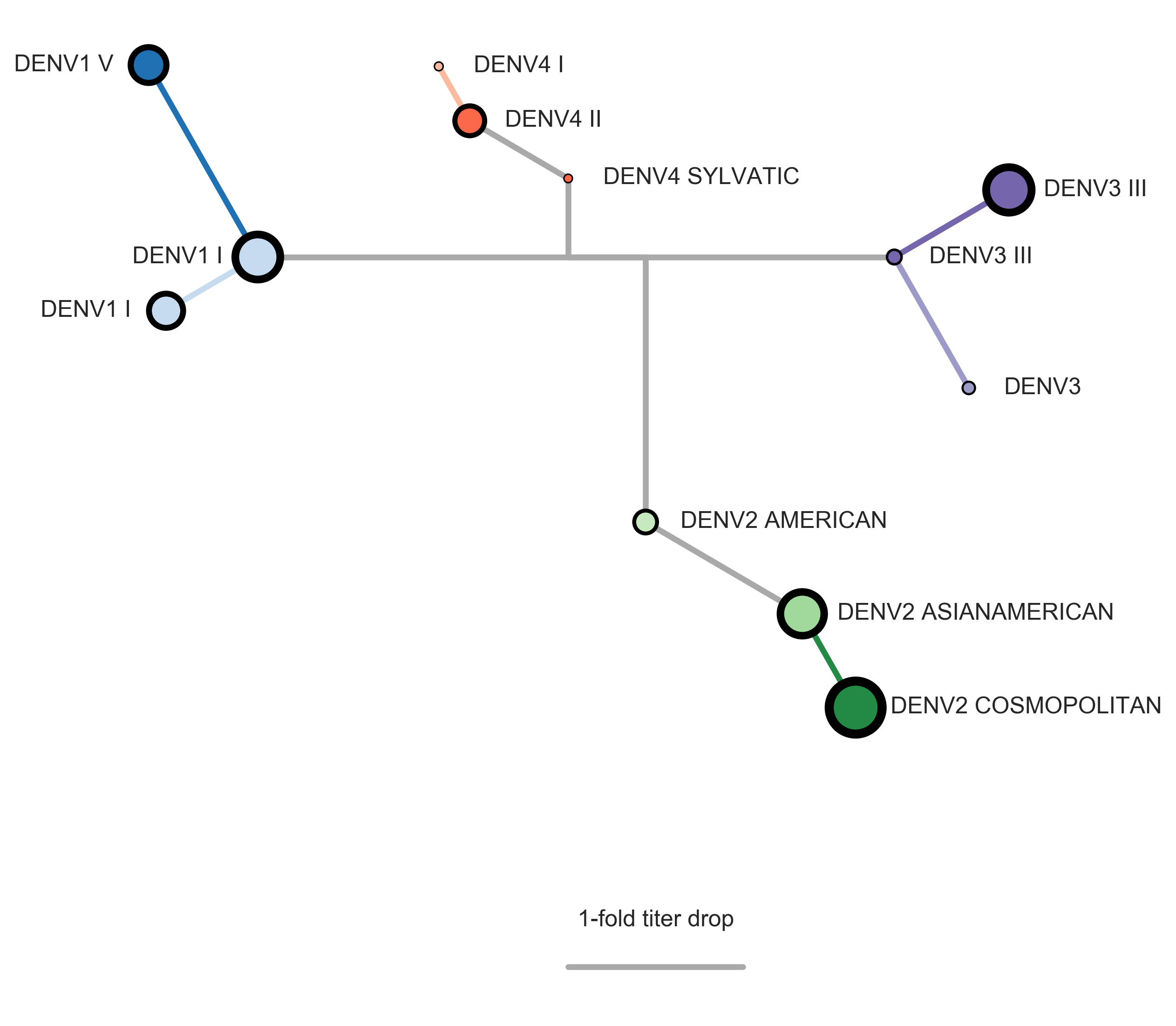
DENV antigenic relationships
are poorly understood
Serotypes are genetically distinct
Serotypes are antigenically distinct
Clades are genetically distinct
Are clades antigenically distinct?
How does
dengue evolve
antigenically?
Models of antigenic evolution
Interserotype hypothesis
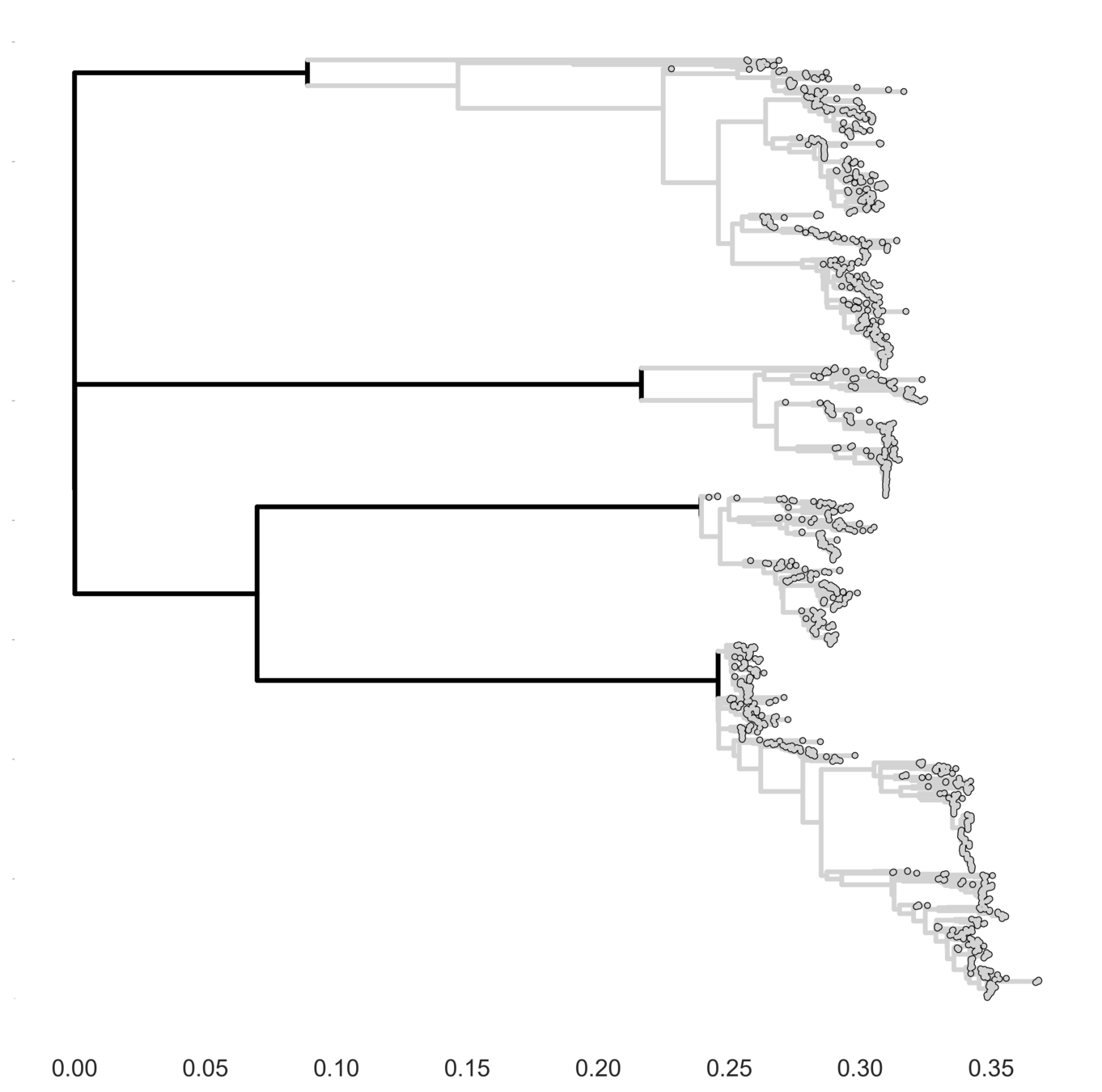
