Influenza evolutionary dynamics and global circulation patterns
Trevor Bedford (@trvrb)
23 Apr 2018
HIV Dynamics and Evolution
Leavenworth, WA
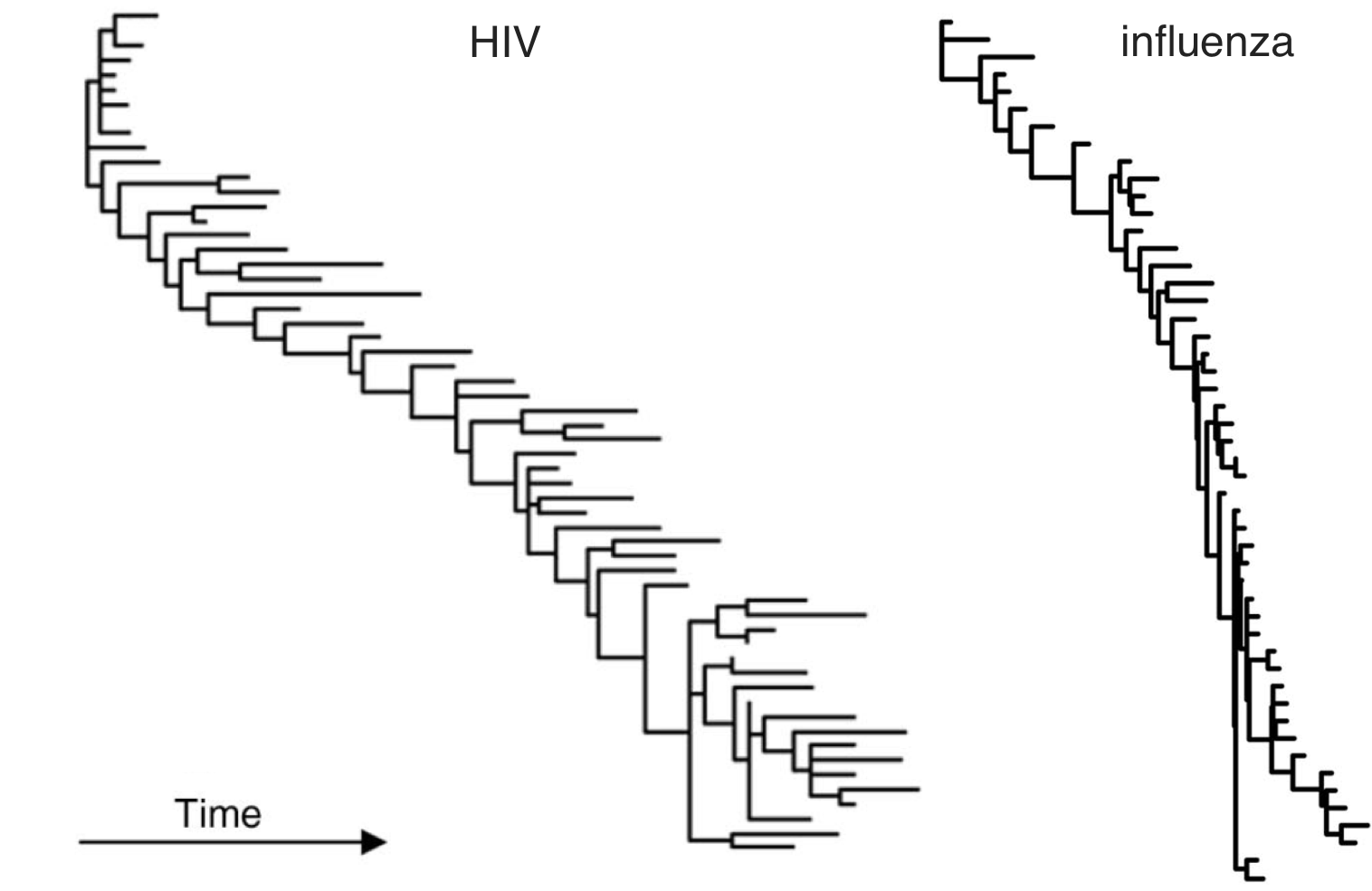
HIV vs influenza
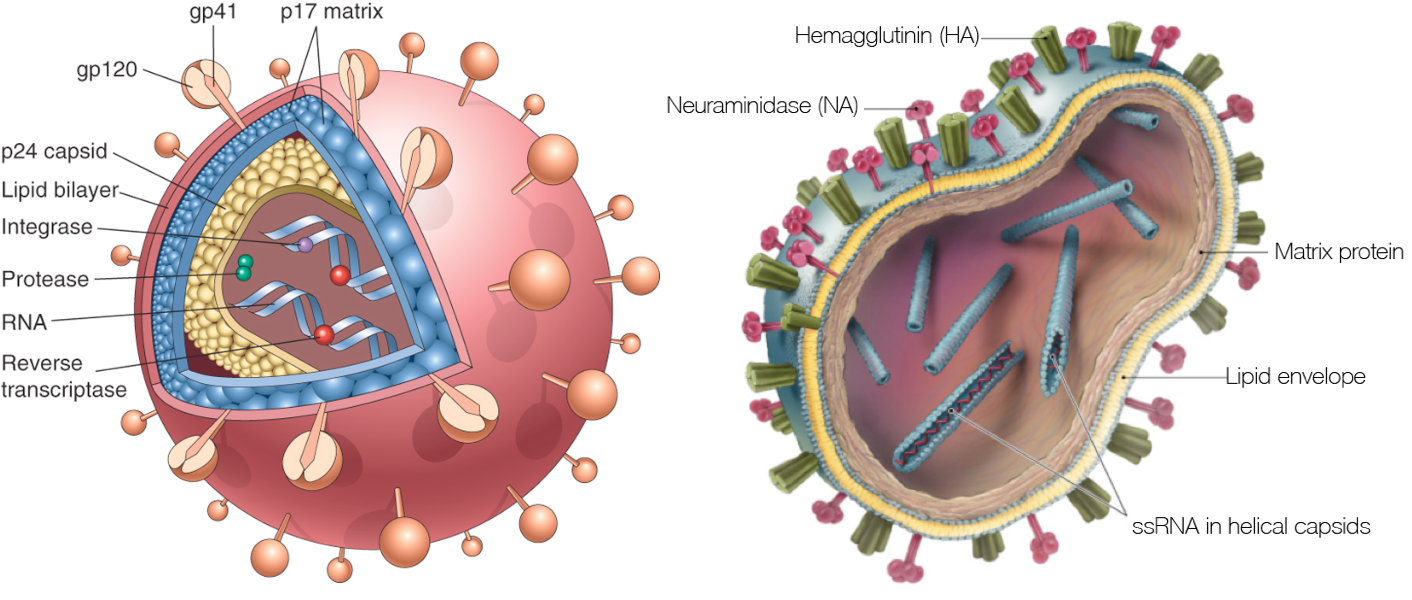
HIV within-host phylogeny vs influenza between-host phylogeny
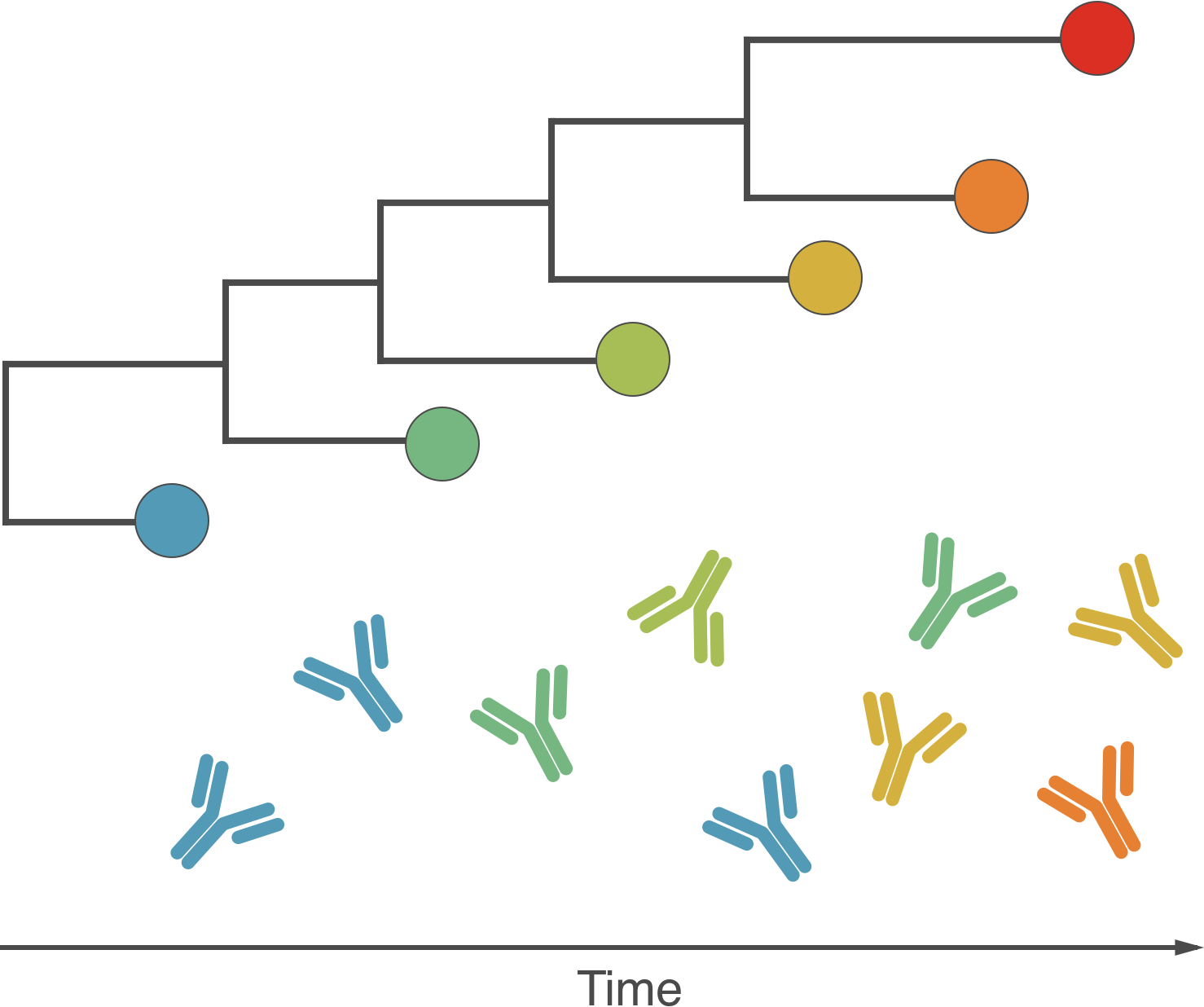
Fundamentally driven by the same process
Antigenic drift driven by acquired immunity
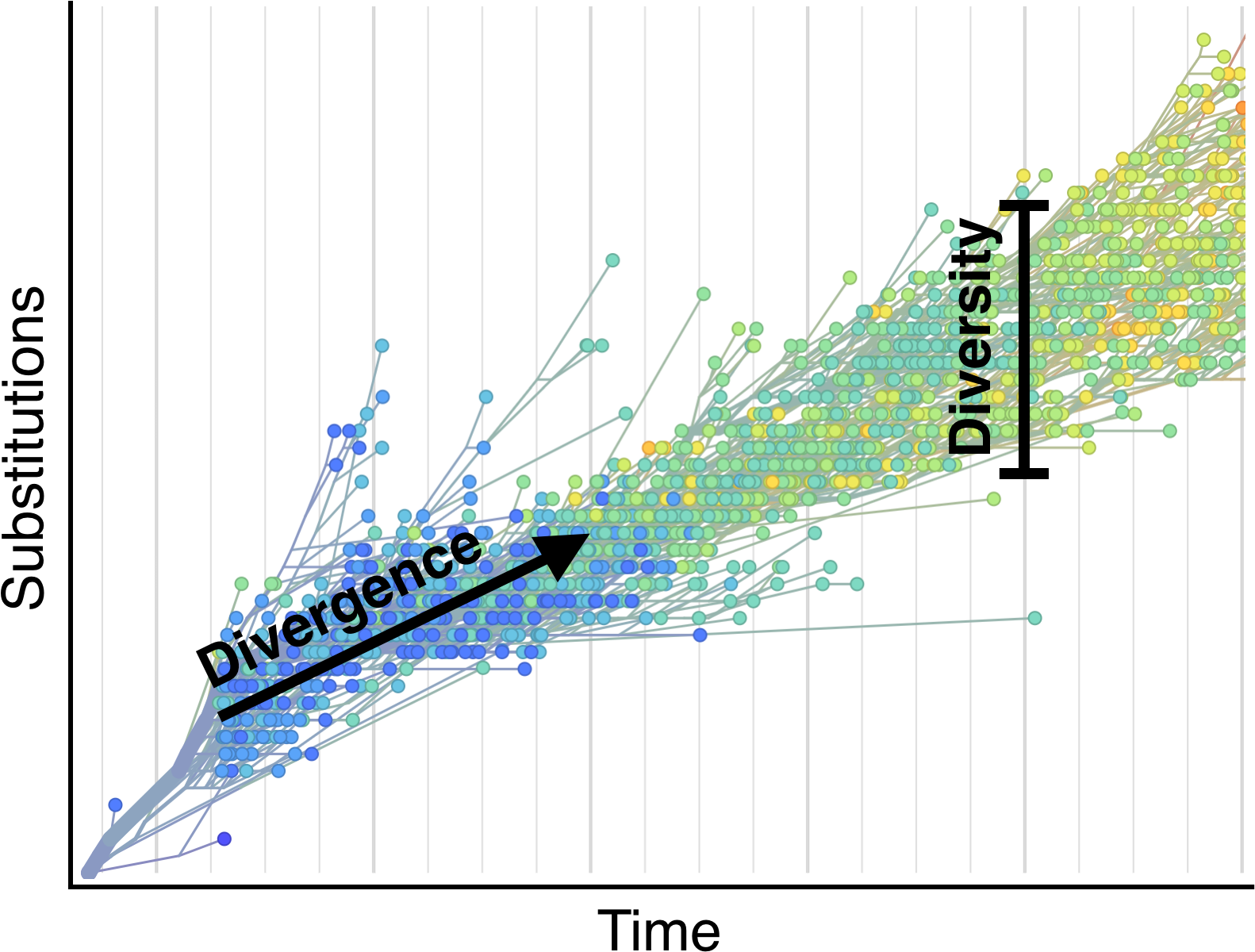
Selection visible in diversity and divergence
