Evolutionary forecasting for seasonal influenza and SARS-CoV-2
Trevor Bedford
Fred Hutchinson Cancer Center / Howard Hughes Medical Institute
14 Dec 2023
Division of Viral Products Seminar Series
FDA
Disclosure
Trevor Bedford has no relevant financial relationships with commercial interests
None of the planners have any financial interests or relationships with a commercial entity
Learning objectives
- Apply fitness models and multinomial logistic regression models to predict changes in viral variant frequencies
- Assess evolutionary forecasts for retrospective accuracy
- Intrepret differences in evolutionary rate between seasonal influenza and SARS-CoV-2
Question
Which factors contributed to the observed high yearly attack rate of SARS-CoV-2 in 2022 and 2023?
- Intrinsic $R_0$ of SARS-CoV-2
- Rapid antigenic evolution of SARS-CoV-2 spike protein
- SARS-CoV-2 is a pandemic virus rather than epidemic virus
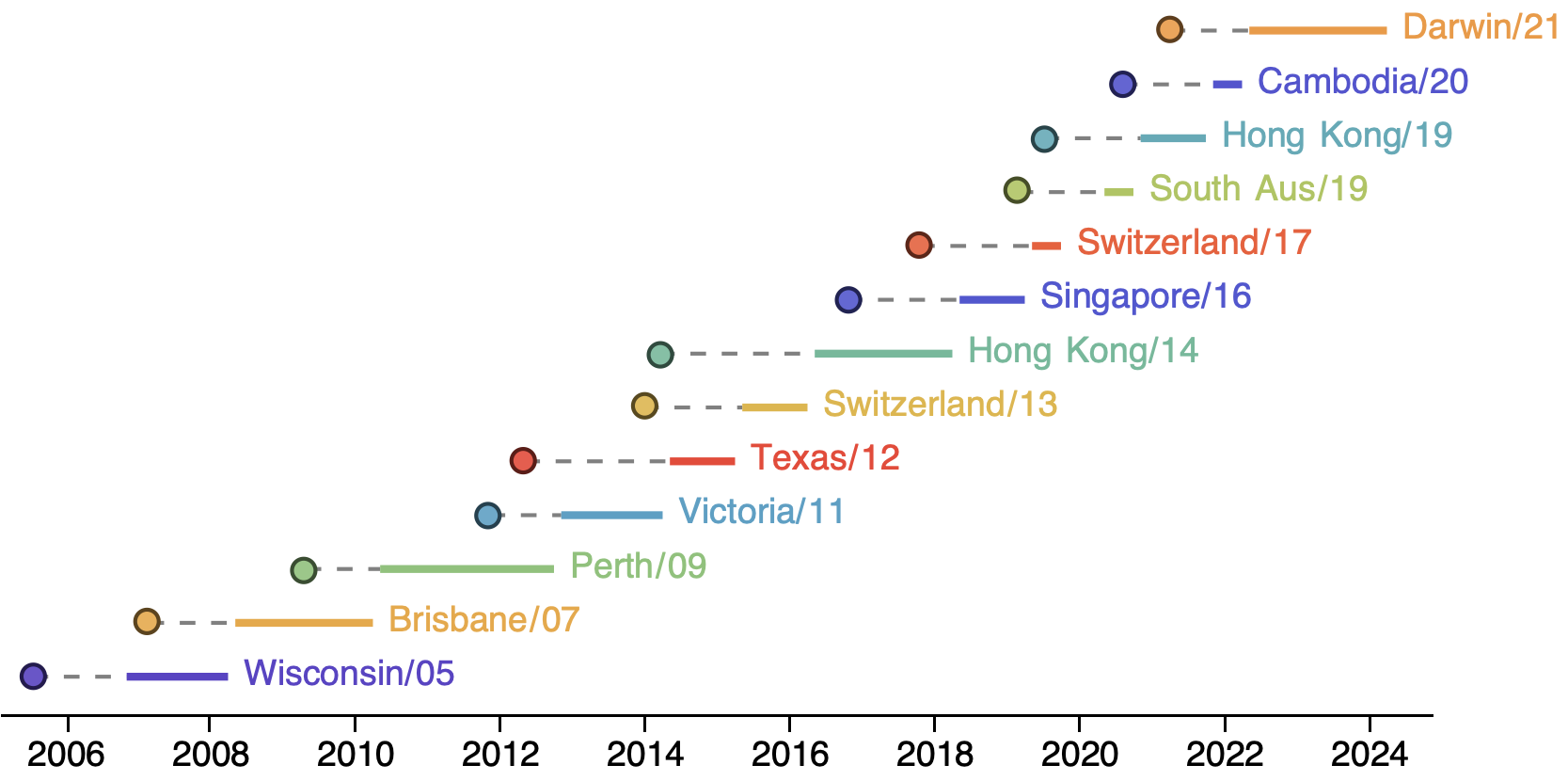
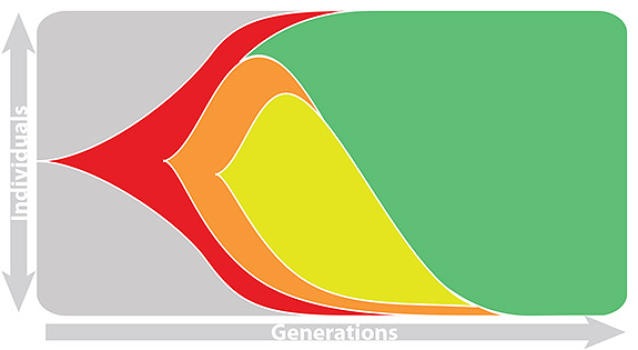
Seasonal influenza
Rapid turnover of the A/H3N2 influenza population
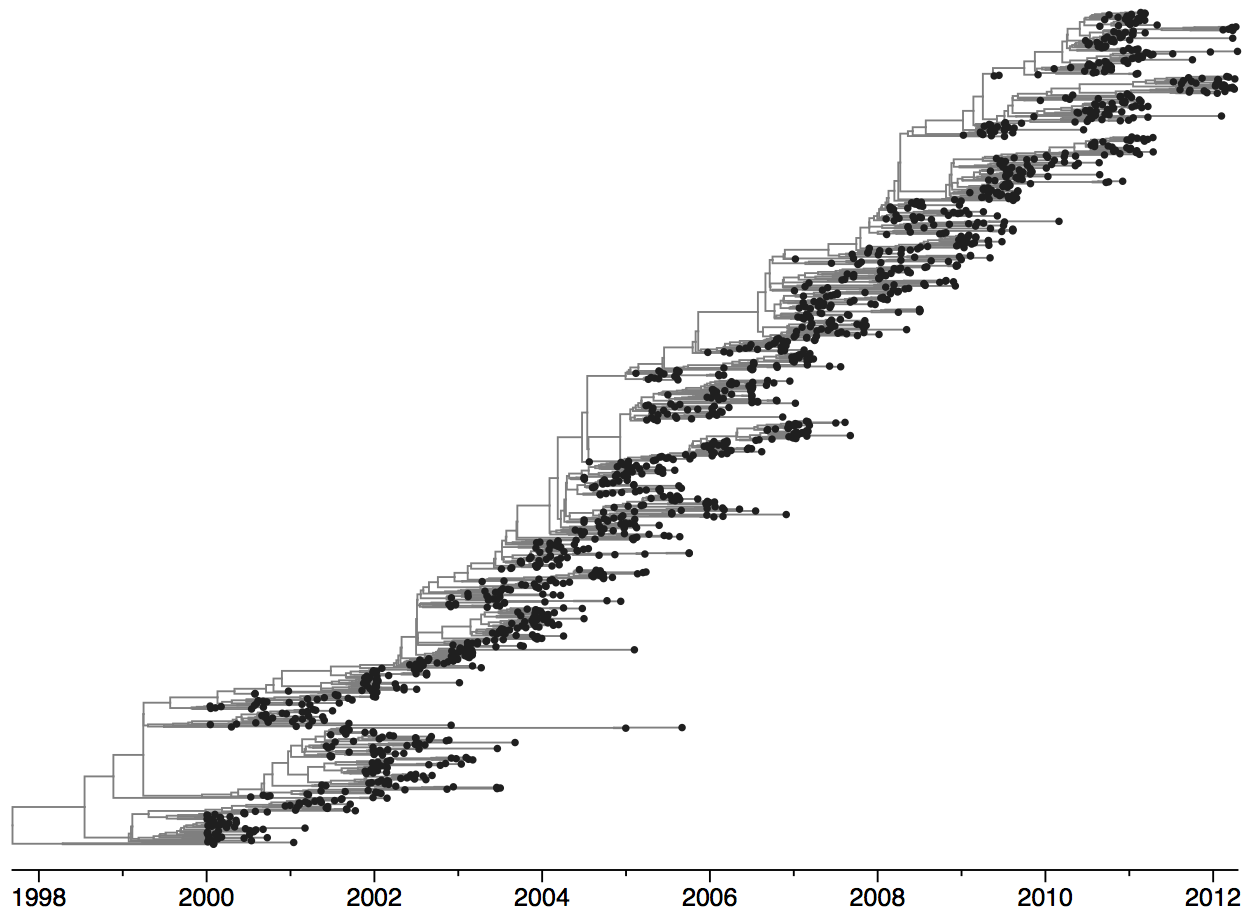
Clades emerge, die out and take over
Clades show rapid turnover

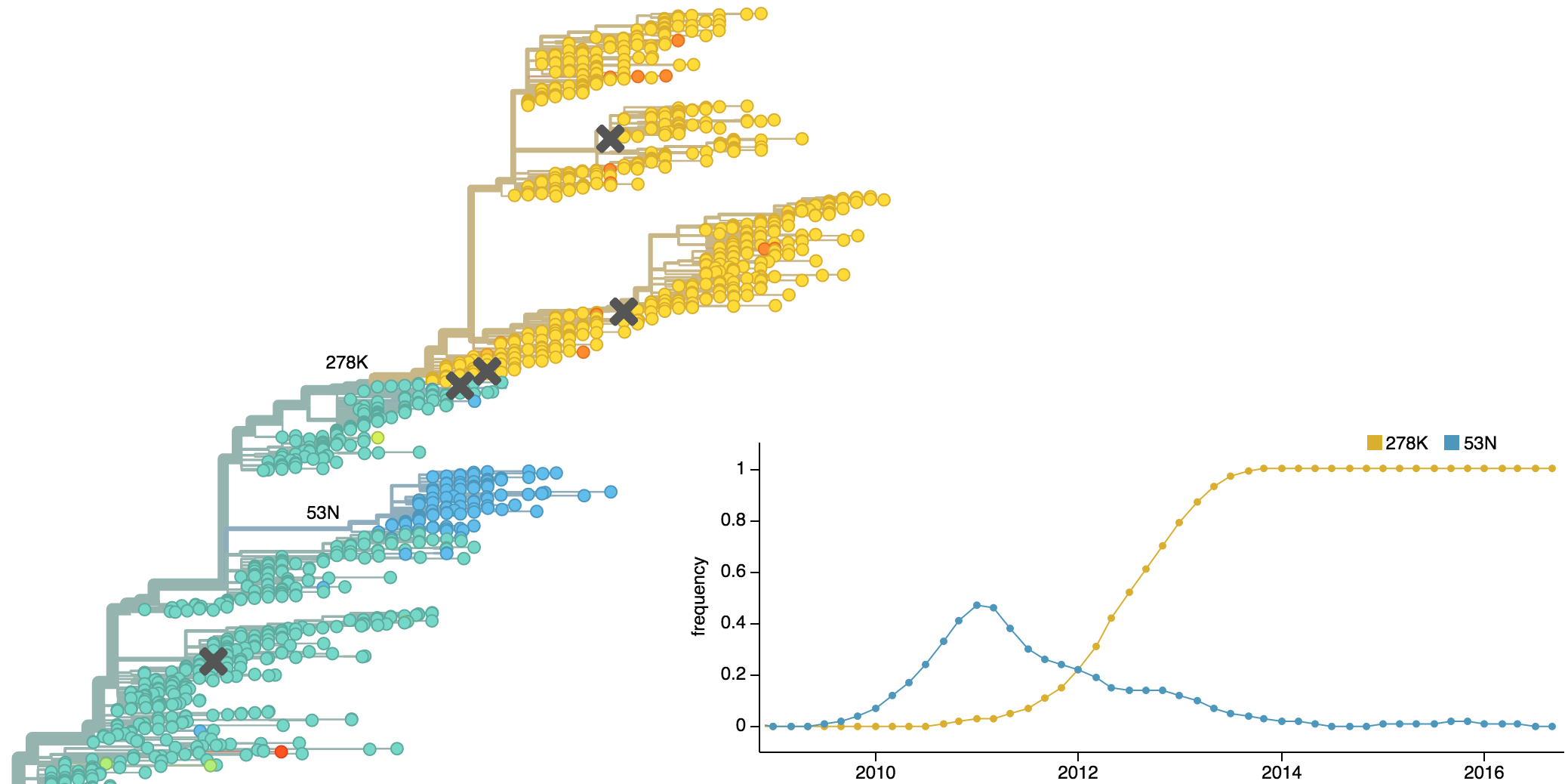