Full tree hypothesis
Dataset
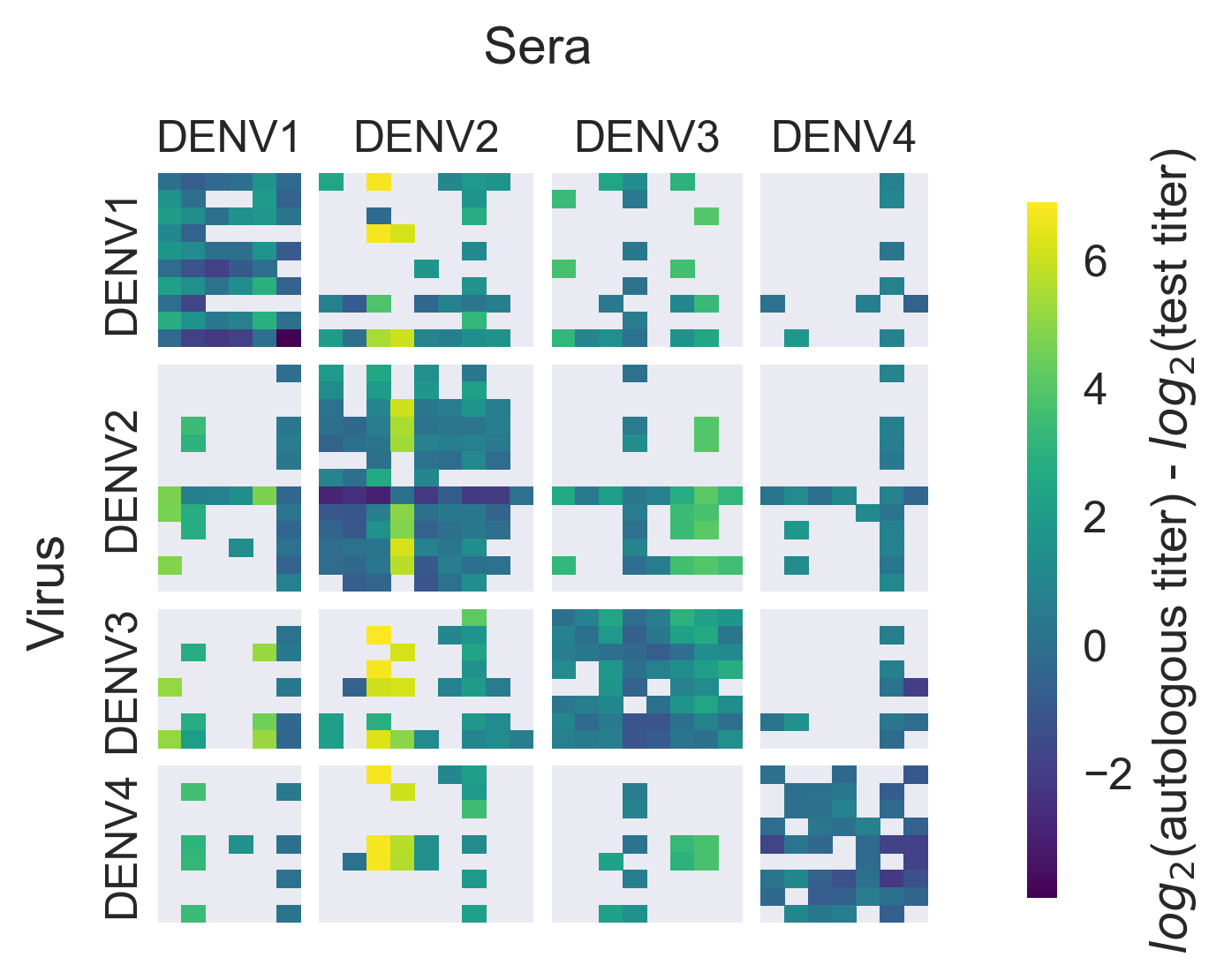
PRNT50 titers from monovalent vaccine trials + nonhuman primates
Within-serotype variation significantly contributes to dengue antigenic phenotypes
Interserotype model
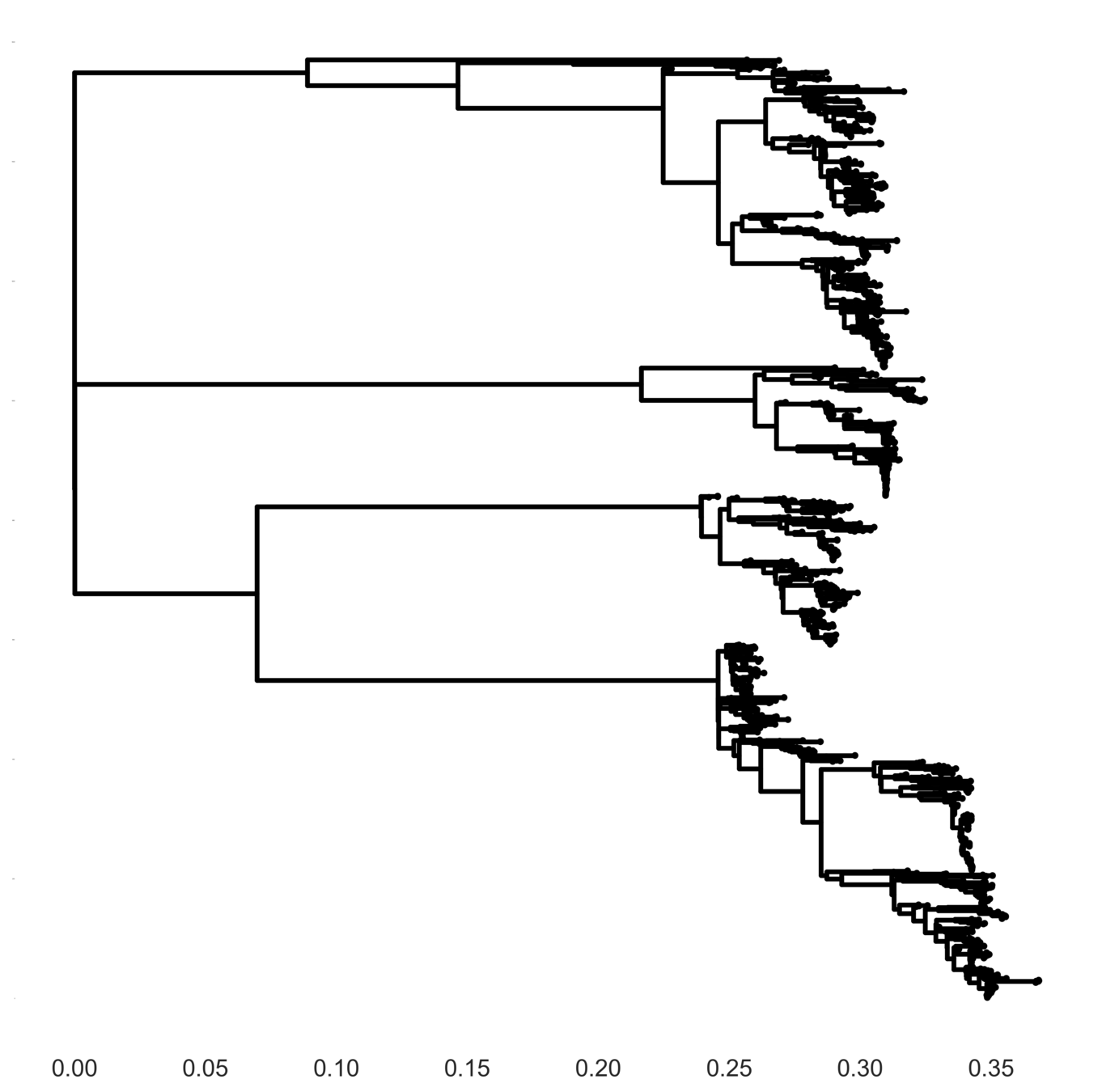
Full tree model
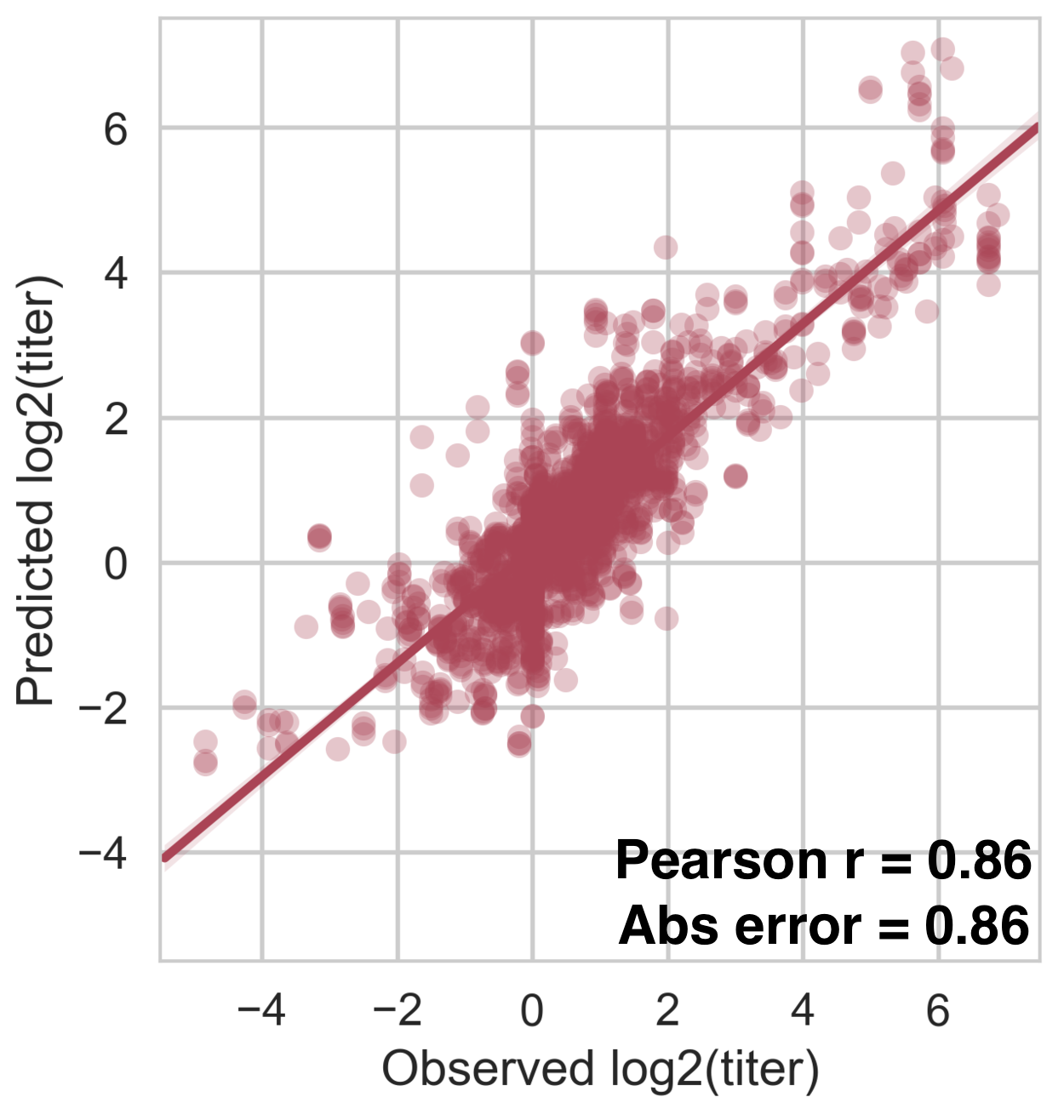
Each serotype of dengue contains multiple