Systematic divergence at gp120 through time
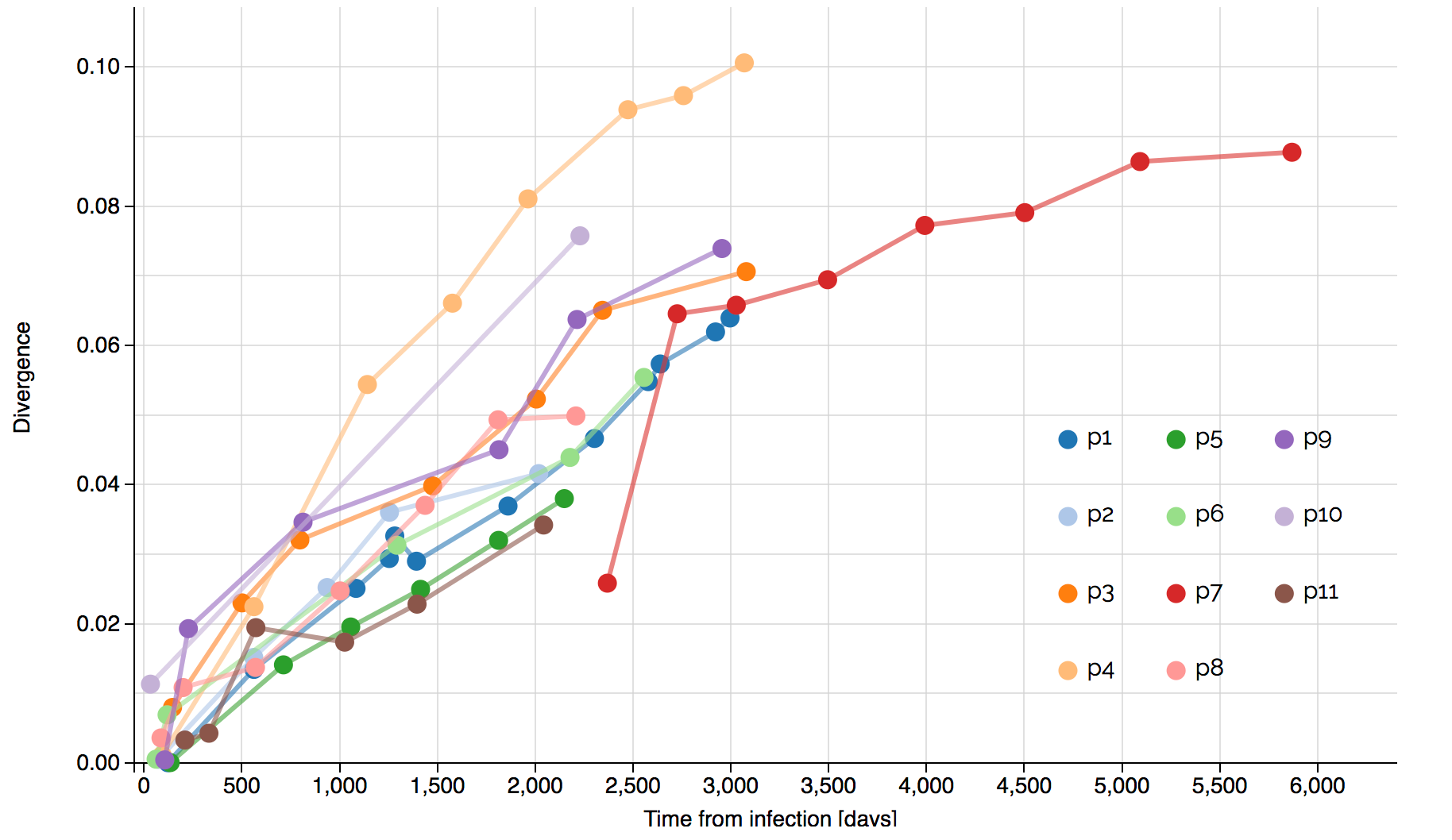
Within-host HIV shows adaptive evolution in envelope and purifying selection in enzymes
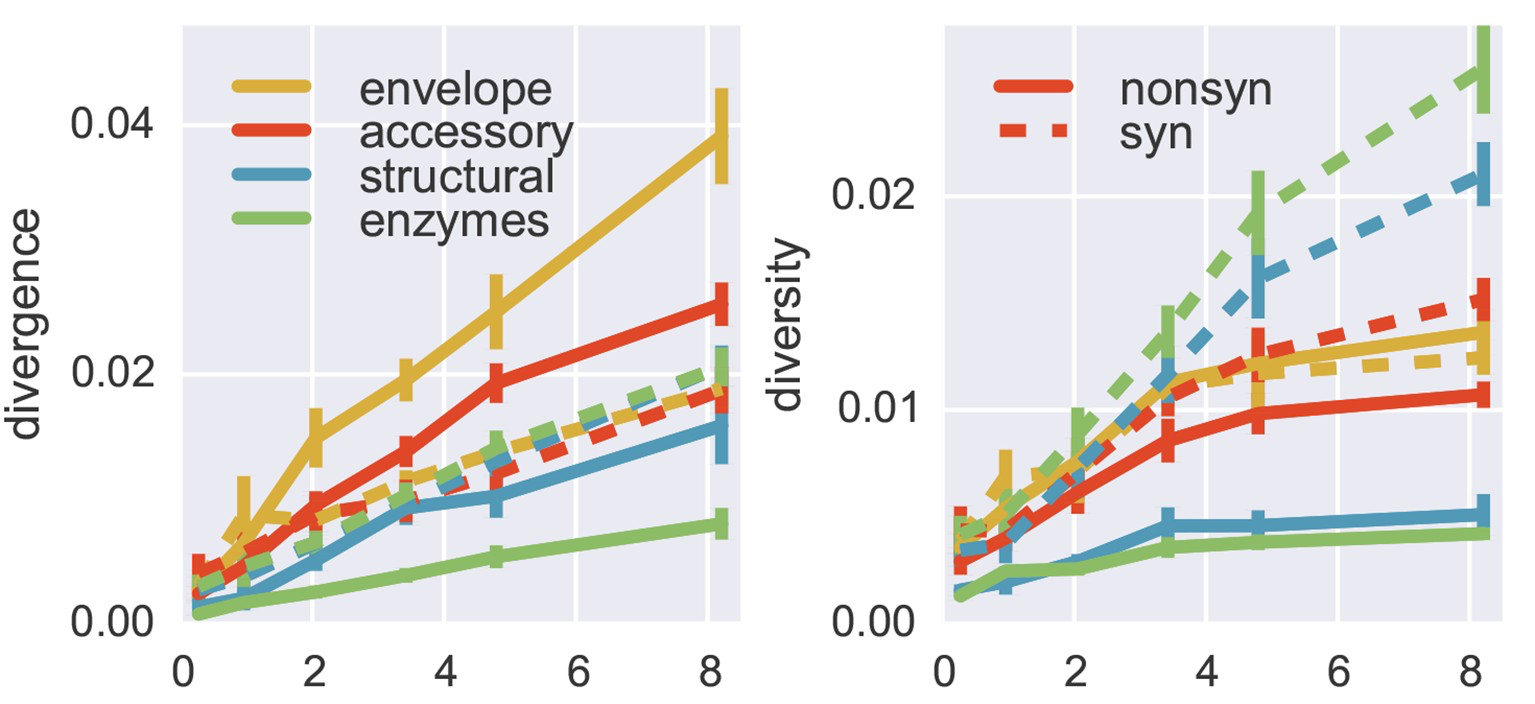
Between-host influenza shows adaptive evolution in surface proteins and purifying selection in internal proteins
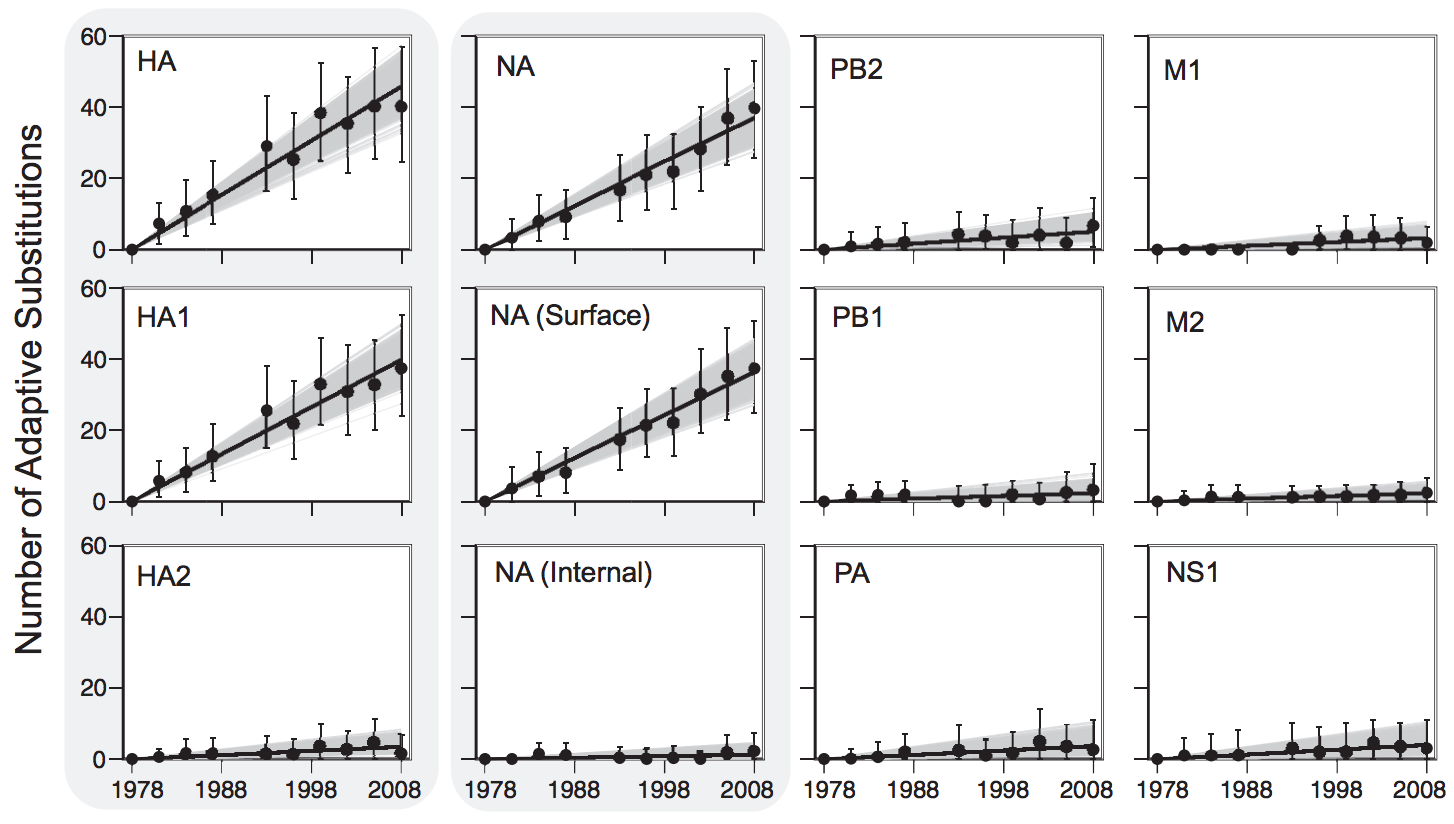
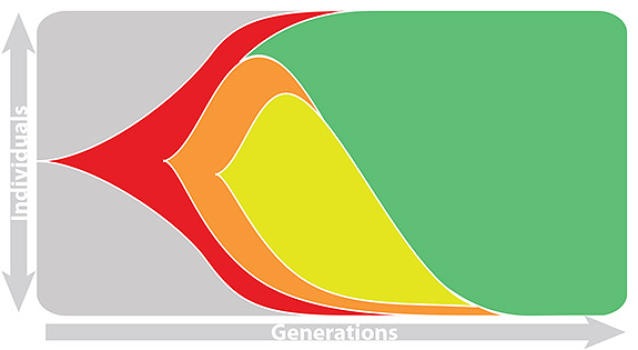
Influenza evolution
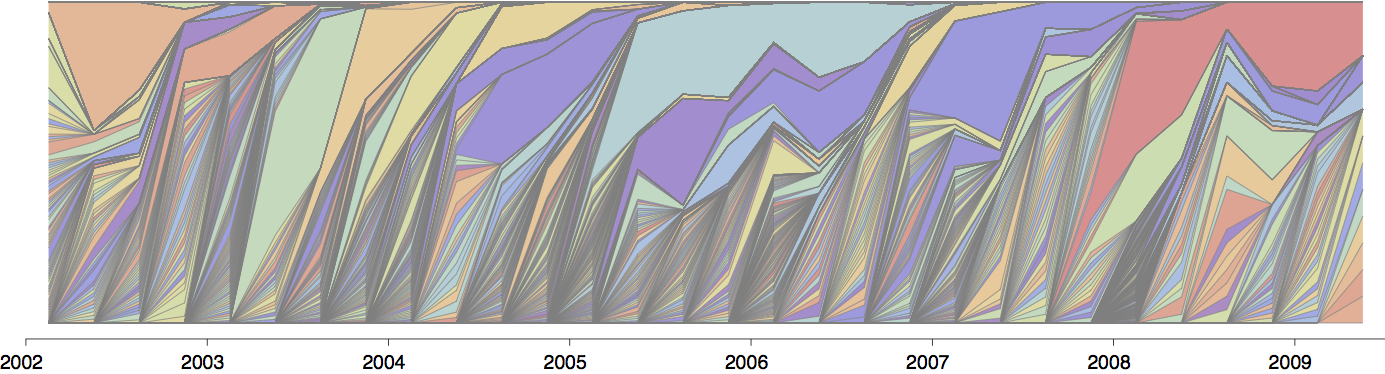
Population turnover is extremely rapid