Dynamics driven by antigenic drift
Drift necessitates vaccine updates
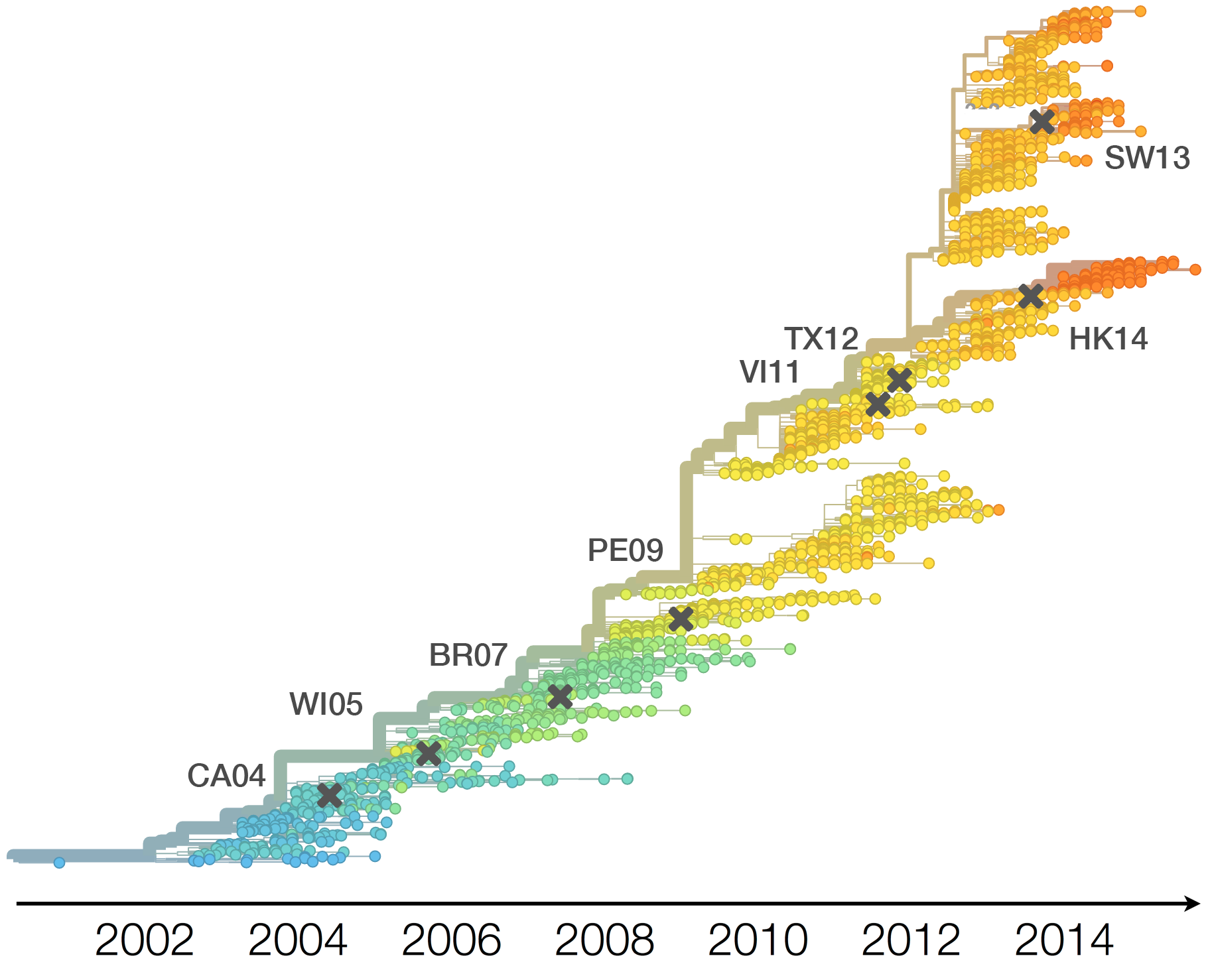
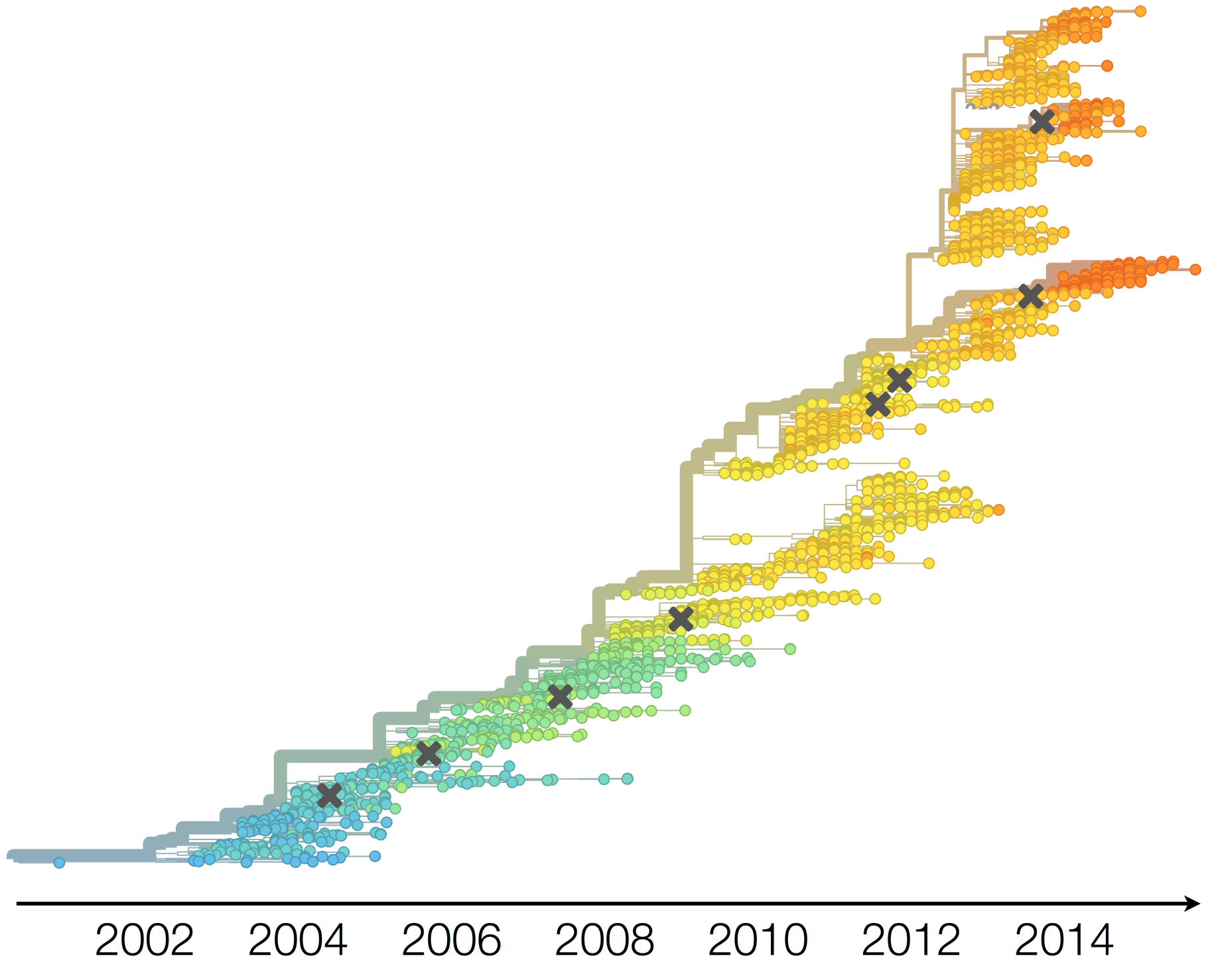
H3N2 vaccine updates occur every 1-2 years
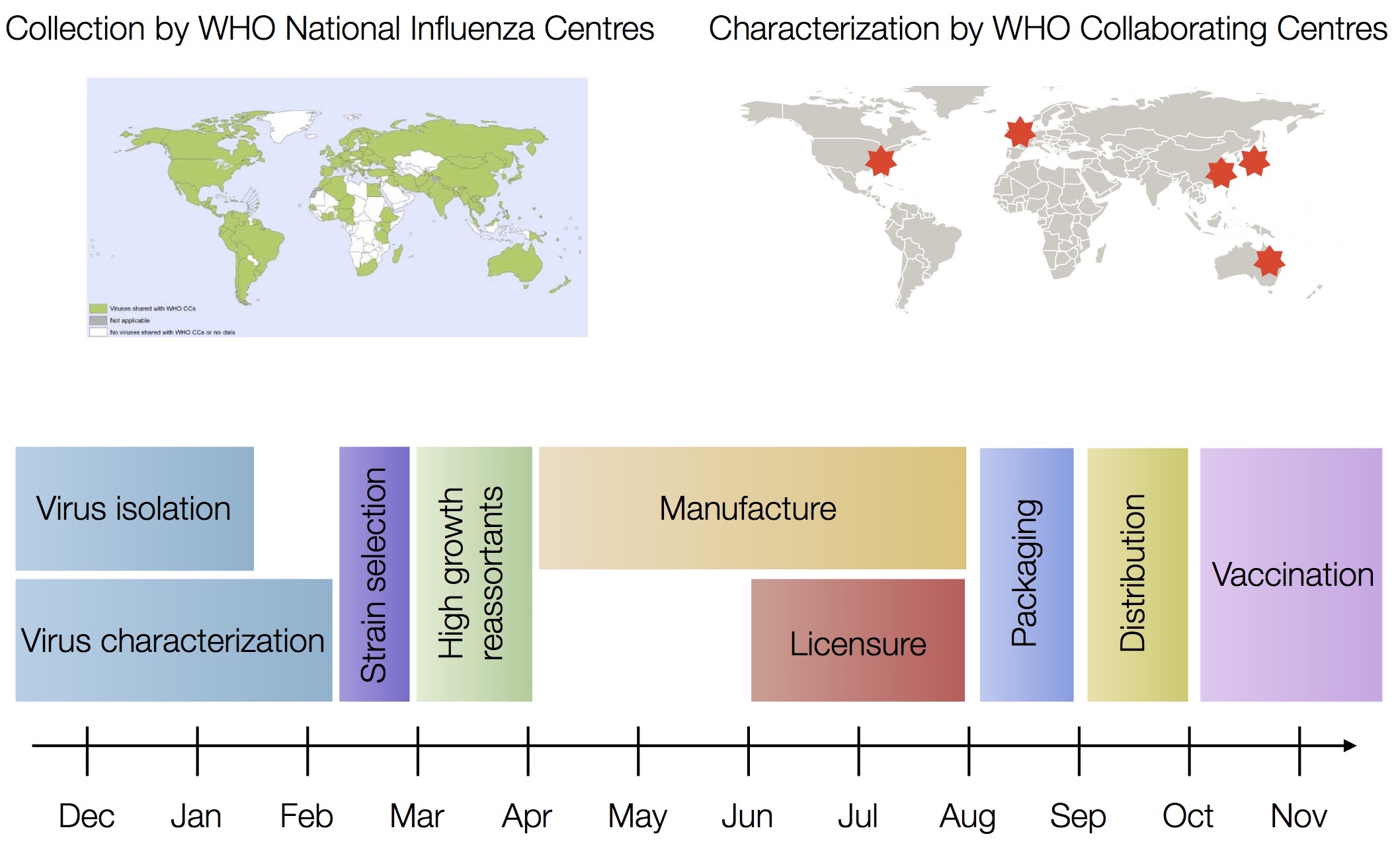
Vaccine strain selection by WHO
Nextflu
Project to provide a real-time view of the evolving influenza population
All in collaboration with  Richard Neher
Richard Neher
Nextflu → Nextstrain
Real-time tracking of pathogen evolution
![]() Richard Neher,
Richard Neher,
![]() Ivan Aksamentov,
Ivan Aksamentov,
![]() Jennifer Chang
Jennifer Chang
![]() James Hadfield,
James Hadfield,