distinct antigenic phenotypes
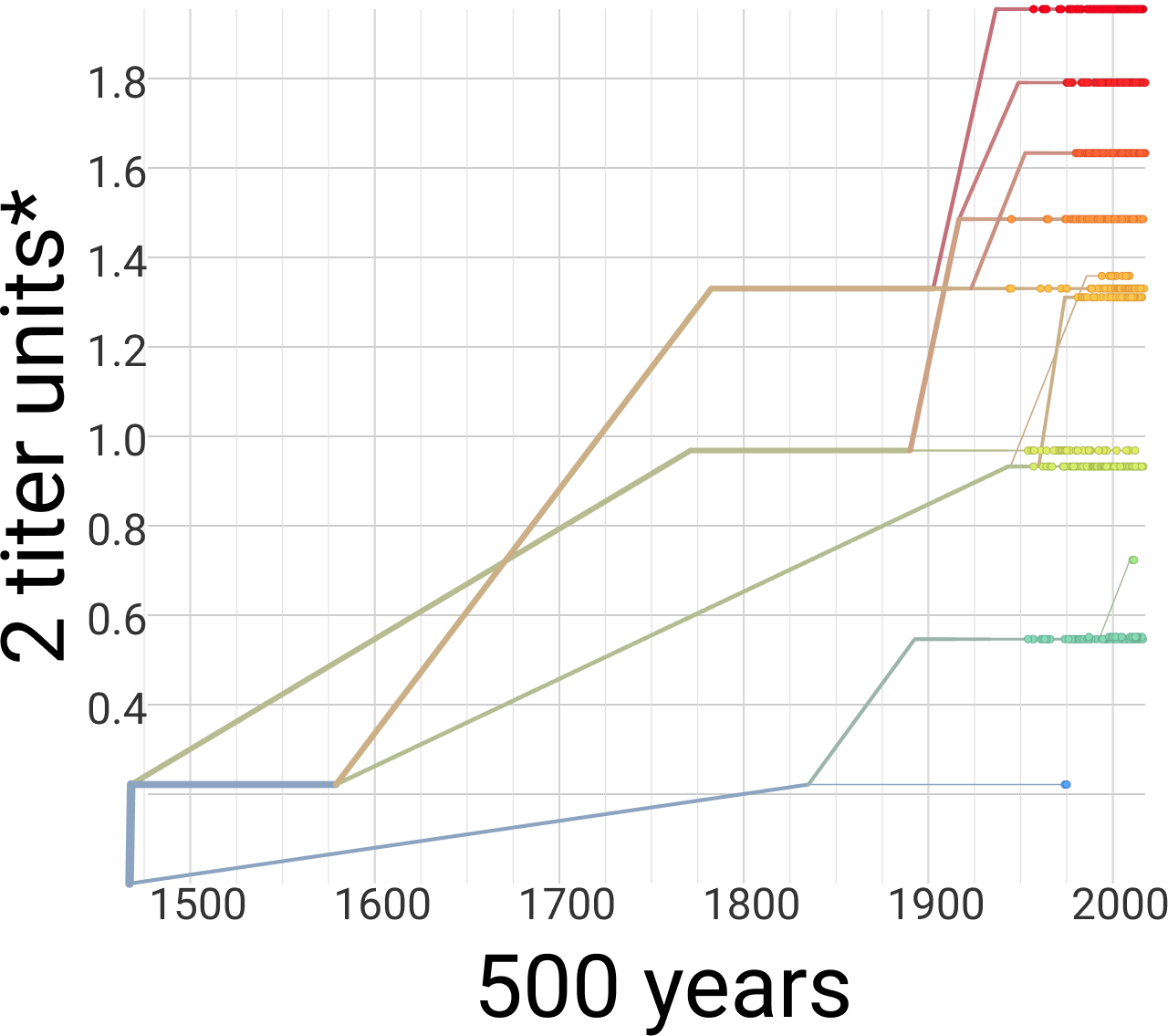
Dengue antigenic evolution is ongoing but slow
Dengue
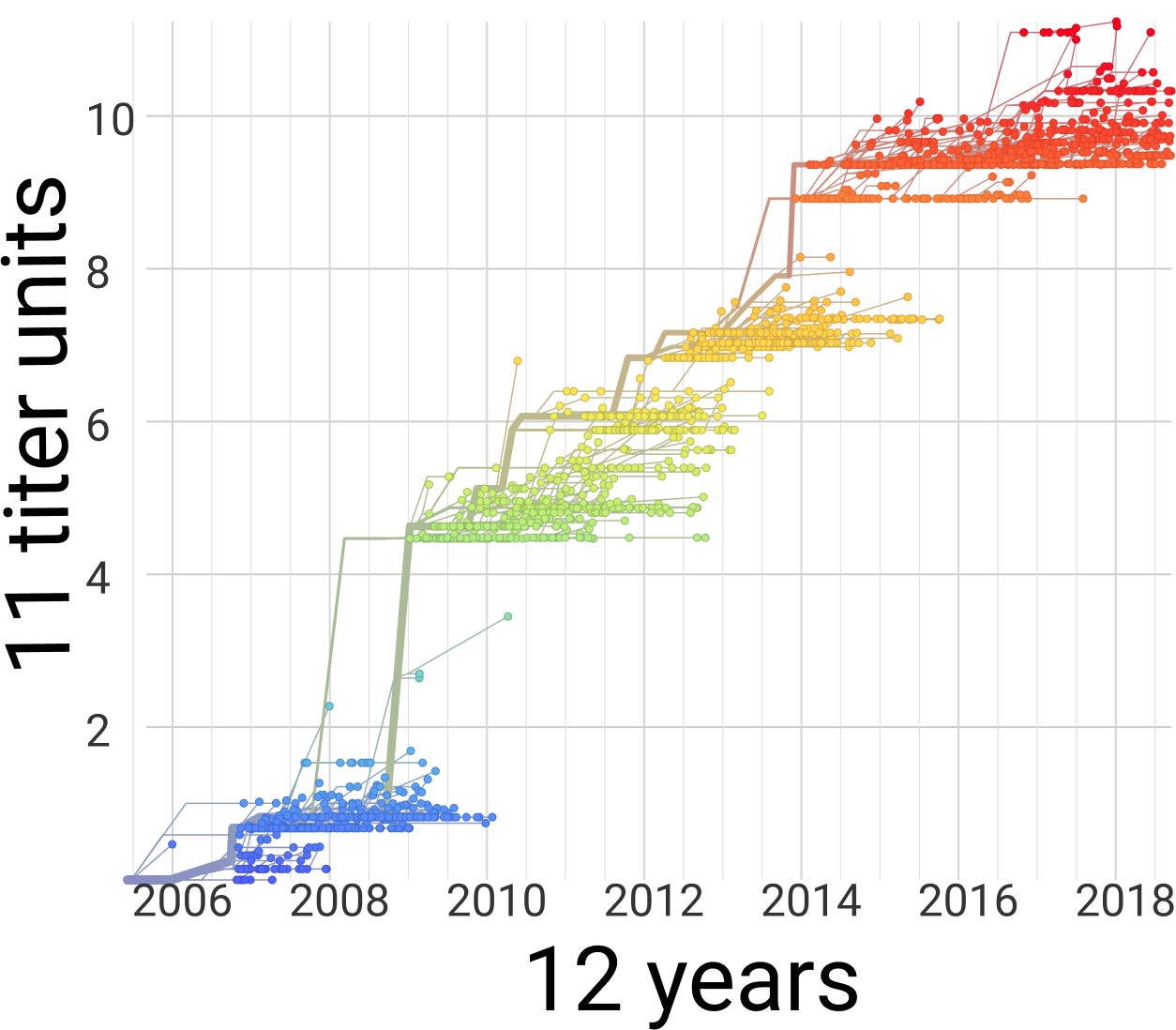
Flu (H3N2)
Does antigenic diversity impact dengue population dynamics?
Serotypes cycle through populations
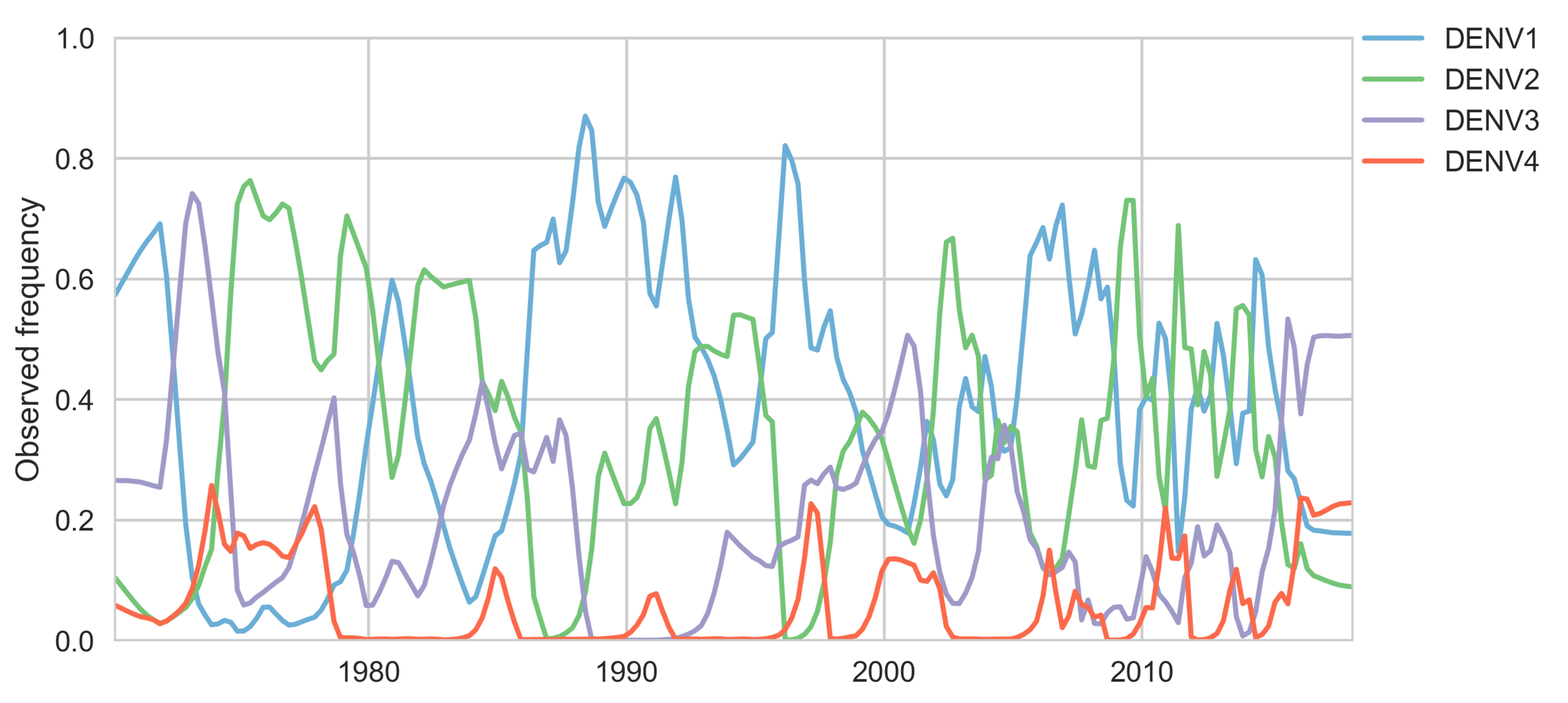
Genotypes cycle through populations