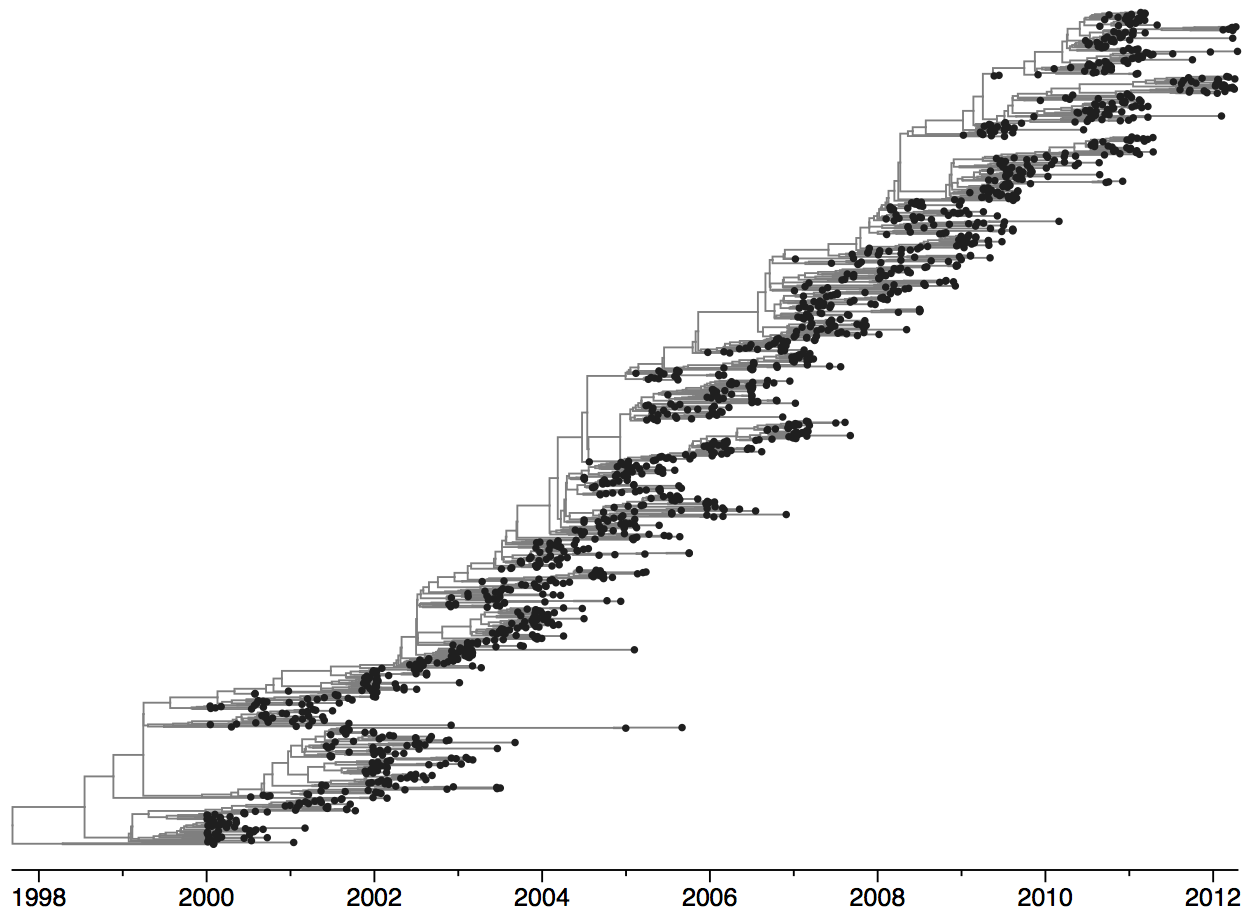
Clades emerge, die out and take over
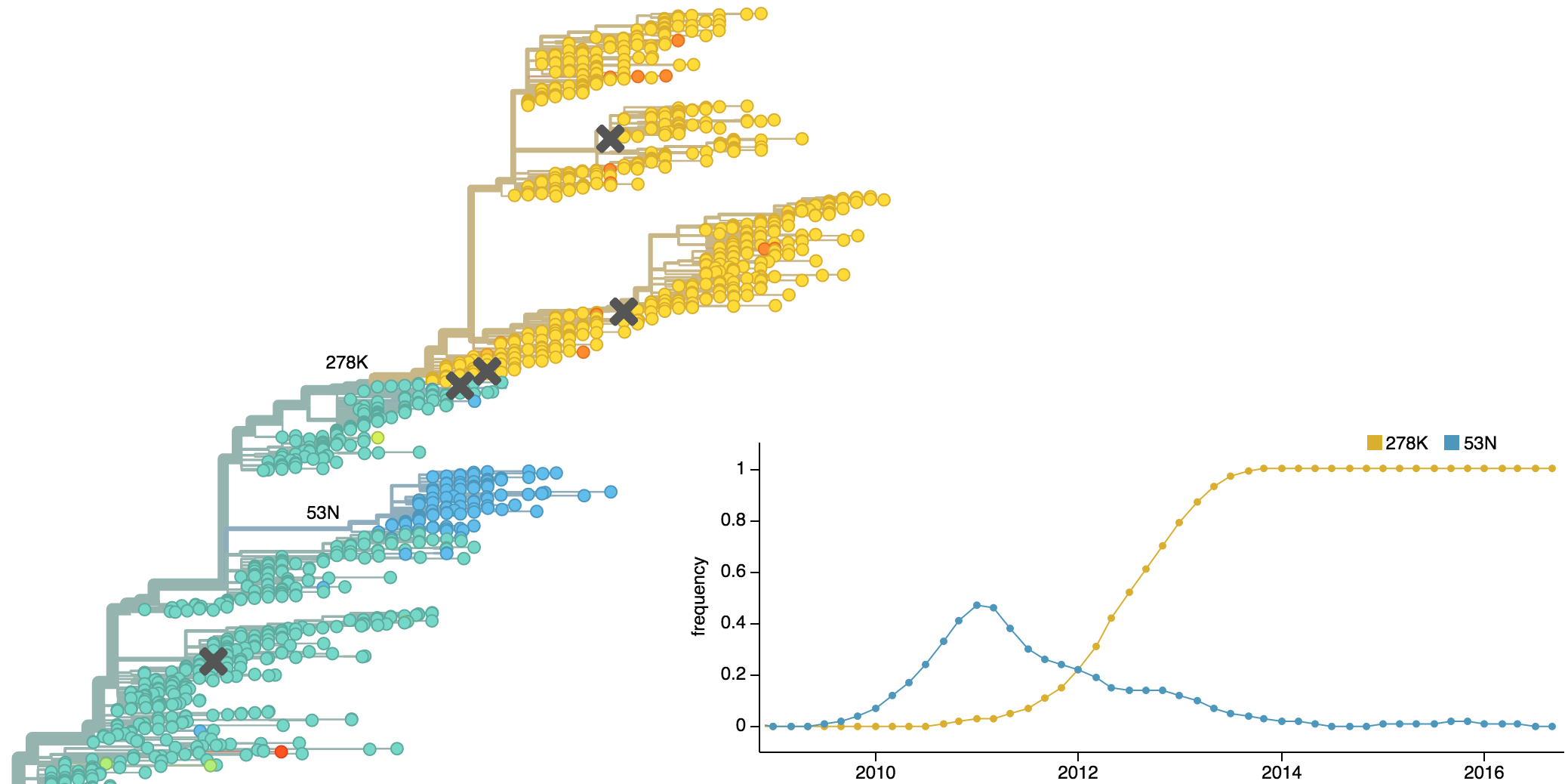
Clades show rapid turnover

Dynamics driven by antigenic drift
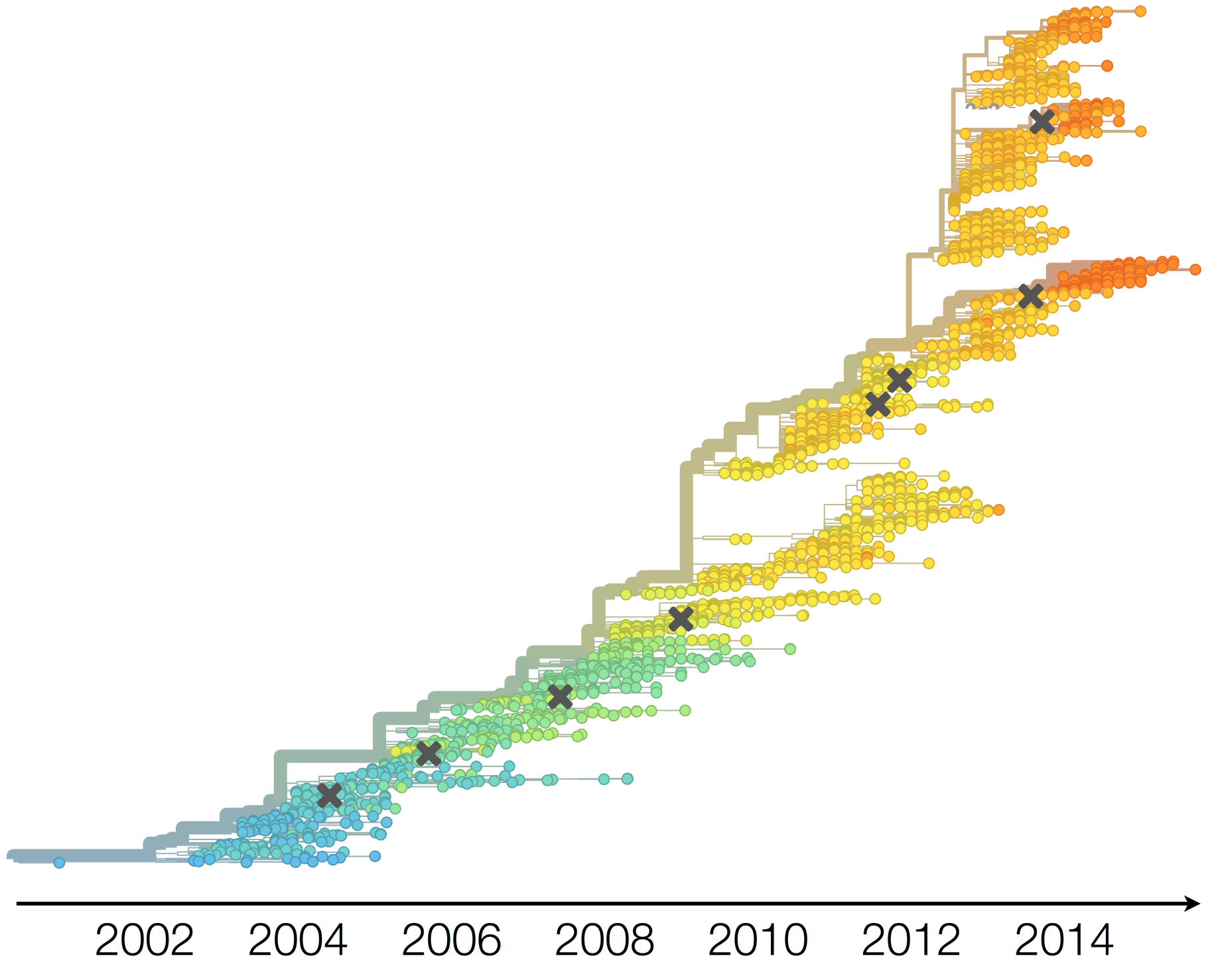
Drift necessitates vaccine updates
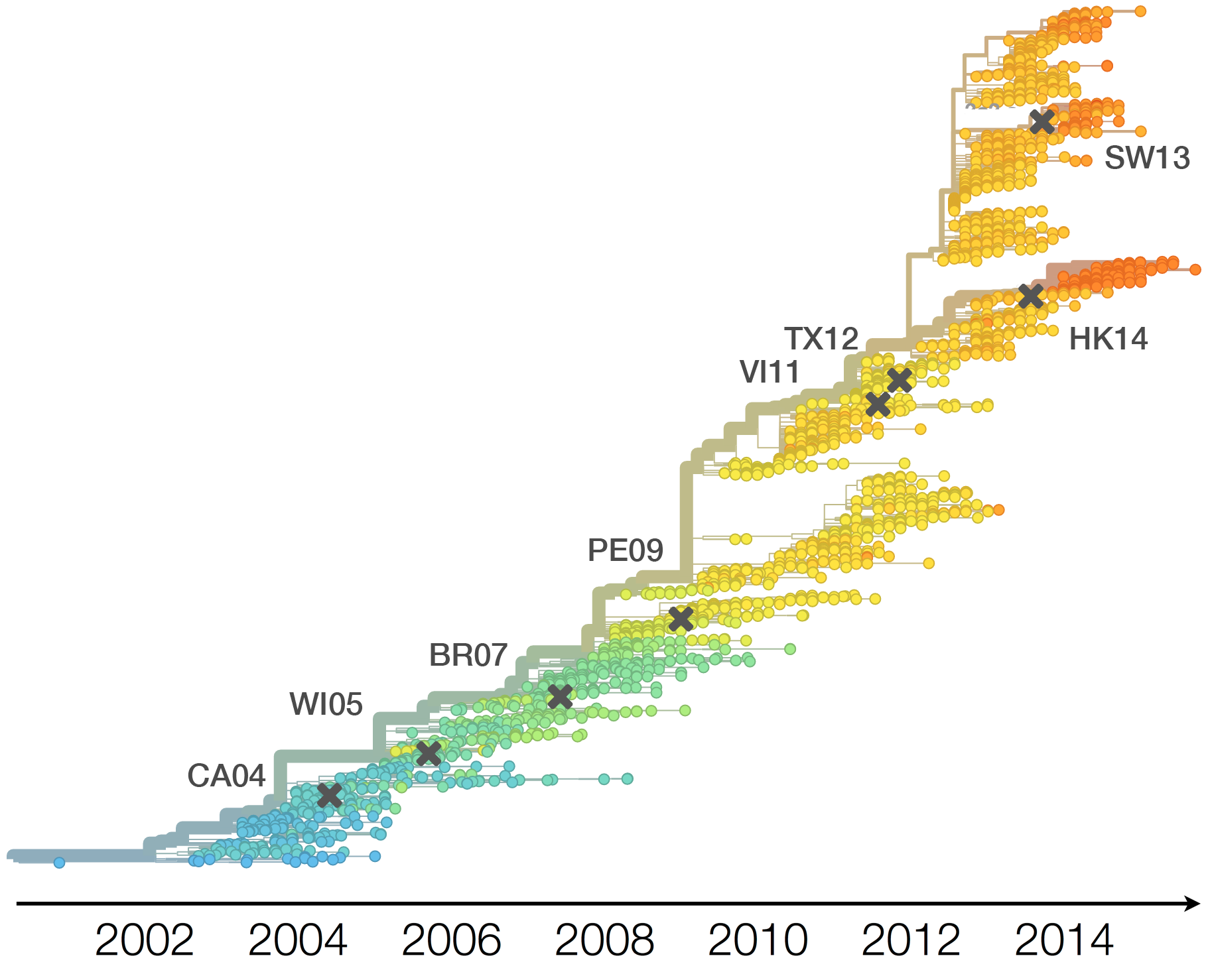
H3N2 vaccine updates occur every ~2 years
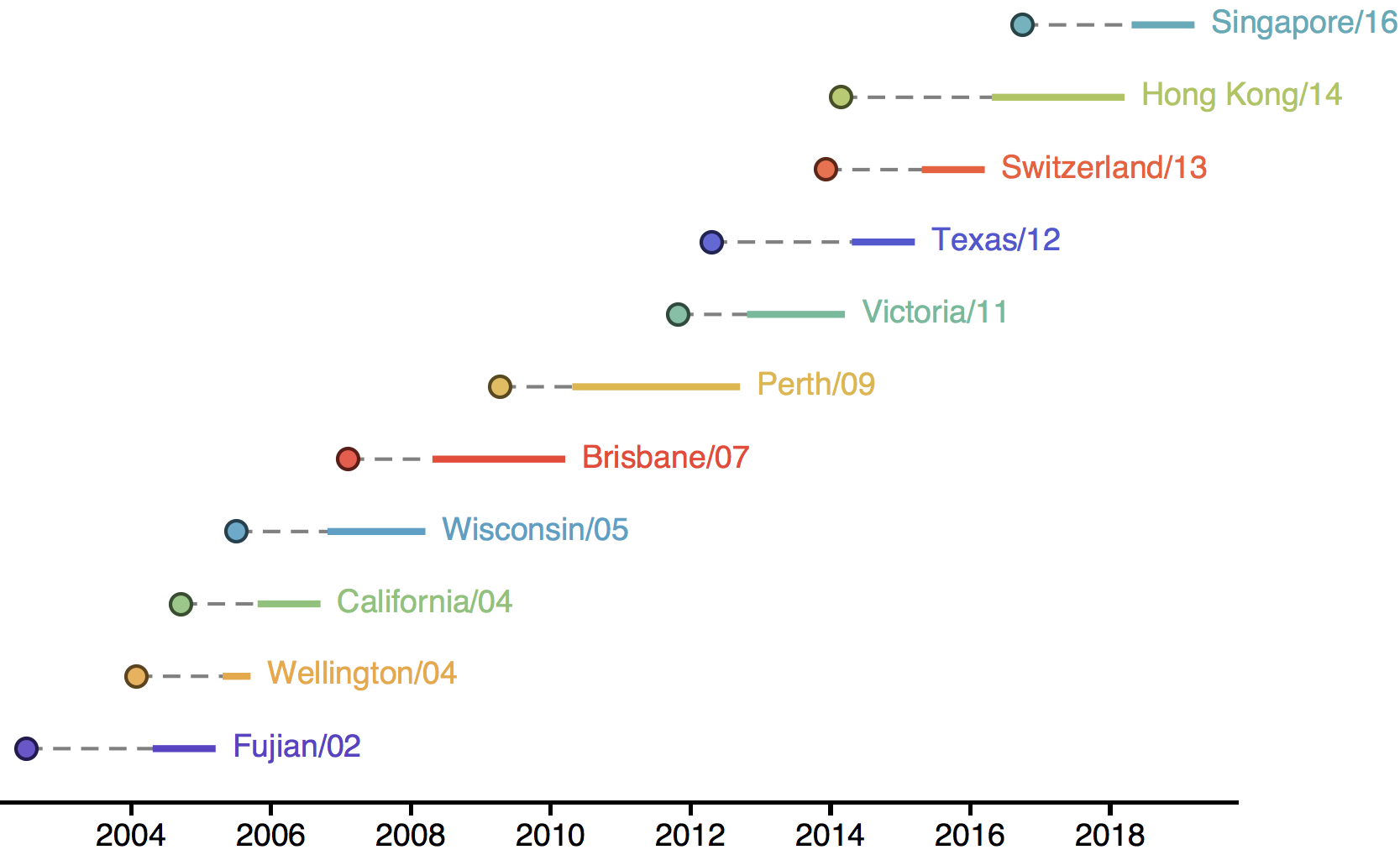
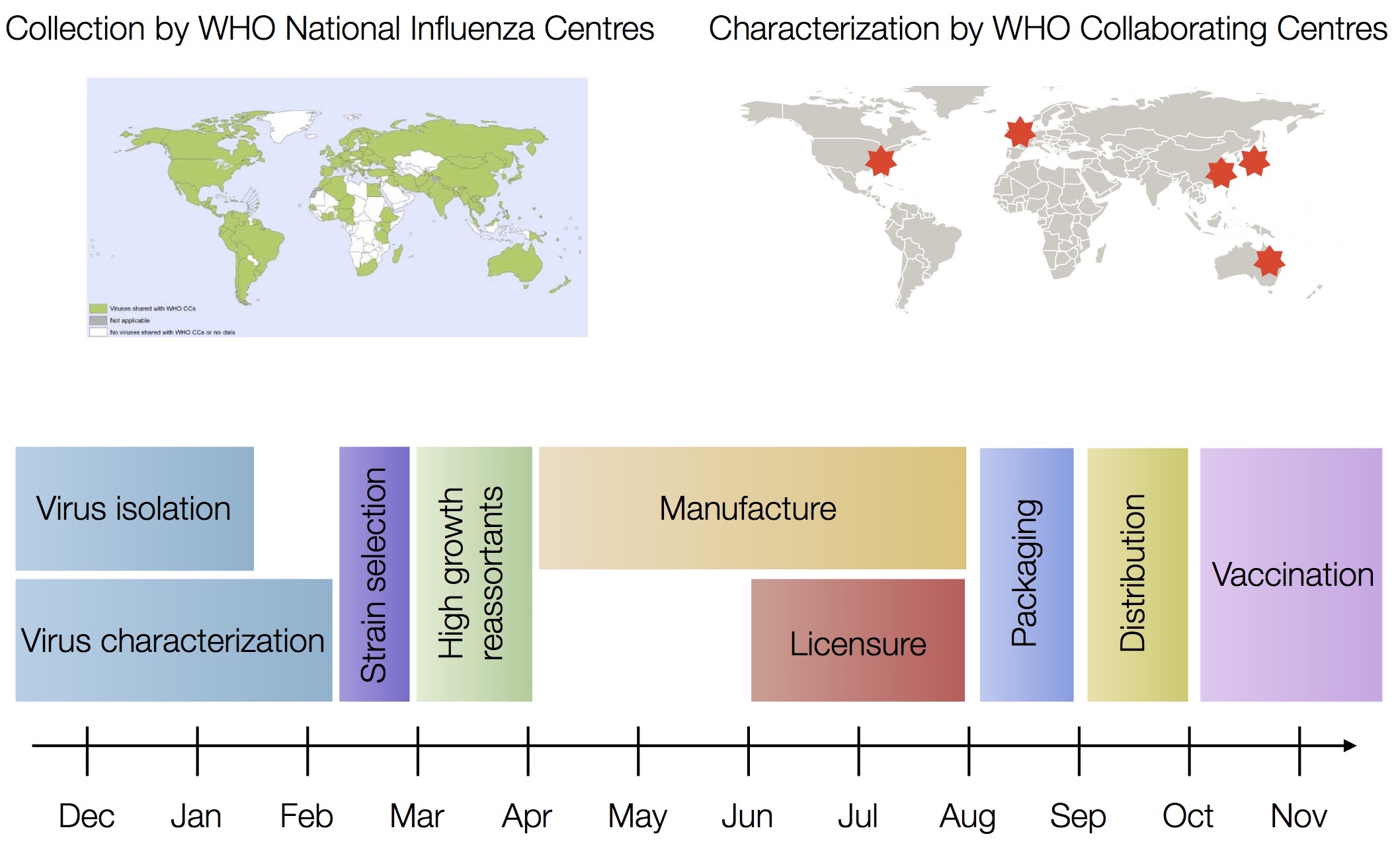
Vaccine strain selection by WHO
Problem of applied evolutionary biology
Every paper in the field...