![]() Emma Hodcroft,
Emma Hodcroft,
![]() John Huddleston,
John Huddleston,
![]() Jover Lee,
Jover Lee,
![]() Victor Lin,
Victor Lin,
![]() Cornelius Roemer,
Cornelius Roemer,
![]() Thomas Sibley
Thomas Sibley
Current view of H3N2 from nextstrain.org/flu
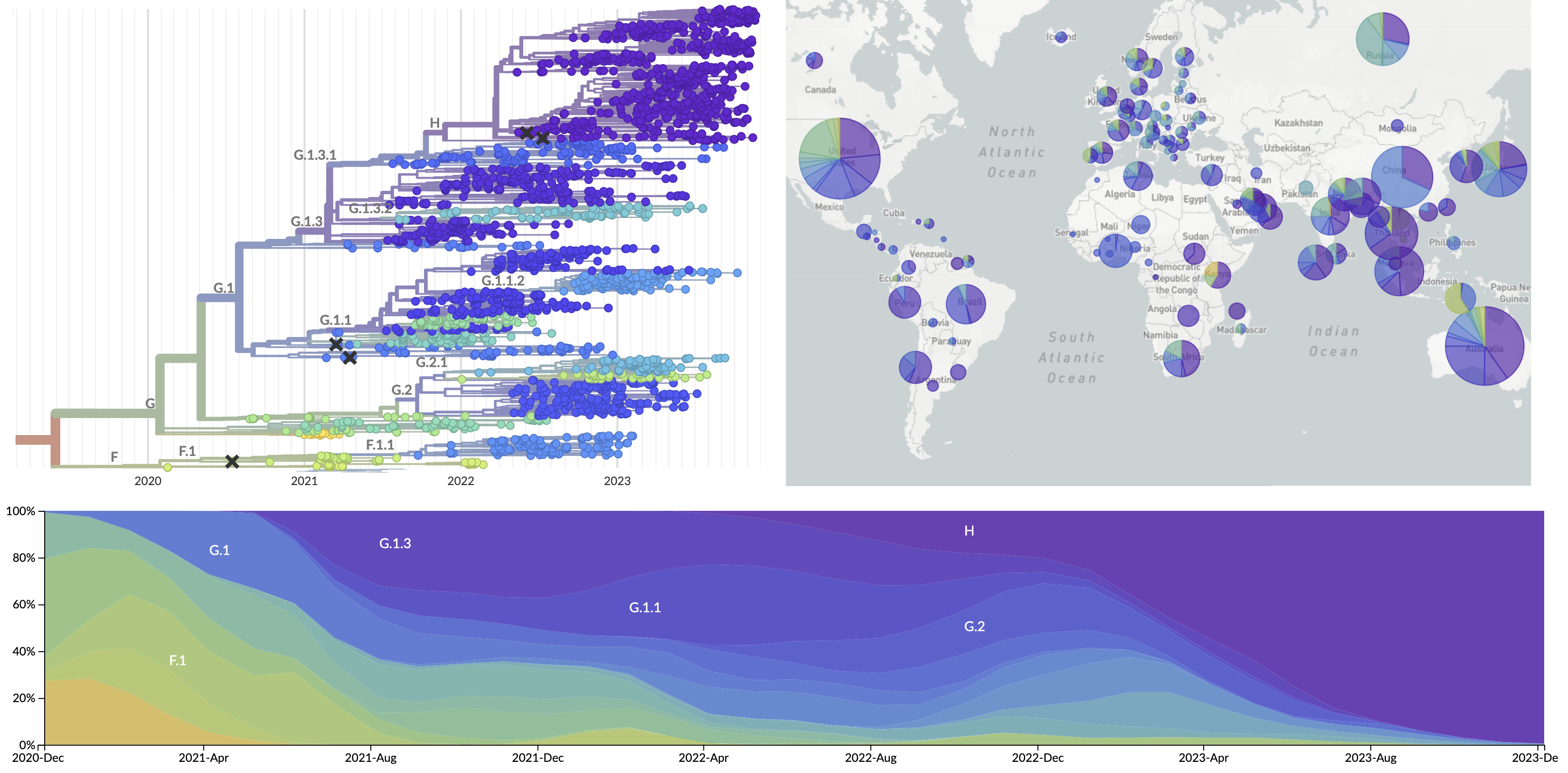
Forecasting seasonal influenza evolution
Fitness models project strain frequencies
Future frequency $x_i(t+\Delta t)$ of strain $i$ derives from strain fitness $f_i$ and present day frequency $x_i(t)$, such that
$$x_i(t+\Delta t) = \frac{1}{Z(t)} \, x_i(t) \, \mathrm{exp}(f_i \, \Delta t)$$