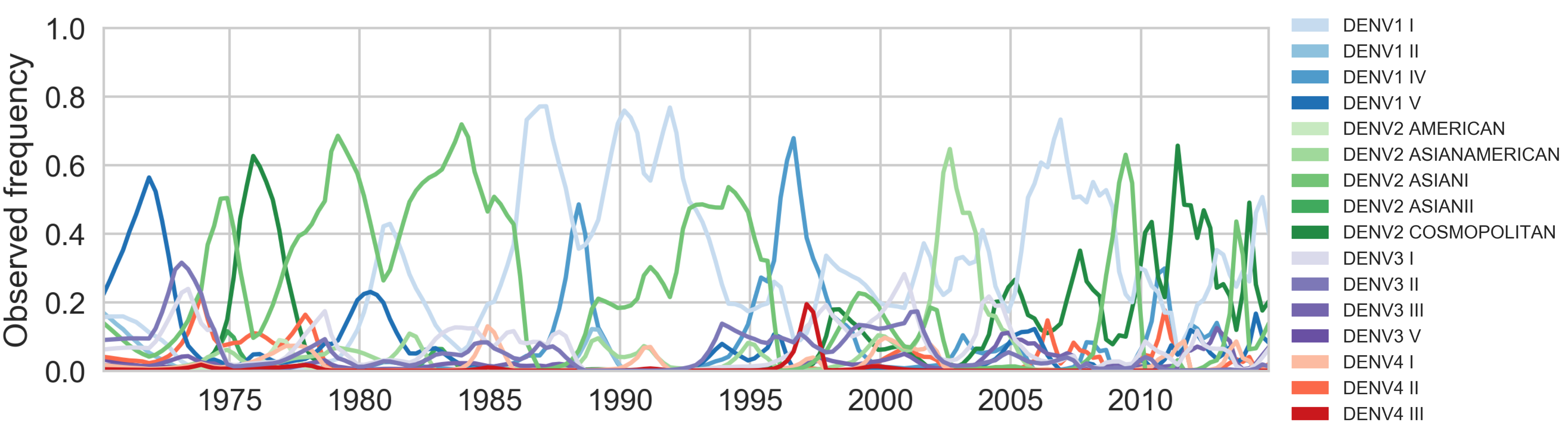
Predict clade growth based on fitness (as before)
Clade frequencies $X$ derive from the clade fitness $f_i$, such that
$$\hat{X}_i(t+\Delta t) = X_i(t) \, \mathrm{exp}(f_i \, \Delta t)$$
Estimate fitness as frequency-weighted antigenic distance*
from recently circulating clades
* estimated from the interserotype or full tree antigenic model.