- "These observations have implications for influenza surveillance and vaccine formulation" (Wolf et al 2006)
- "Our results have implications for the design of vaccines to combat rapidly mutating viral diseases" (Gupta et al 2006)
- "These results may have important implications for influenza vaccine and antiviral research" (Bhatt et al 2011)
- "Needless to say, these results have important implications for the updating of vaccines against influenza" (Zinder et al 2013)
Disconnect between evolutionary studies and information needed by WHO
- WHO needs specific advice, ie this strain is likely to take off, this strain is likely to die out
- Problems of generality and timeliness
Microbial evolution workshop at the Kavli Institute for Theoretical Physics
Decided to tackle this head on and build something that
- Charts behavior of specific strains
- Can be kept continually up to date
Nextflu
Project to provide a real-time view of the evolving influenza population
Nextflu
Project to provide a real-time view of the evolving influenza population
All in collaboration with Richard Neher

Nextflu pipeline
- Download all recent HA sequences from GISAID
- Filter to remove outliers
- Subsample across time and space
- Align sequences
- Build tree
- Estimate clade frequencies
- Infer antigenic phenotypes
- Export for visualization
Subsequently wrapped into larger efforts to do real-time analysis across pathogens with the Nextstrain project
with
![]() Richard Neher,
Richard Neher,
![]() James Hadfield,
James Hadfield,
![]() Colin Megill,
Colin Megill,
![]() Sidney Bell,
Sidney Bell,
![]() John Huddleston,
John Huddleston,
![]() Barney Potter,
Barney Potter,
![]() Charlton Callender,
Charlton Callender,
![]() Emma Hodcroft
Emma Hodcroft
Up-to-date analysis publicly available at:
nextstrain.org/flu
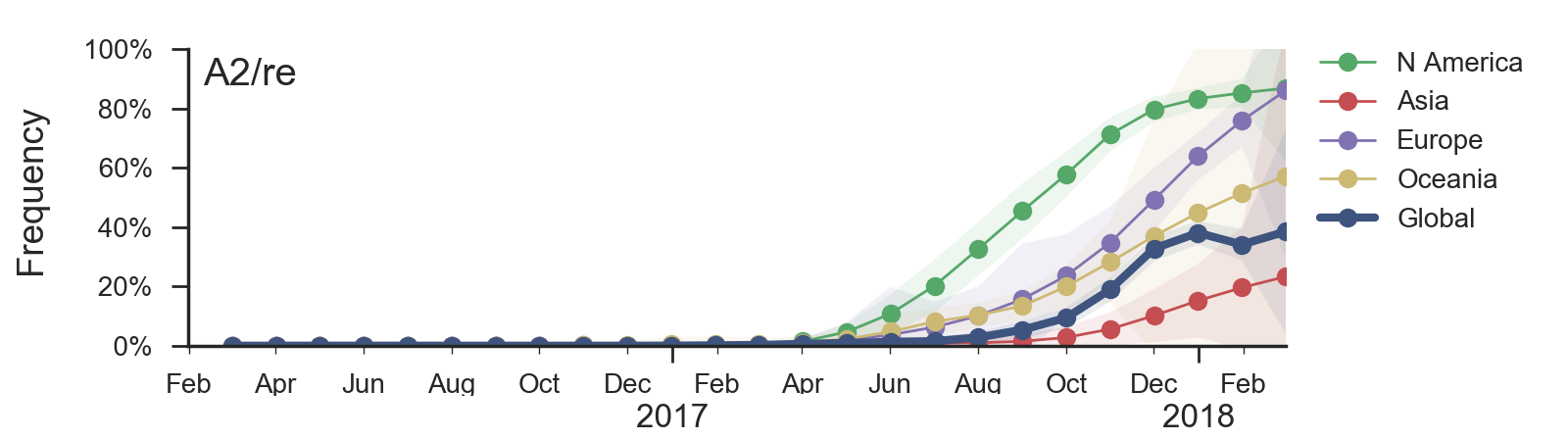
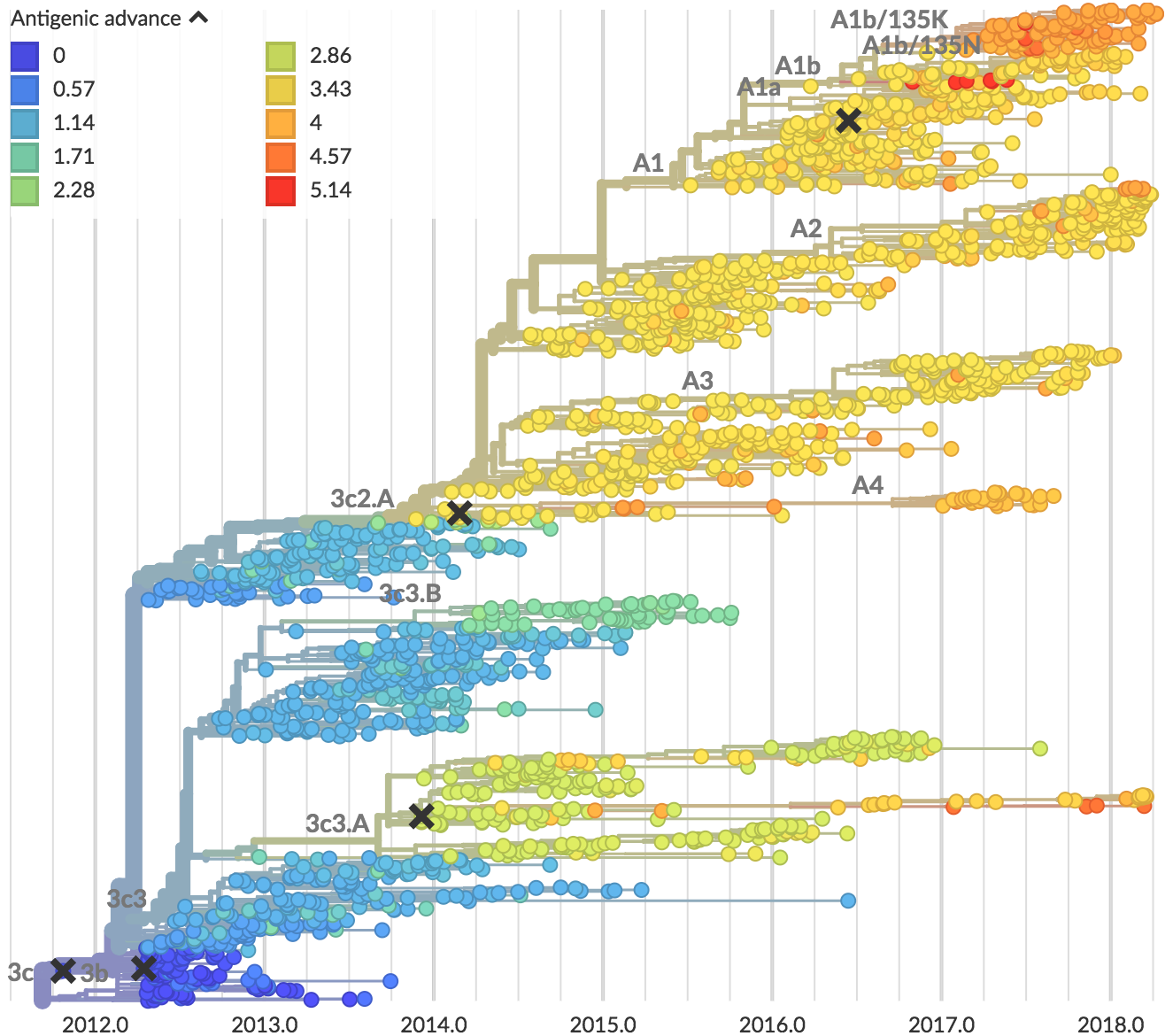
Current H3N2 diversity
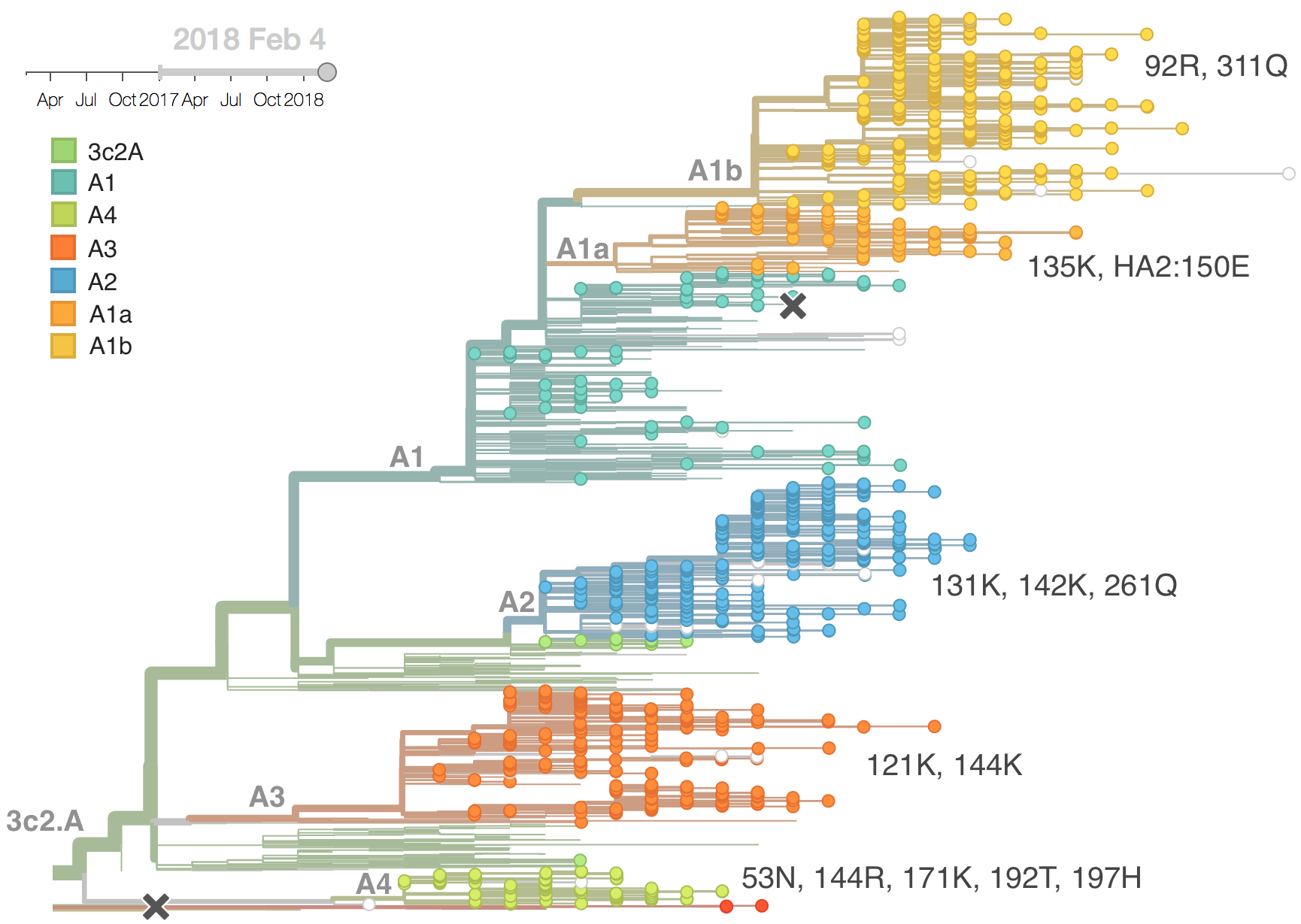
Current H3N2 diversity
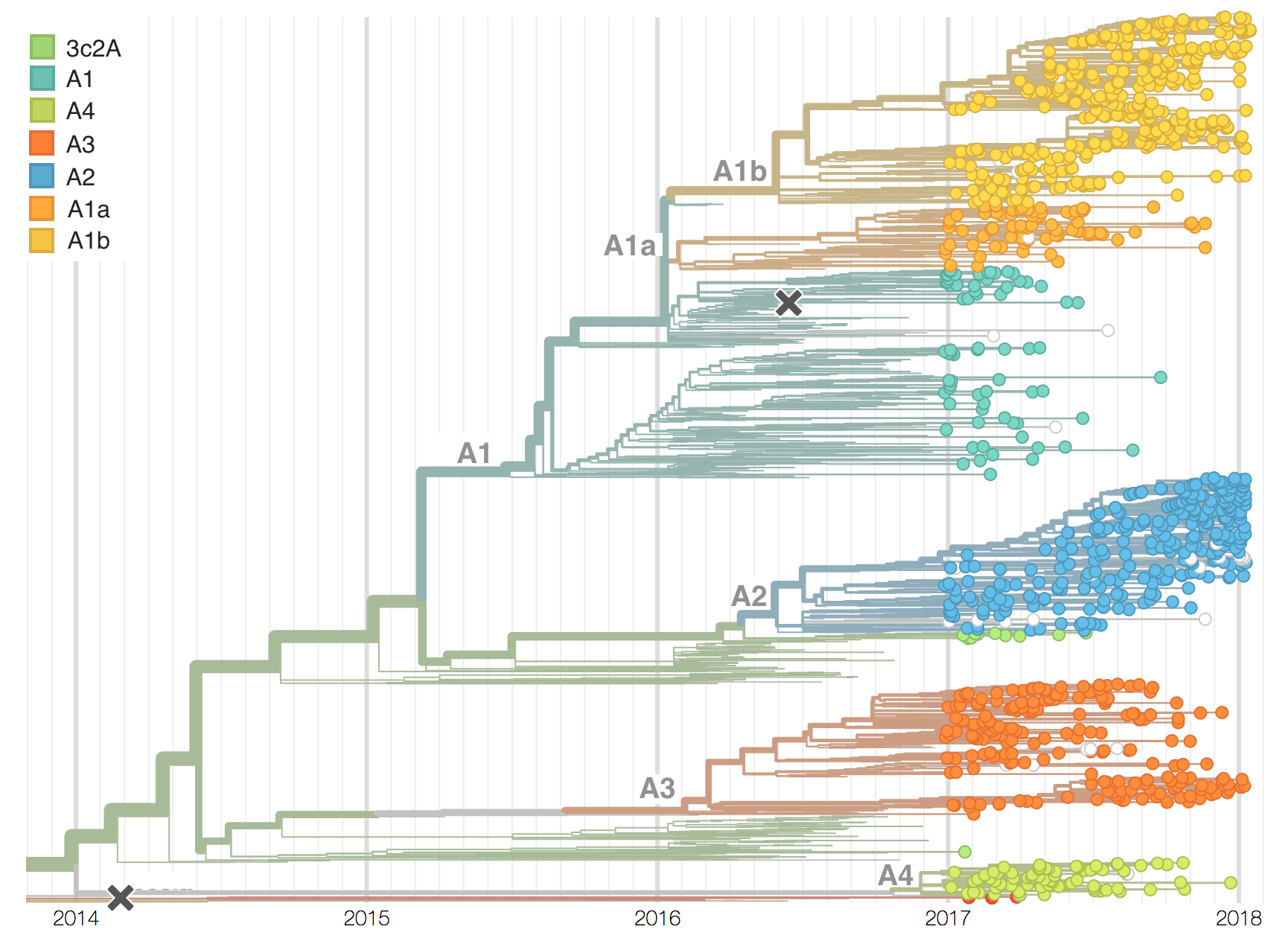
Two clades have been growing rapidly

Clade A2 more recently increasing
Reassortment event appears to drive success of clade A2

Reassortment event appears to drive success of clade A2
Antigenic analysis
Influenza hemagglutination inhibition (HI) assay
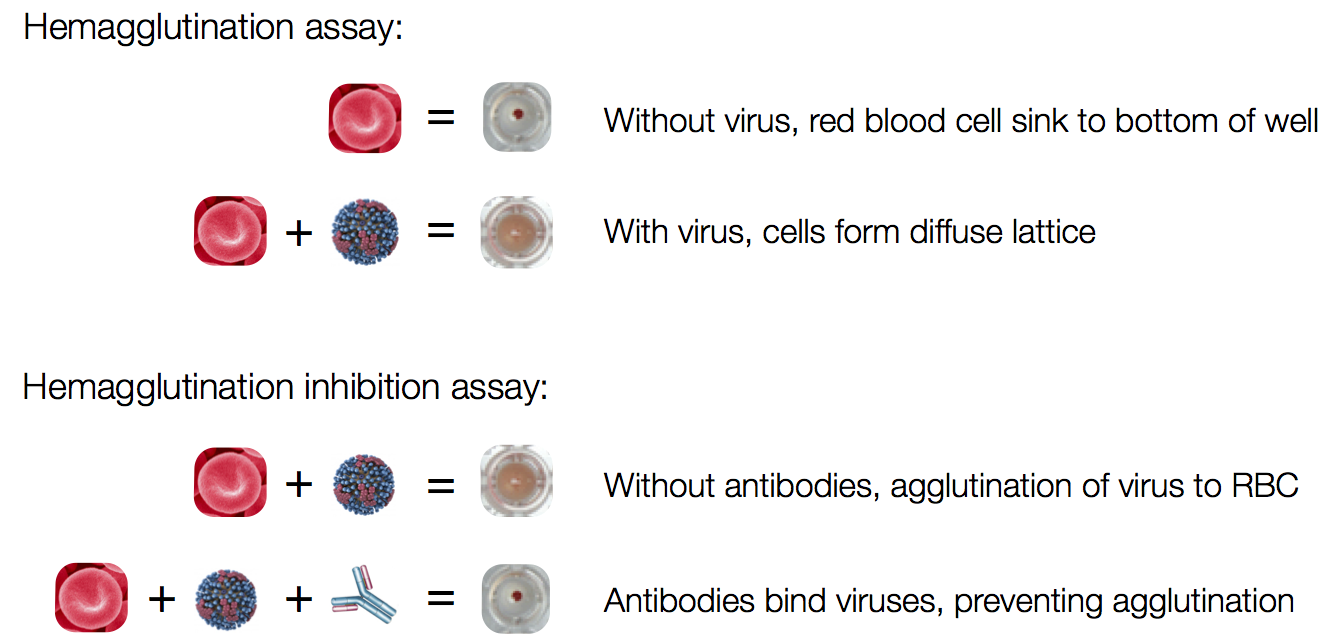
HI measures cross-reactivity across viruses
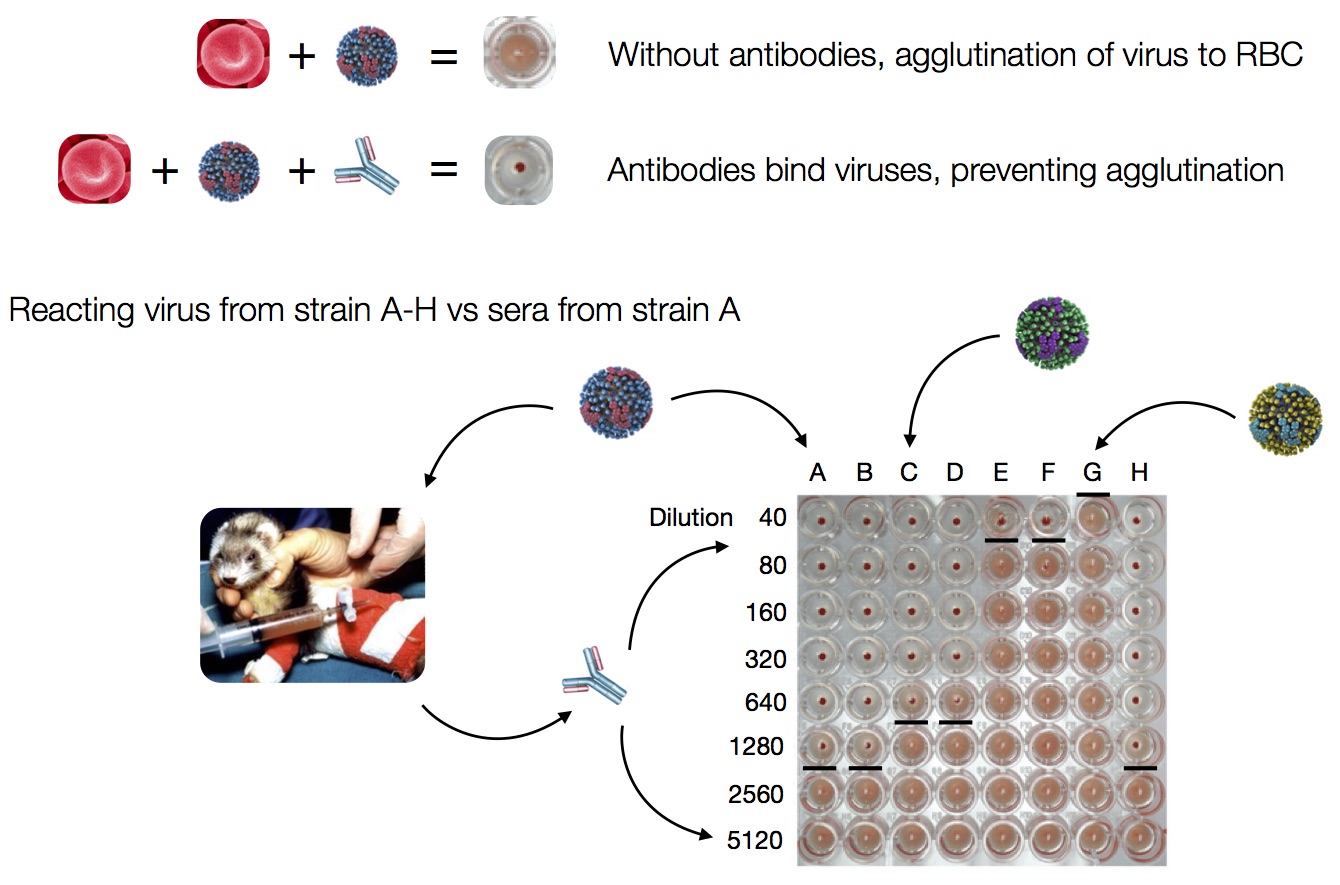
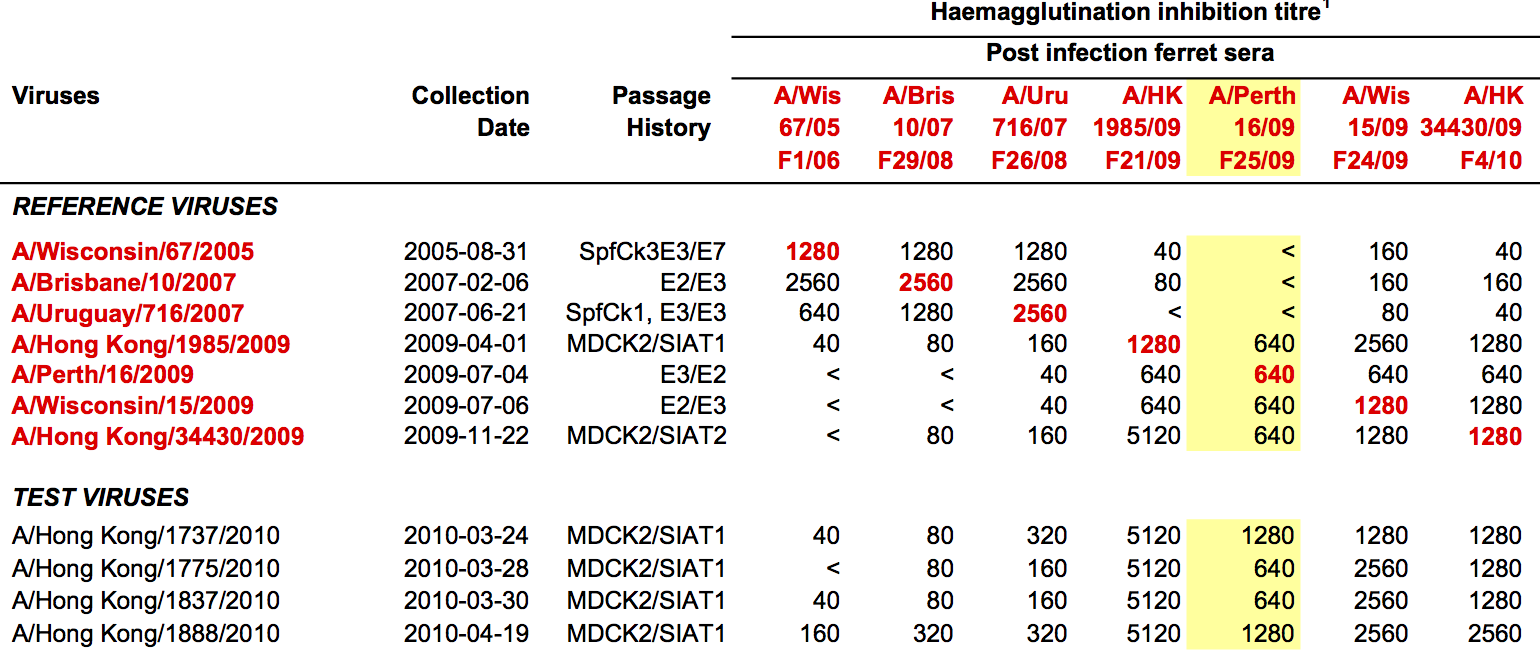
Data in the form of table of maximum inhibitory titers
Antigenic cartography compresses HI measurements into an interpretable diagram
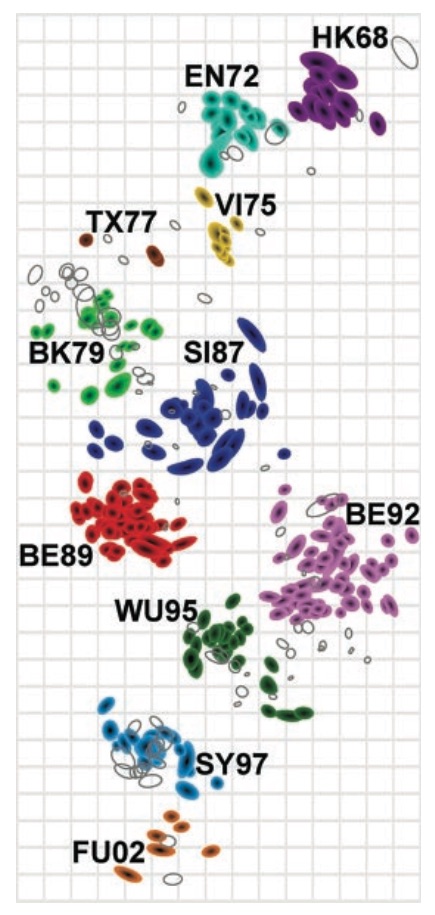