Strain frequencies at each timepoint are normalized by total frequency $Z(t)$. This captures clonal interference between competing lineages.
Integrating genotypes and phenotypes improves long-term forecasts of seasonal influenza A/H3N2 evolution
with ![]() John Huddleston, Richard Neher, Dave Wentworth, Becky Kondor, John McCauley, Hideki Hasegawa, Kanta Subbarao and others
John Huddleston, Richard Neher, Dave Wentworth, Becky Kondor, John McCauley, Hideki Hasegawa, Kanta Subbarao and others
Match strain forecast to retrospective circulation
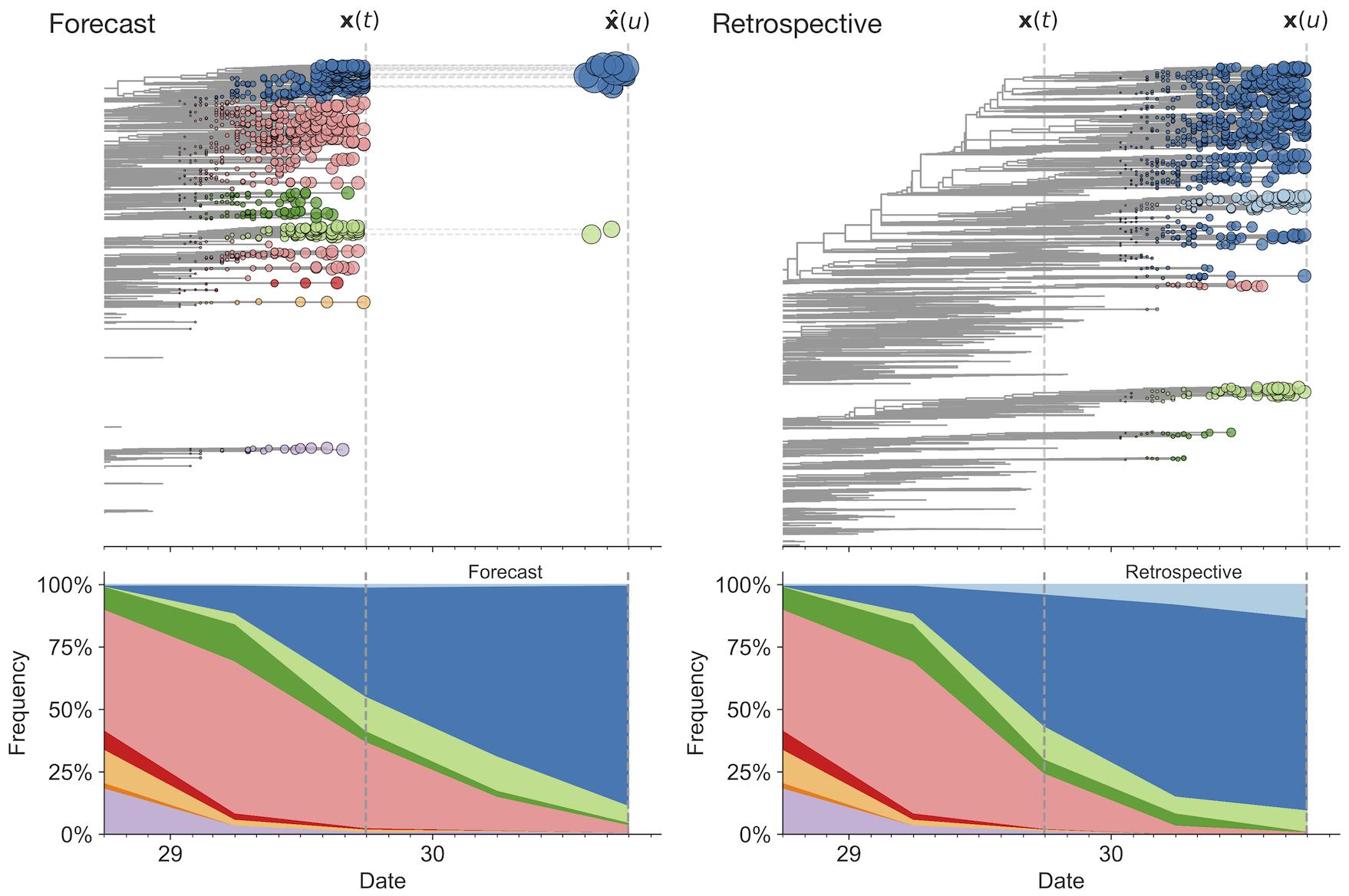
Two inputs