Fitness based on antigenic distance from
standing population immunity
Circulating clades:
Population immunity:
Population susceptibility:
Clade growth:
Serotype antigenic relationships
drive serotype dynamics
Serotype antigenic relationships
drive genotype dynamics
Interserotype model
Full tree model
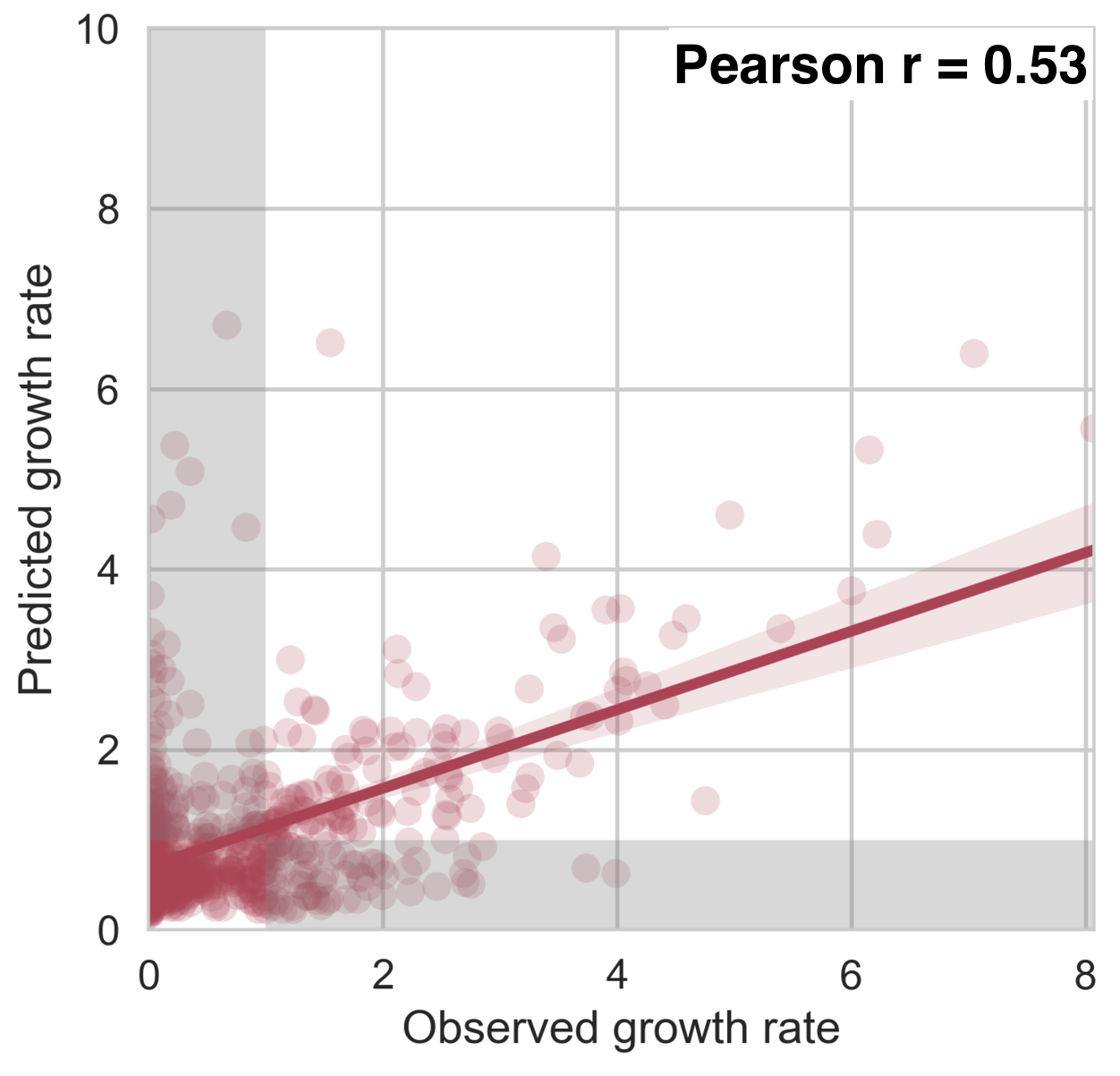