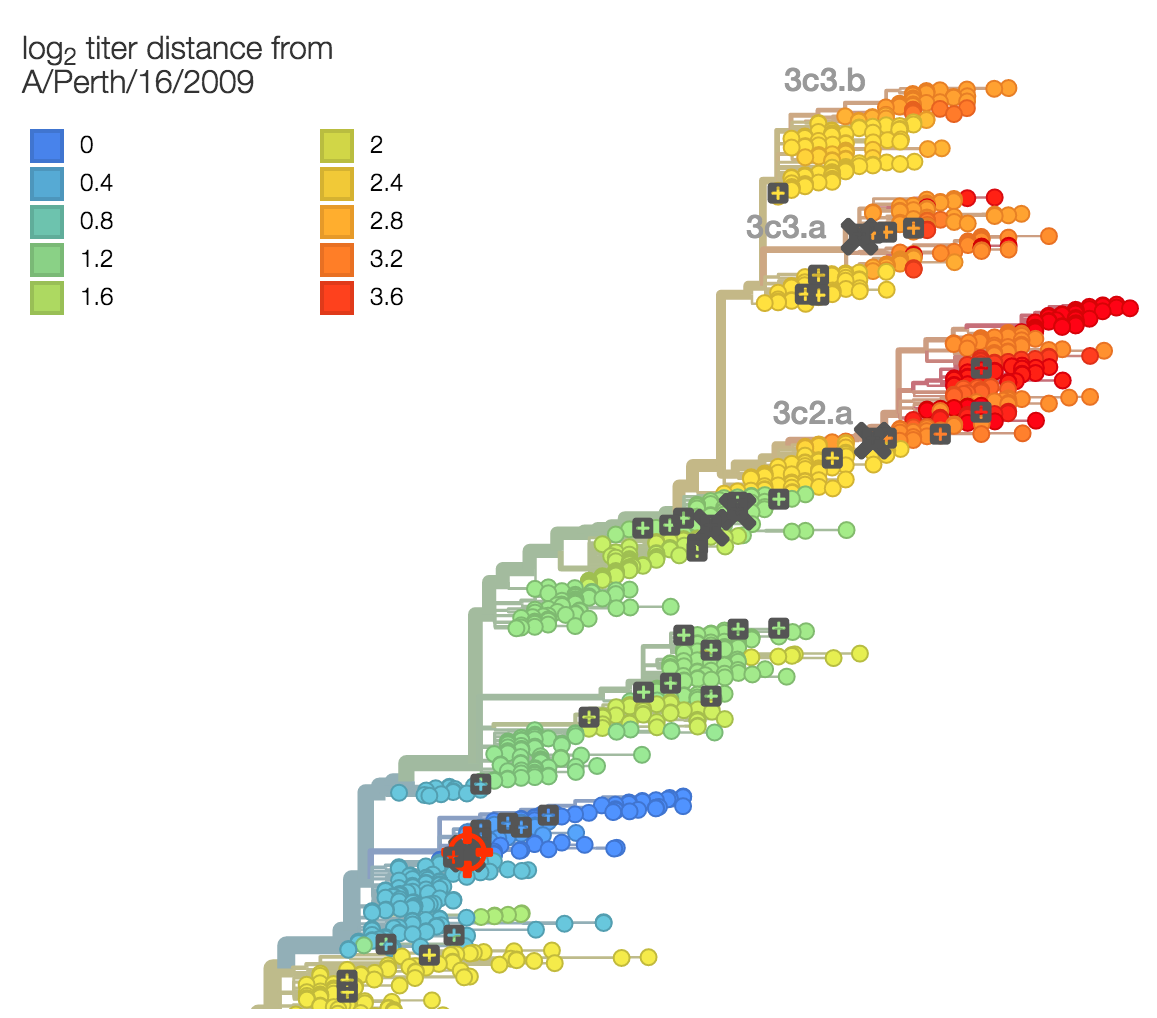
Evolutionary model of HI titer data in which particular substitutions drop titer
Model can be used to interpolate across tree and predict phenotype of untested viruses
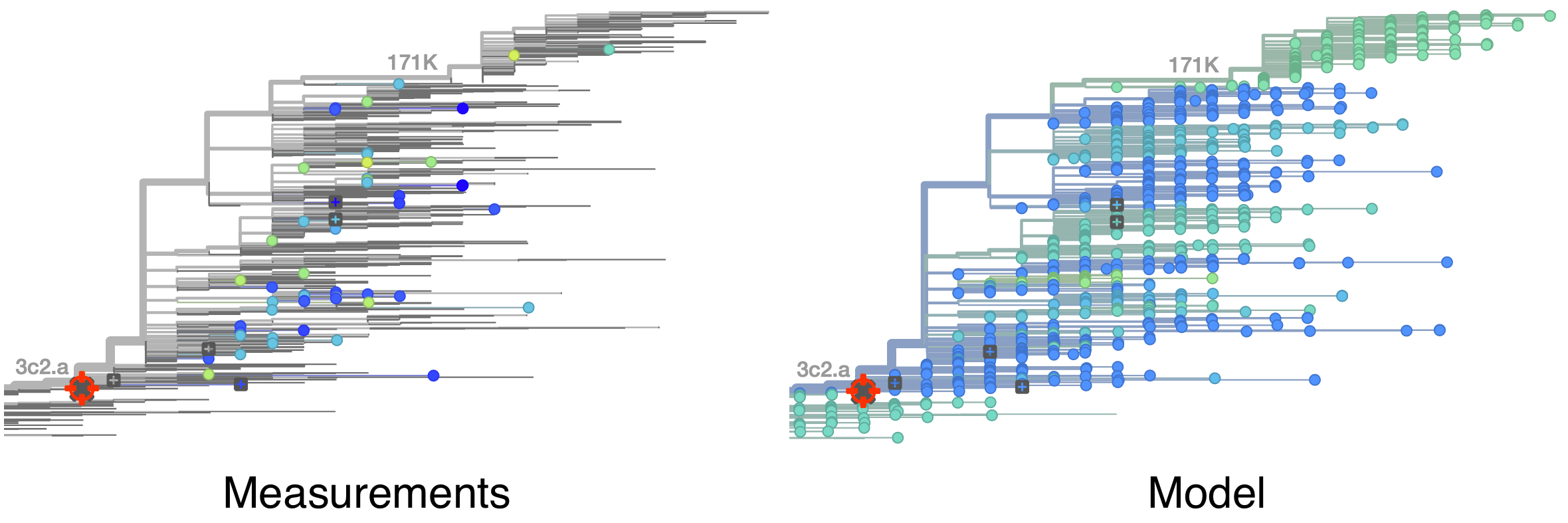
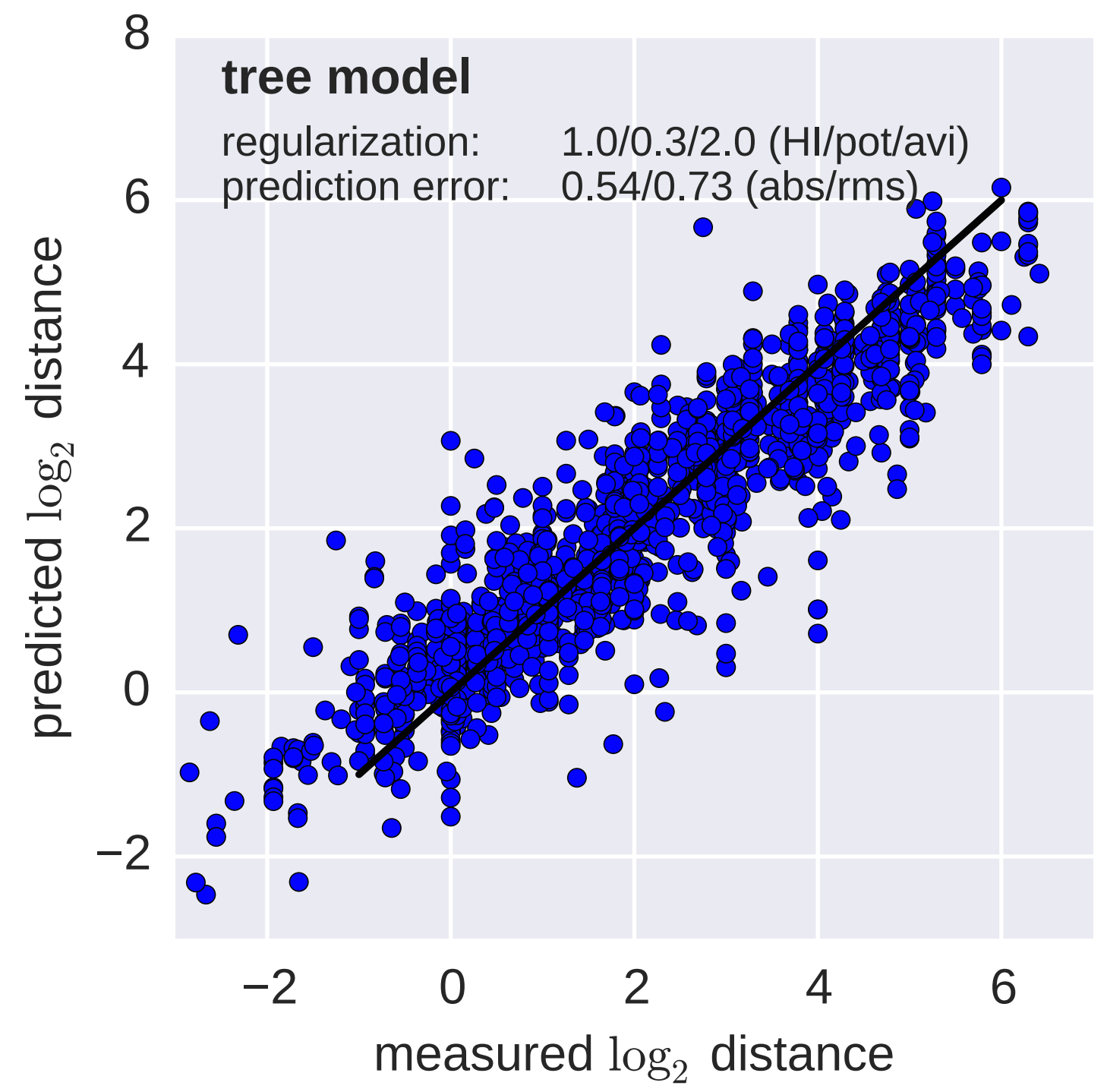
Model is highly predictive of missing titer values
Incorporate HI data from WHO Collaborating Centers
Recent H3N2 antigenic evolution
Forecasting
"The future is here, it's just not evenly distributed yet"
— William Gibson
USA music industry, 2011 dollars per capita
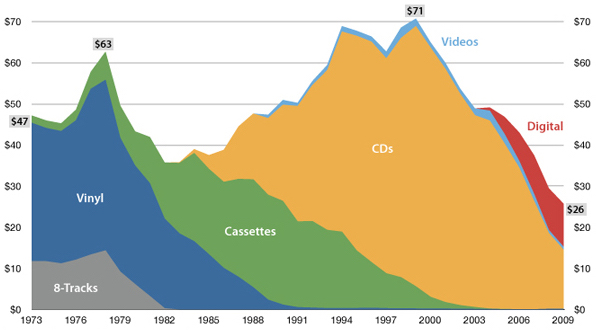
Influenza population turnover
Vaccine strain selection timeline
Seek to explain change in clade frequencies over 1 year
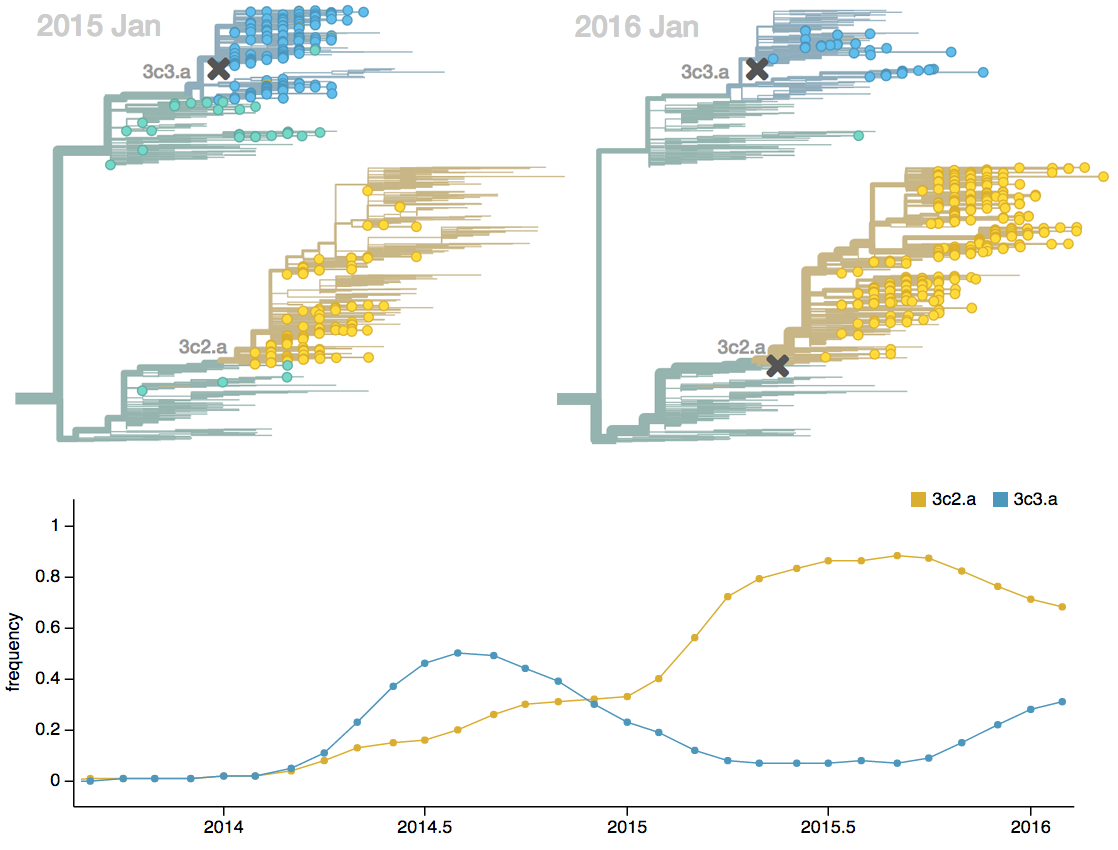
Fitness models can project clade frequencies
Clade frequencies $X$ derive from the fitnesses $f$ and frequencies $x$ of constituent viruses, such that
$$\hat{X}_v(t+\Delta t) = \sum_{i:v} x_i(t) \, \mathrm{exp}(f_i \, \Delta t)$$
This captures clonal interference between competing lineages
The question of forecasting becomes: how do we accurately estimate fitnesses of circulating viruses?
Fortunately, there's lots of training data and previously successful strains have had:
- Amino acid changes at epitope sites