- Estimate of present-day strain frequencies $x_i(t)$
- Estimate of present-day strain fitnesses $f_i$
Strain frequency estimated via region-weighted KDE
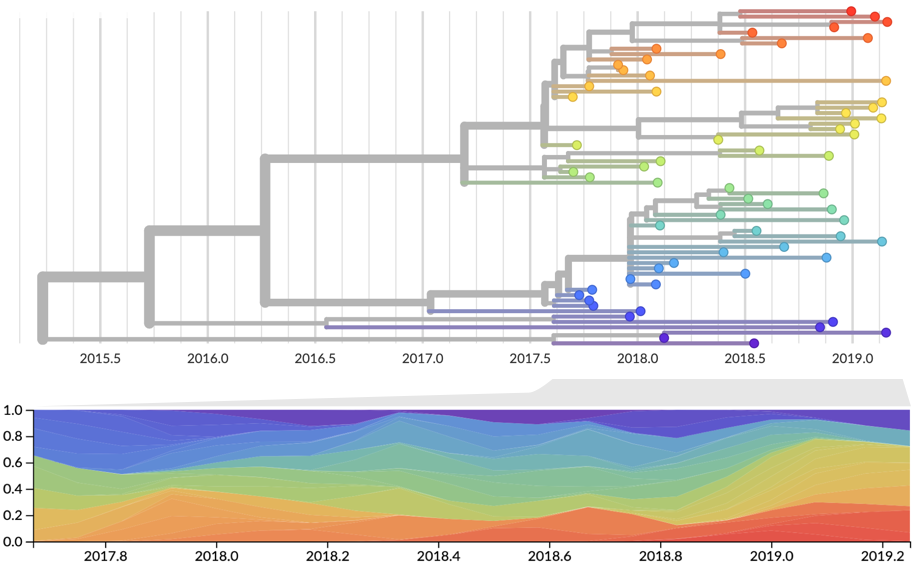
Strain fitness estimated from viral attributes
The fitness $f$ of strain $i$ is estimated as
$$f_i = \beta^\mathrm{A} \, f_i^\mathrm{A} + \beta^\mathrm{B} \, f_i^\mathrm{B} + \ldots$$
where $f^A$, $f^B$, etc... are different standardized viral attributes and $\beta^A$, $\beta^B$, etc... coefficients are trained based on historical evolution
| Antigenic drift | Intrinsic fitness | Recent growth |
|---|---|---|