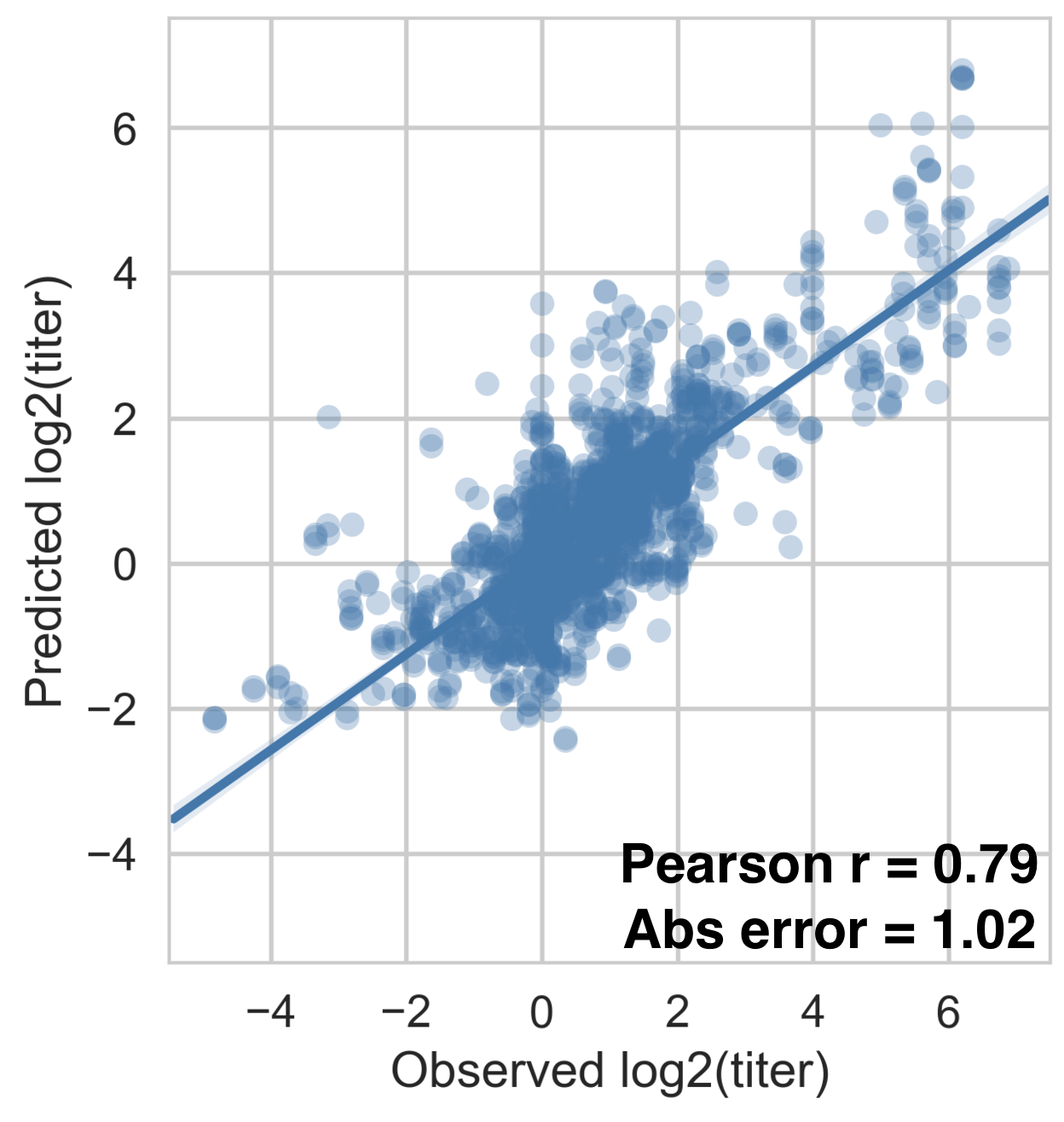
Antigenic fitness drives clade growth & decline
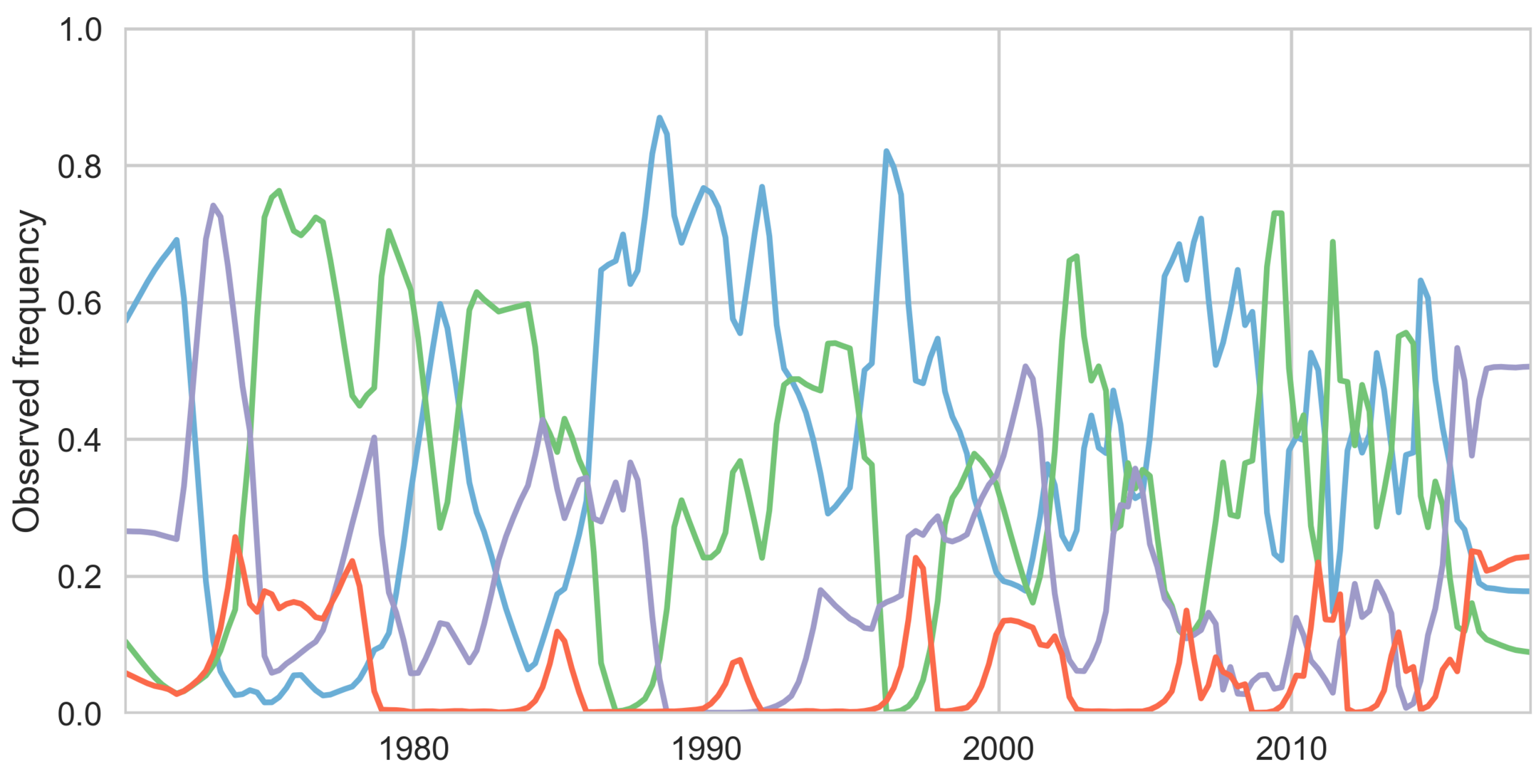

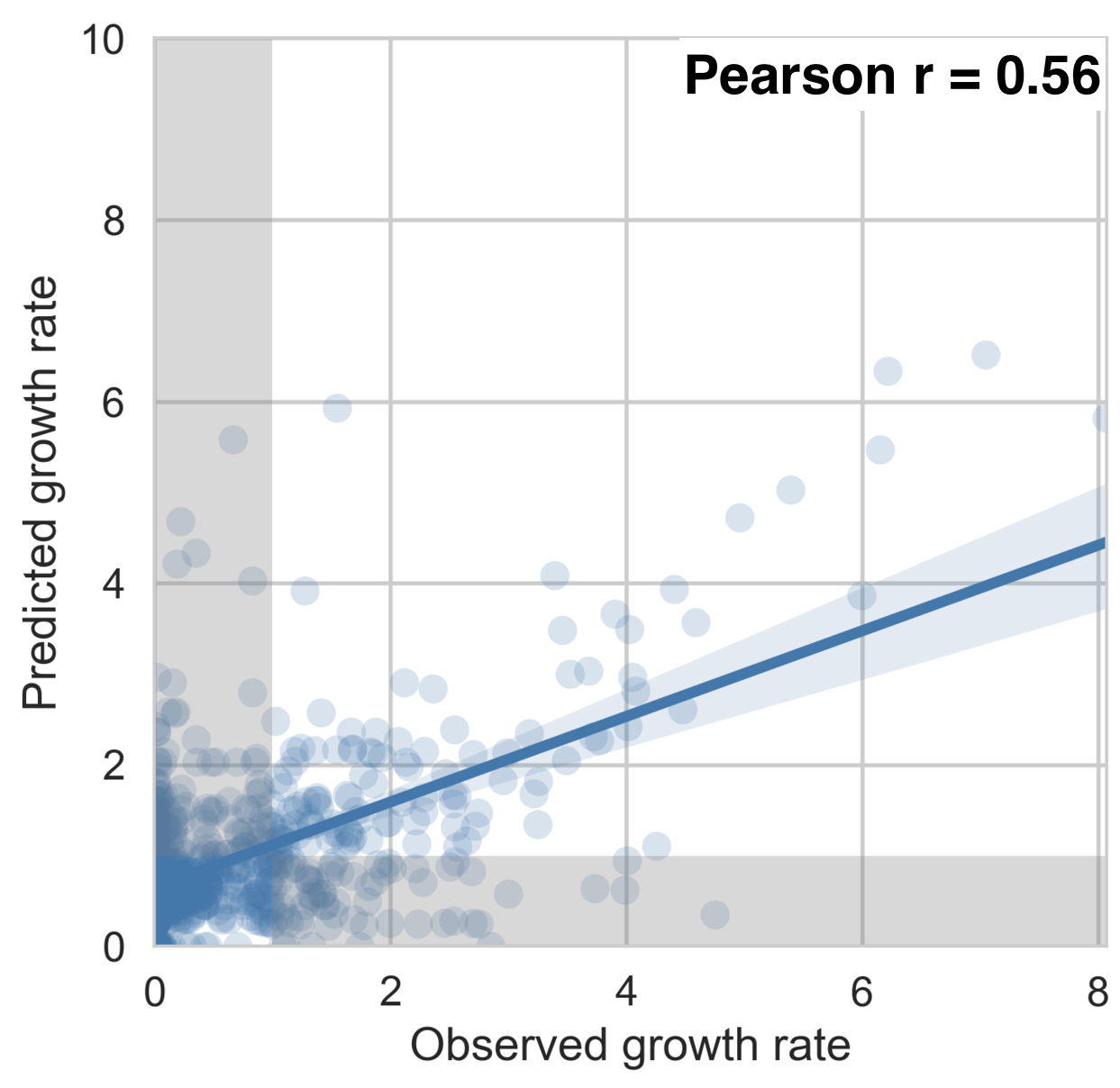
Dengue serotype flux
62% of variation explained
5 year windows
Flu clade turnover
53% of variation explained
1 year windows
Conclusions
Similar models of antigenic evolution are effective
for pathogens with very different evolutionary dynamics
Prospective modeling can reveal relative contributions to viral fitness
Acknowledgements
Bedford Lab:
 Alli Black,
Alli Black,
 Sidney Bell,
Sidney Bell,
 John Huddleston,
John Huddleston,
 Barney Potter,
Barney Potter,
 James Hadfield,
James Hadfield,
 Louise Moncla,
Louise Moncla,
 Tom Sibley,
Tom Sibley,
 Maya Lewinsohn
Maya Lewinsohn
Influenza: WHO Global Influenza Surveillance Network, GISAID, Richard Neher, John Huddleston, Barney Potter, James Hadfield, Rod Daniels, Boris Shraiman, Colin Russell, Andrew Rambaut, Dave Wentworth, Becky Garten, Jackie Katz, Marta Łuksza, Michael Lässig, Richard Reeve
Dengue: Leah Katzelnick, Molly O'hainle, Richard Neher, Paul Edlefsen, Michal Juraska