- Antigenic novelty based on HI
- Rapid phylogenetic growth
We predict fitness based on a simple formula
where the fitness $f$ of virus $i$ is estimated as
$$\hat{f}_i = \beta^\mathrm{HI} \, f_i^\mathrm{HI} + \beta^\mathrm{freq} \, f_i^\mathrm{freq}$$
where $f_i^\mathrm{HI}$ measures antigenic drift via HI and $f_i^\mathrm{freq}$ measures clade growth/decline
We learn coefficients and validate model based on previous 15 H3N2 seasons
Clade growth rate is well predicted (ρ = 0.66)
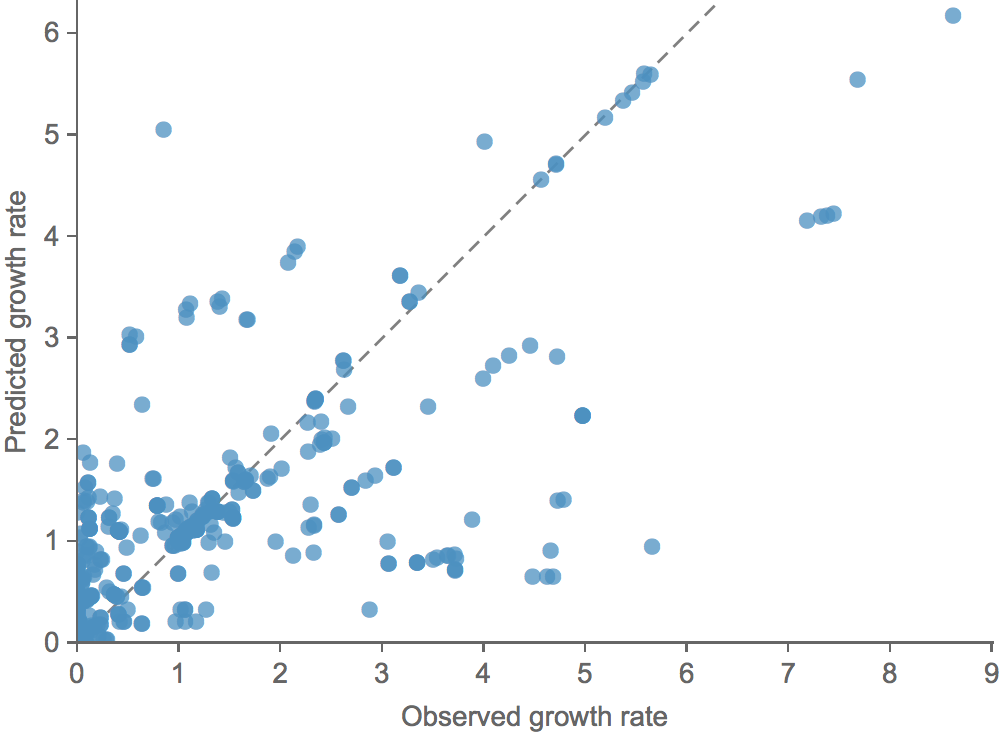
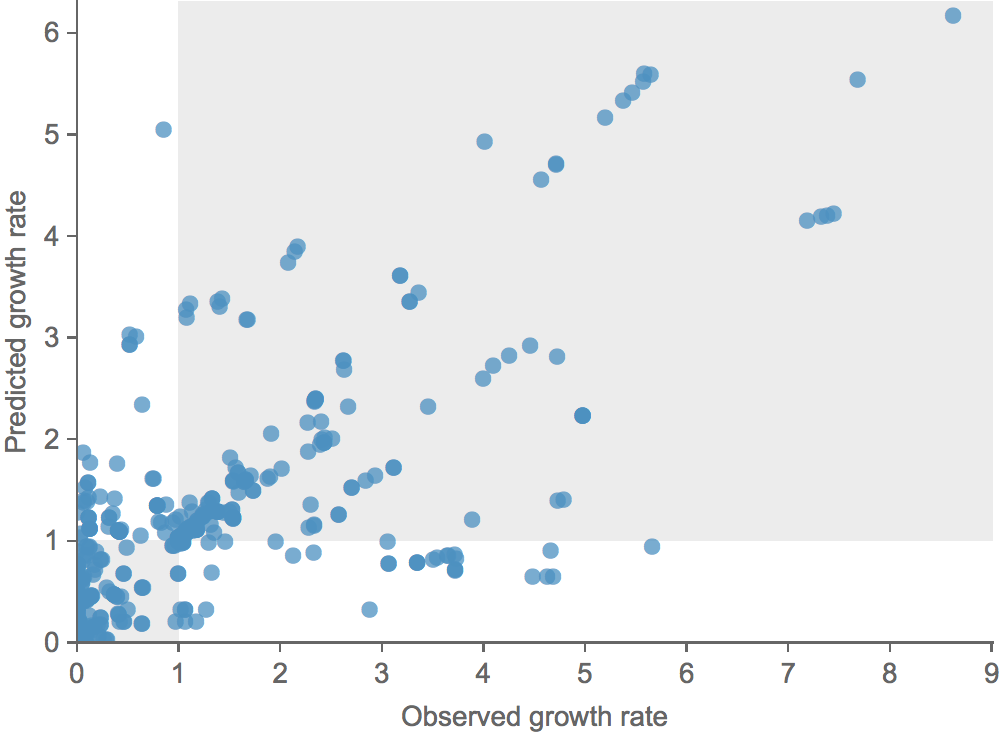
Growth vs decline correct in 84% of cases
Trajectories show more detailed congruence
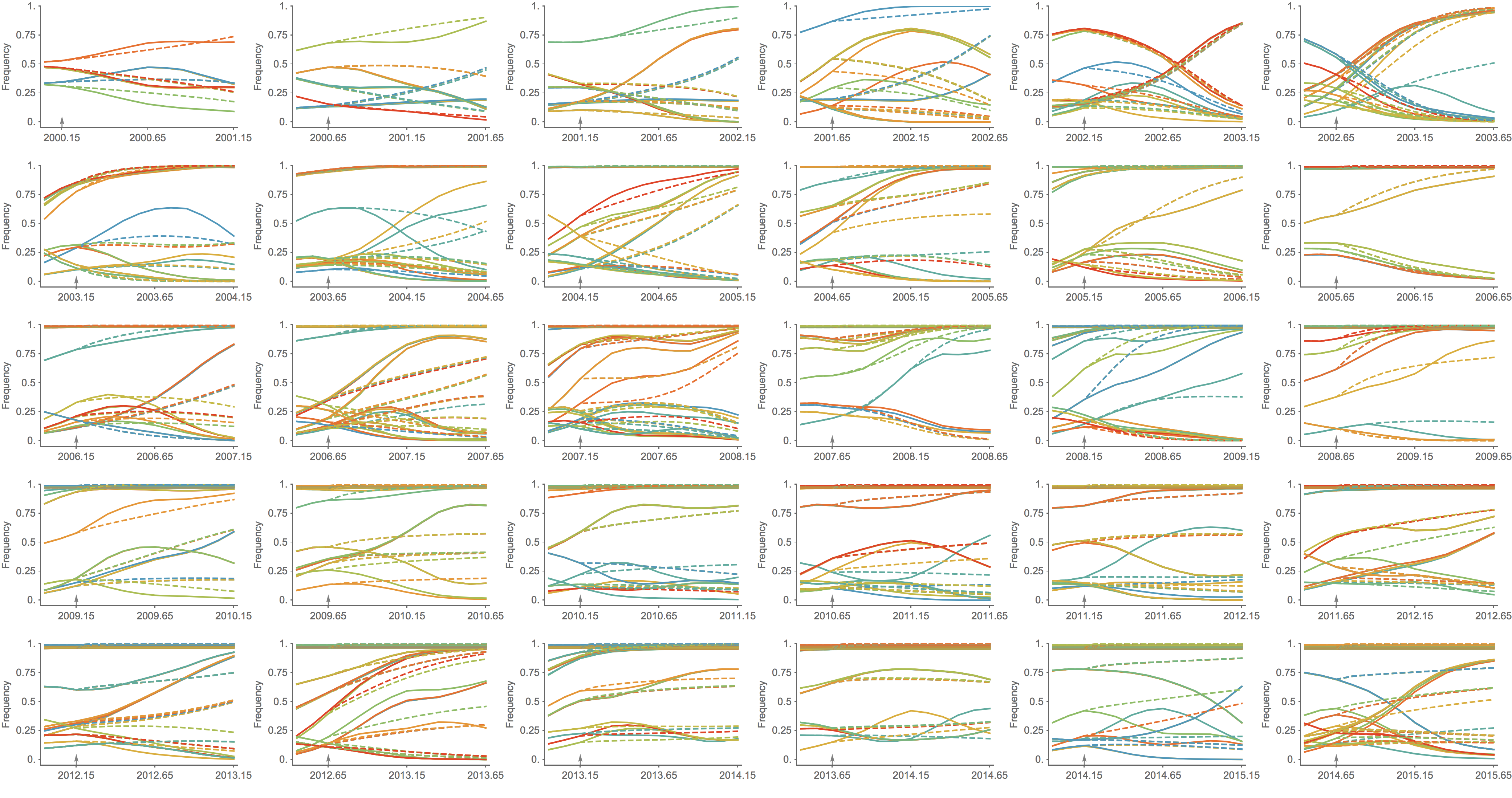
Trajectories show more detailed congruence
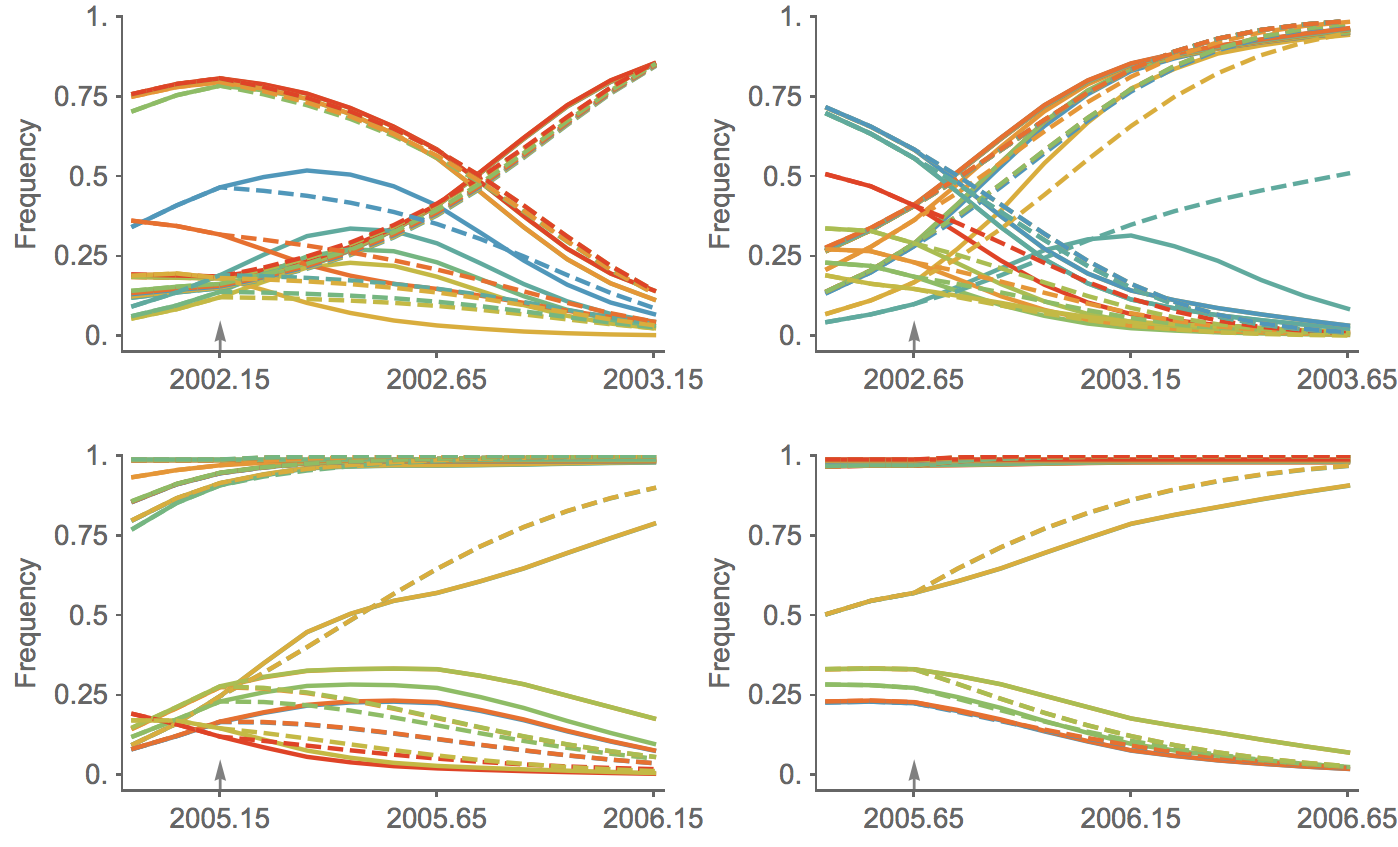
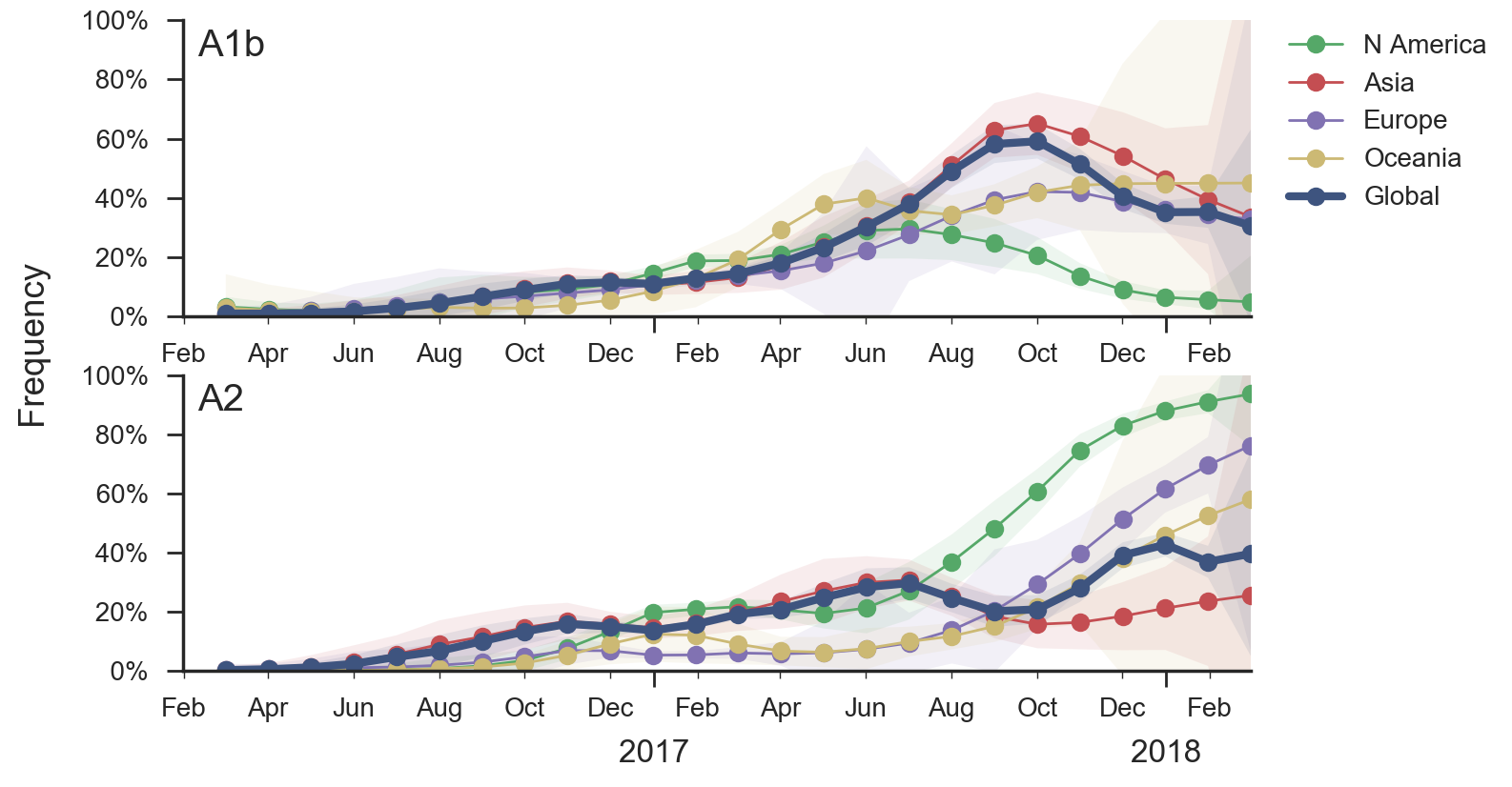
Current forecast favors drifted A1b clade
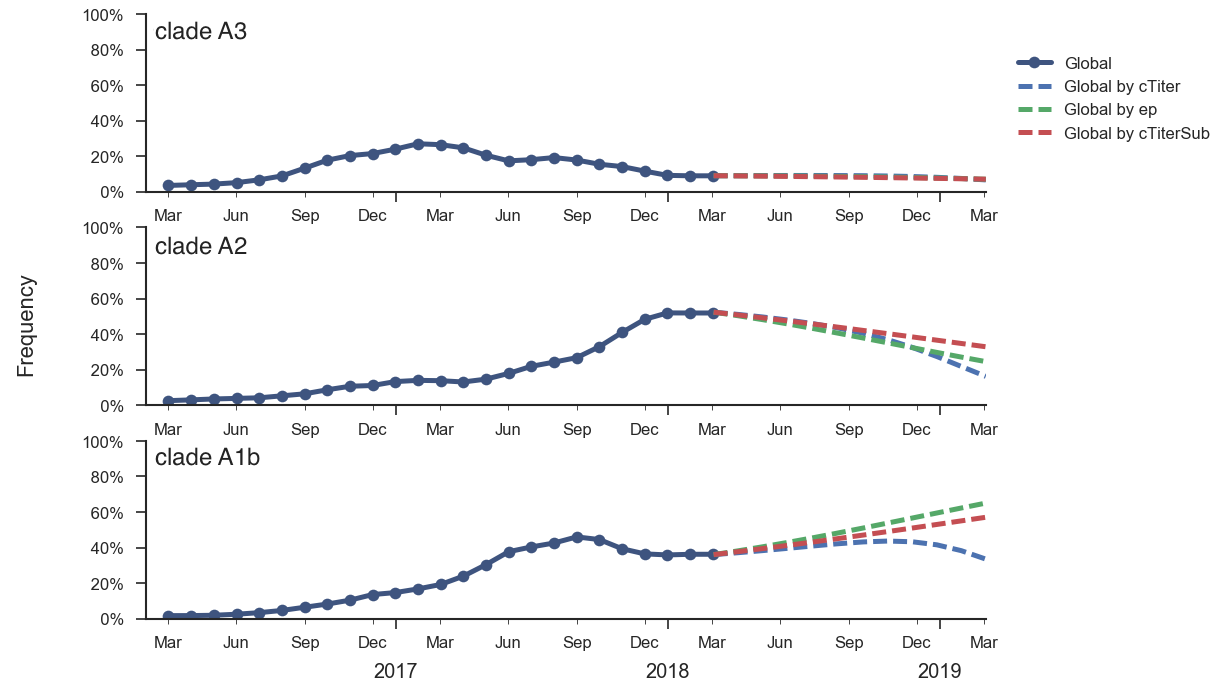
Unlikely to replace human intuition, but could automate serum and VCV selection
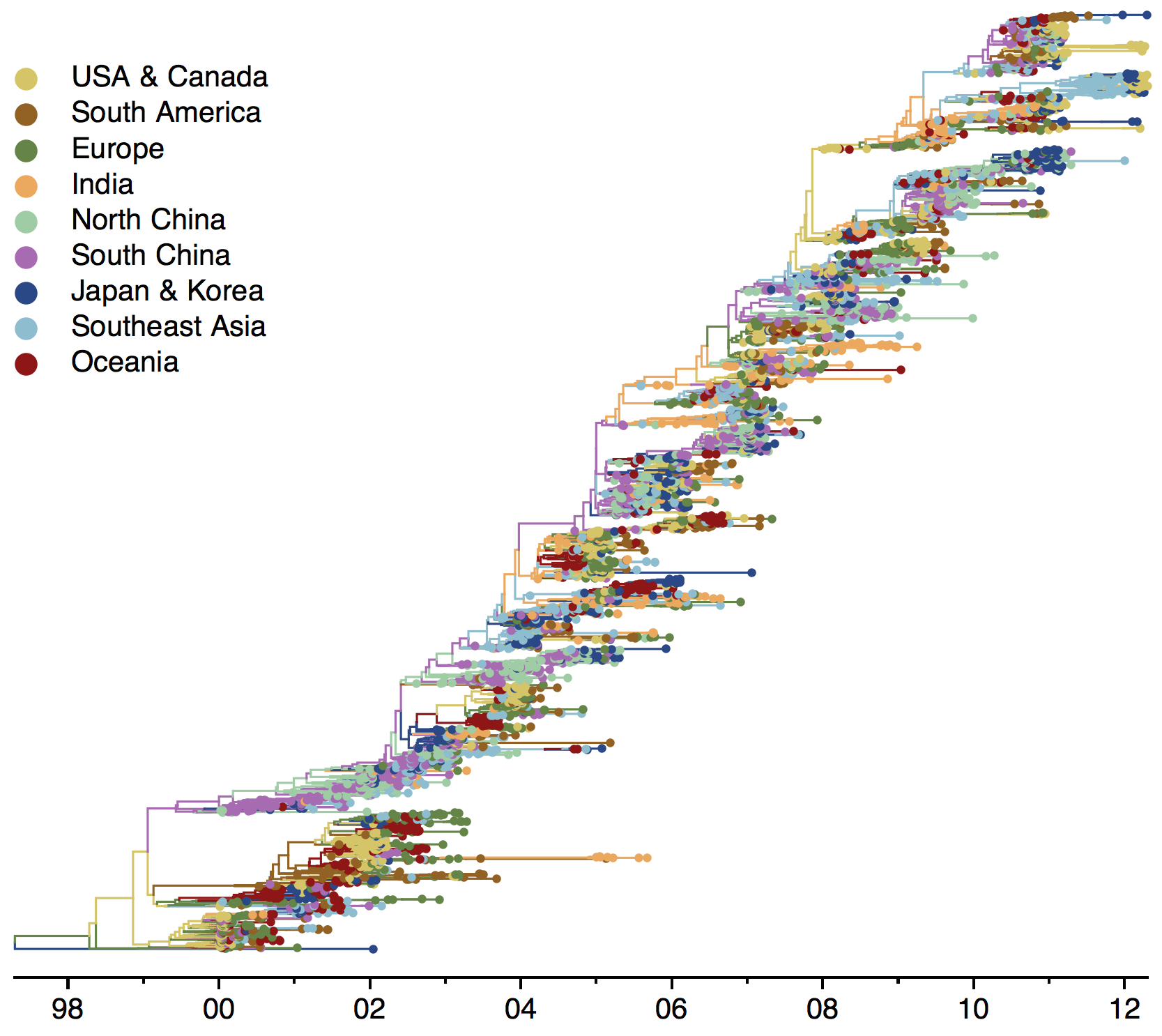
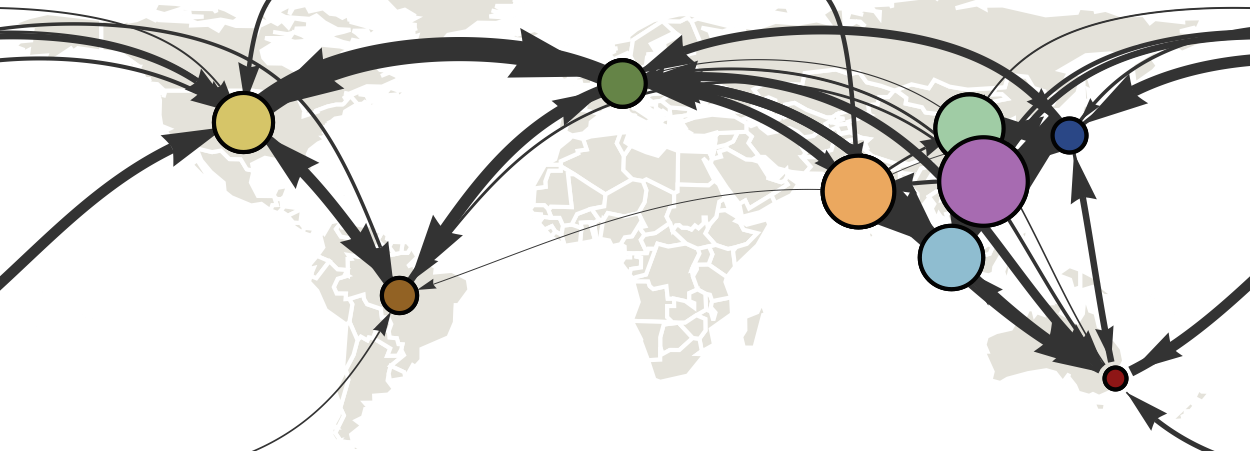
Geographic circulation
Geographic circulation patterns drive evolutionary outcomes
Clades commonly show region-specific circulation patterns
Phylogeny of H3 with geographic history
Infer geographic transition matrix
Geographic location of phylogeny trunk