| epitope mutations | non-epitope mutations | local branching index |
| HI titers | DMS data (via Bloom lab) | delta frequency |
Future population depends on frequency and fitness
Forecast assessed based on weighted distance match to observed future population (earth mover's distance)
Poor fit
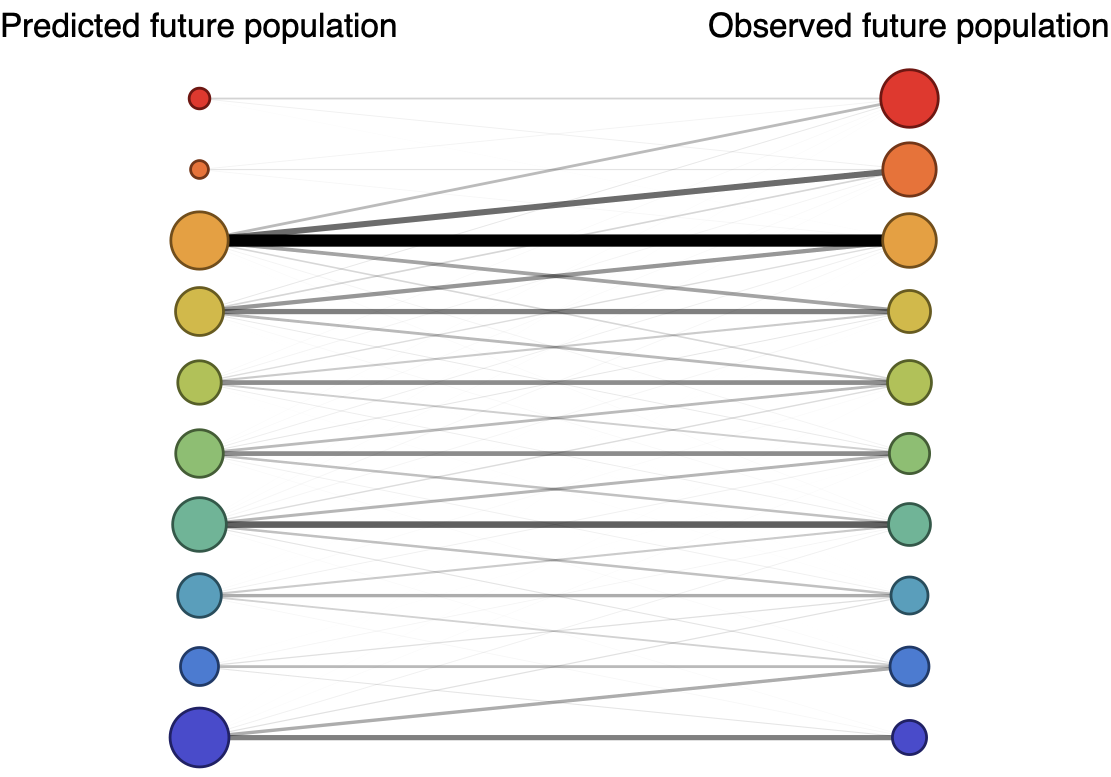
Good fit
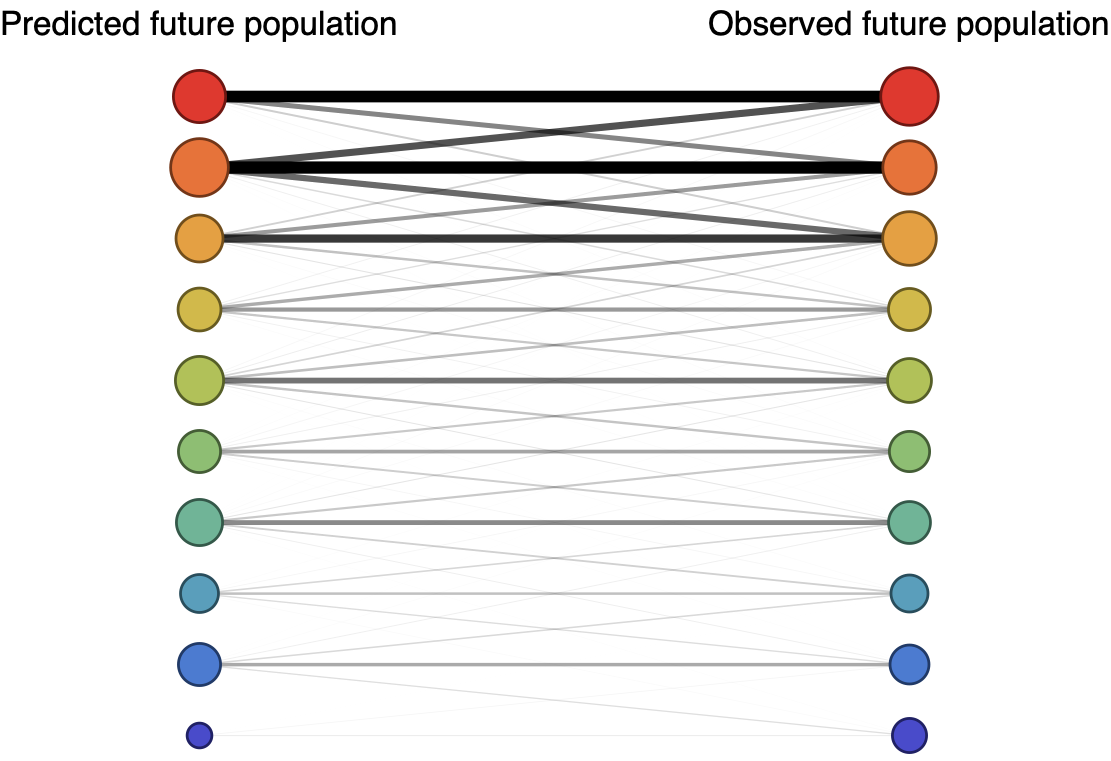
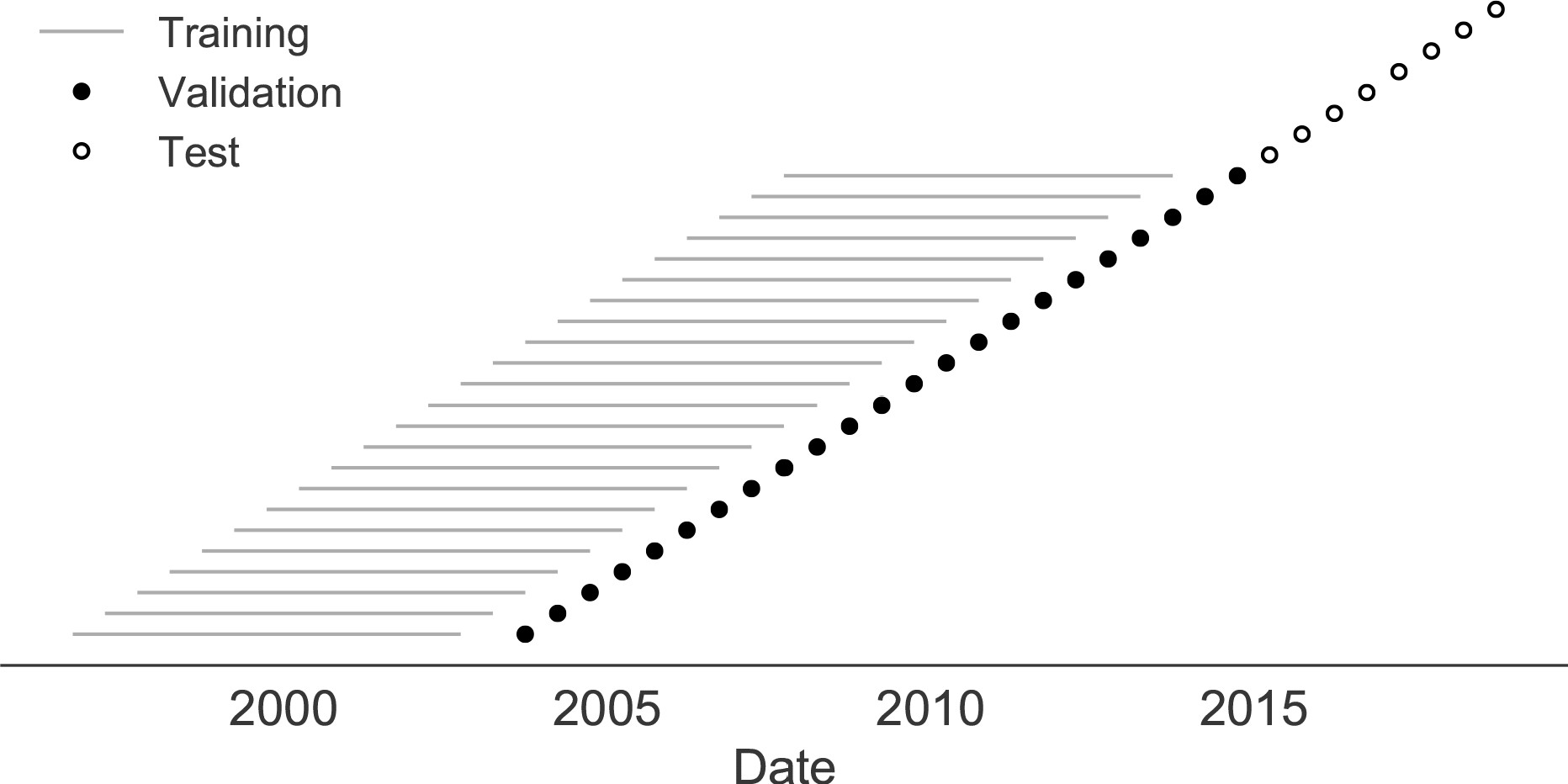
Train in 6-year sliding windows from 1995 to 2015 with most recent years held out as test
Composite models favor combinations of HI drift, local branching index and non-epitope fitness
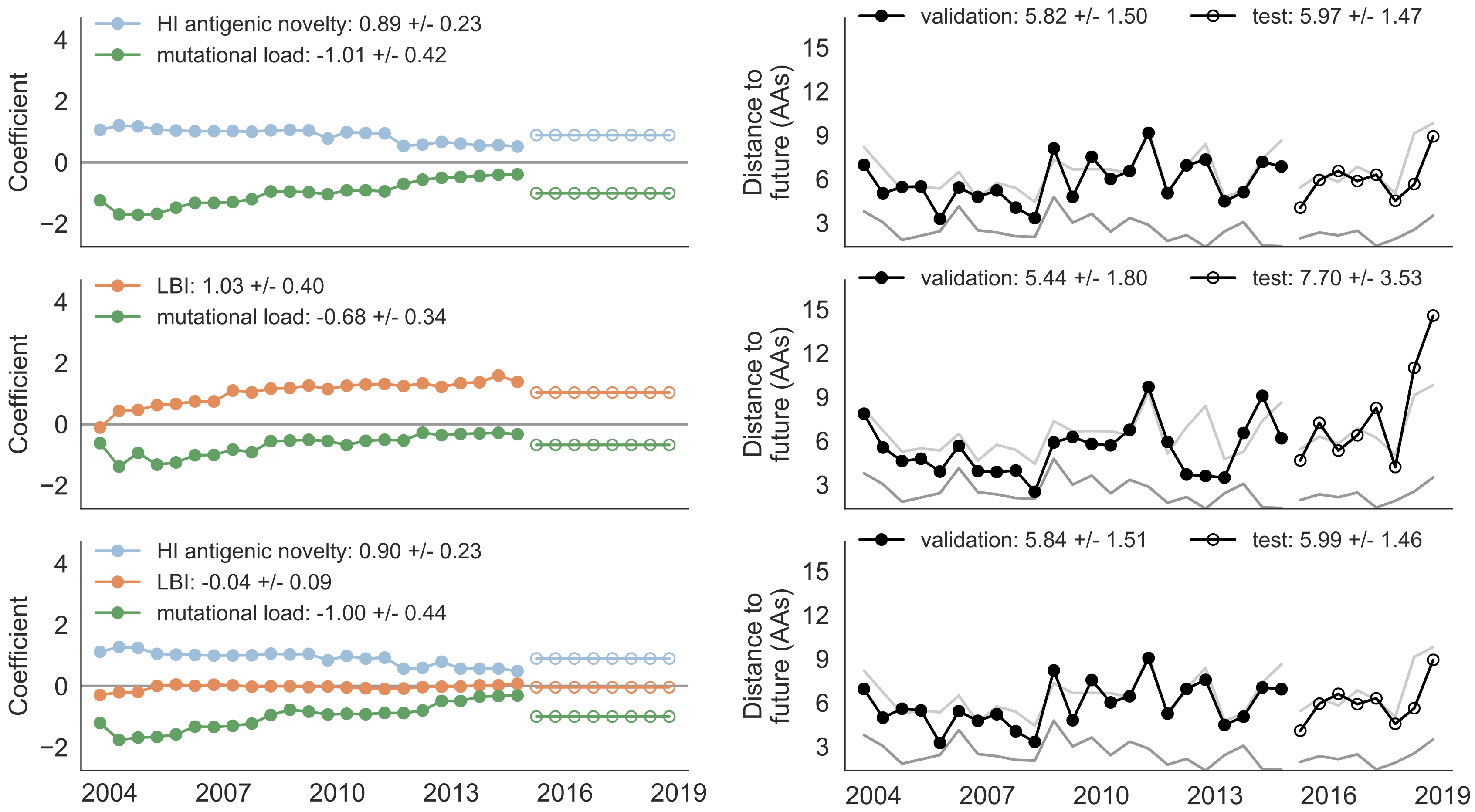