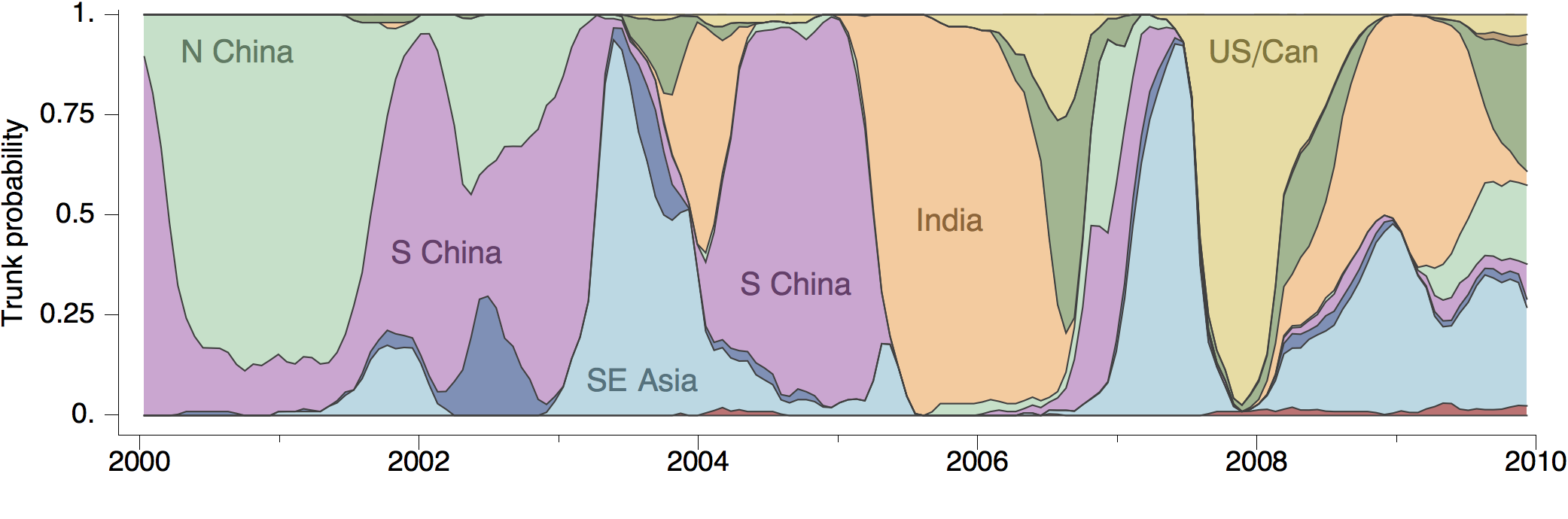
Region-specific ancestry
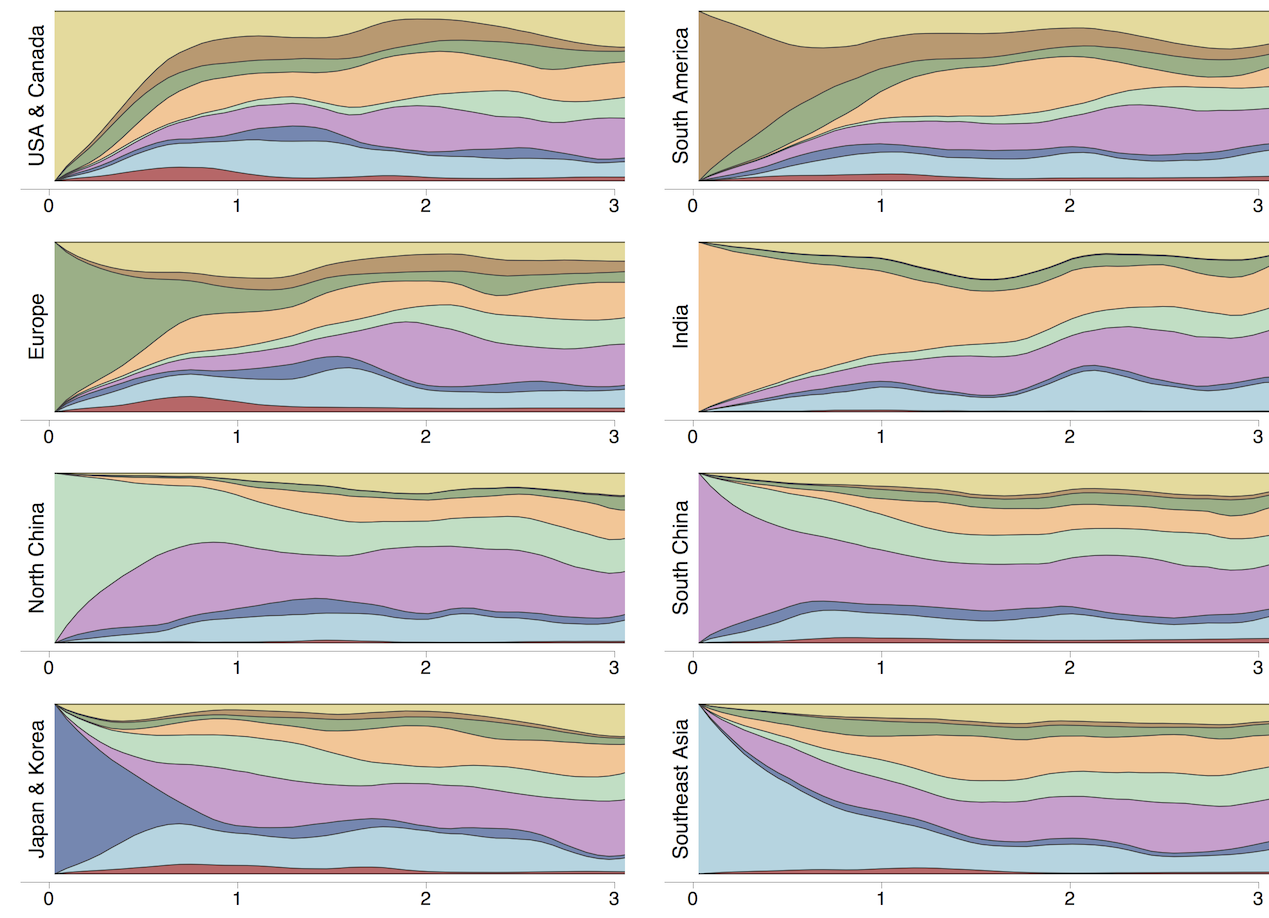
Which regions are most predictive of future outcomes?
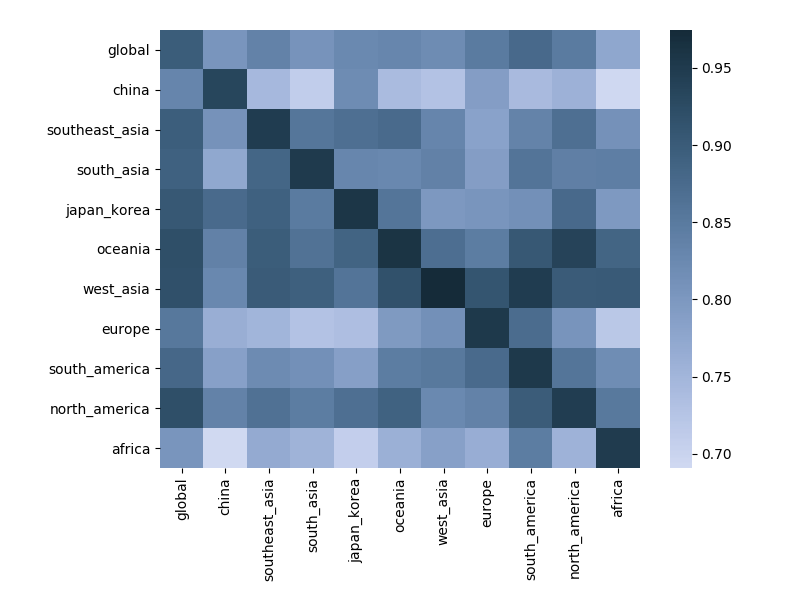
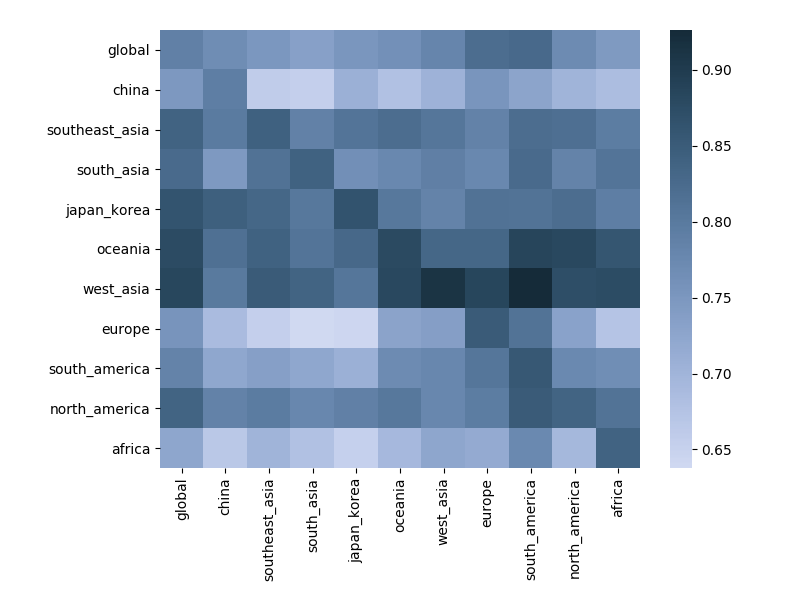
Correlation between clade frequencies
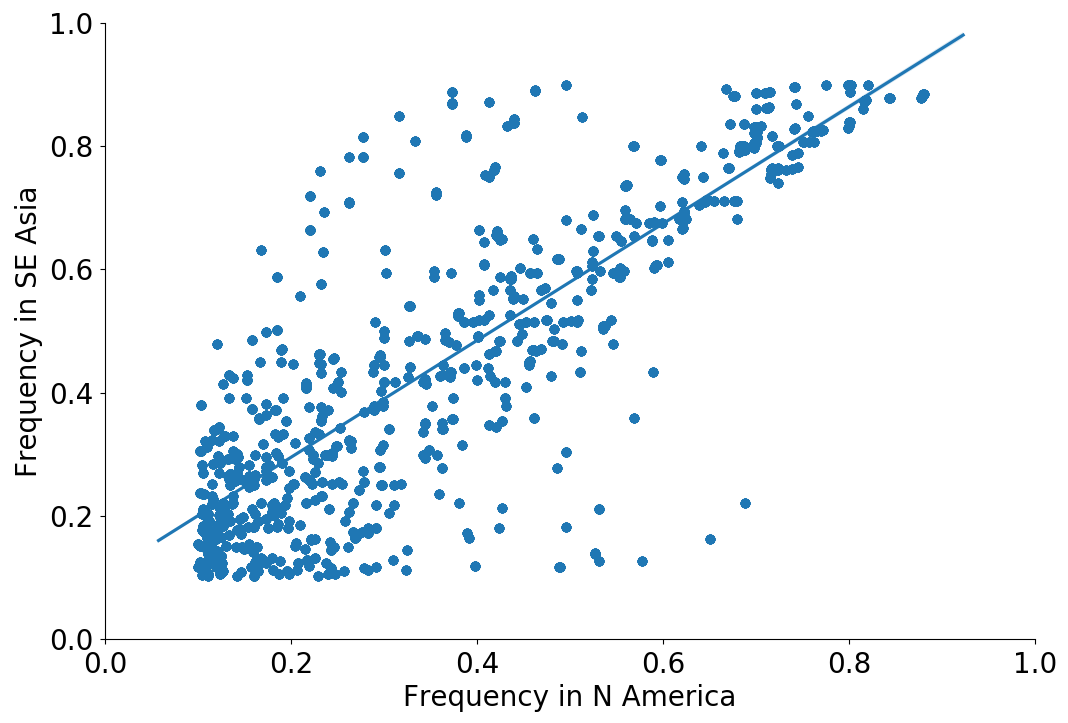
Correlations across regions
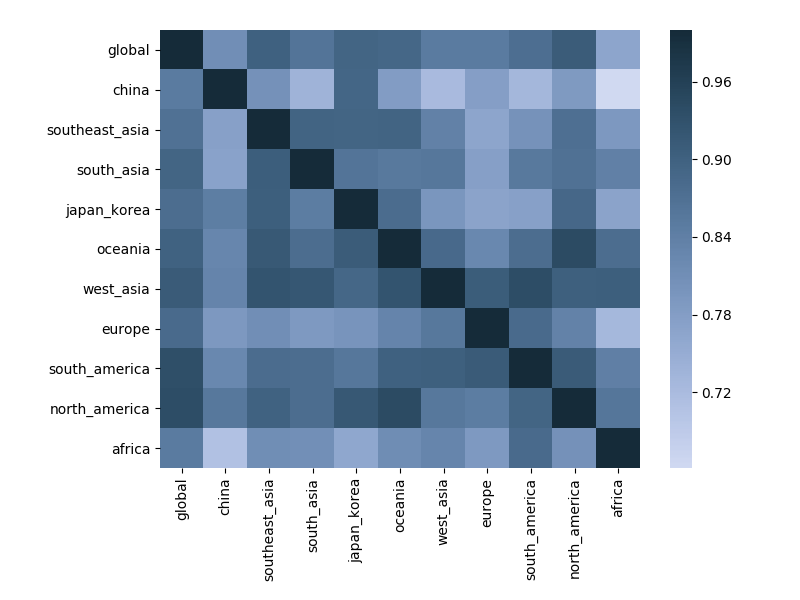
Correlations across regions with 6-month lag
Correlations across regions with 12-month lag
Next steps
- Systematically test fitness predictors
- Include geographic component to fitness model
- Incorporate information from other segments
- Integrate human serology to estimate selective landscape
Acknowledgements
Bedford Lab:
 Alli Black,
Alli Black,
 Sidney Bell,
Sidney Bell,
 Gytis Dudas,
Gytis Dudas,
 John Huddleston,
John Huddleston,
 Barney Potter,
Barney Potter,
 James Hadfield,
James Hadfield,
 Louise Moncla
Louise Moncla
Influenza: WHO Global Influenza Surveillance Network, GISAID, Richard Neher, Barney Potter, John Huddleston, James Hadfield, Colin Russell, Andrew Rambaut, Dave Wentworth, Becky Garten, Jackie Katz, Marta Łuksza, Michael Lässig, Richard Reeve
Nextstrain: Richard Neher, James Hadfield, Colin Megill, Sidney Bell, Charlton Callender, Barney Potter, John Huddleston, Emma Hodcroft