Model successfully predicts clade growth and best pick from model is generally close to best possible retrospective pick
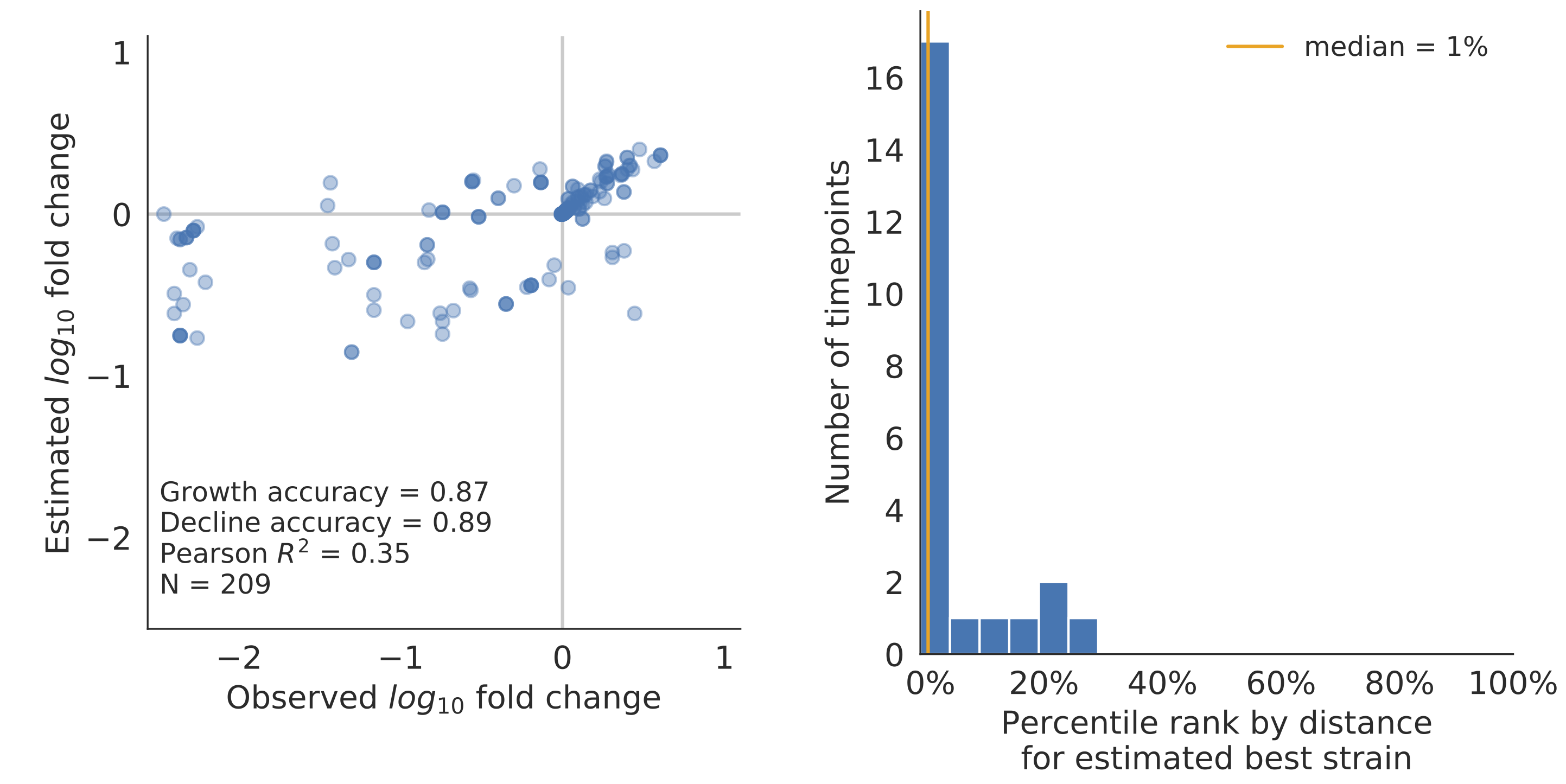
Two main issues
- We swapped from assessing clade frequencies to earth mover's distance because our clade assignments were not stable across trees built at different timepoints, while clade frequencies are the more natural metric.
- Strain fitness $f_i$ is largely fixed by the "fundamentals" of the strain rather than being learned from frequency behavior.
SARS-CoV-2
SARS-CoV-2 continues to show remarkable capacity for evolution
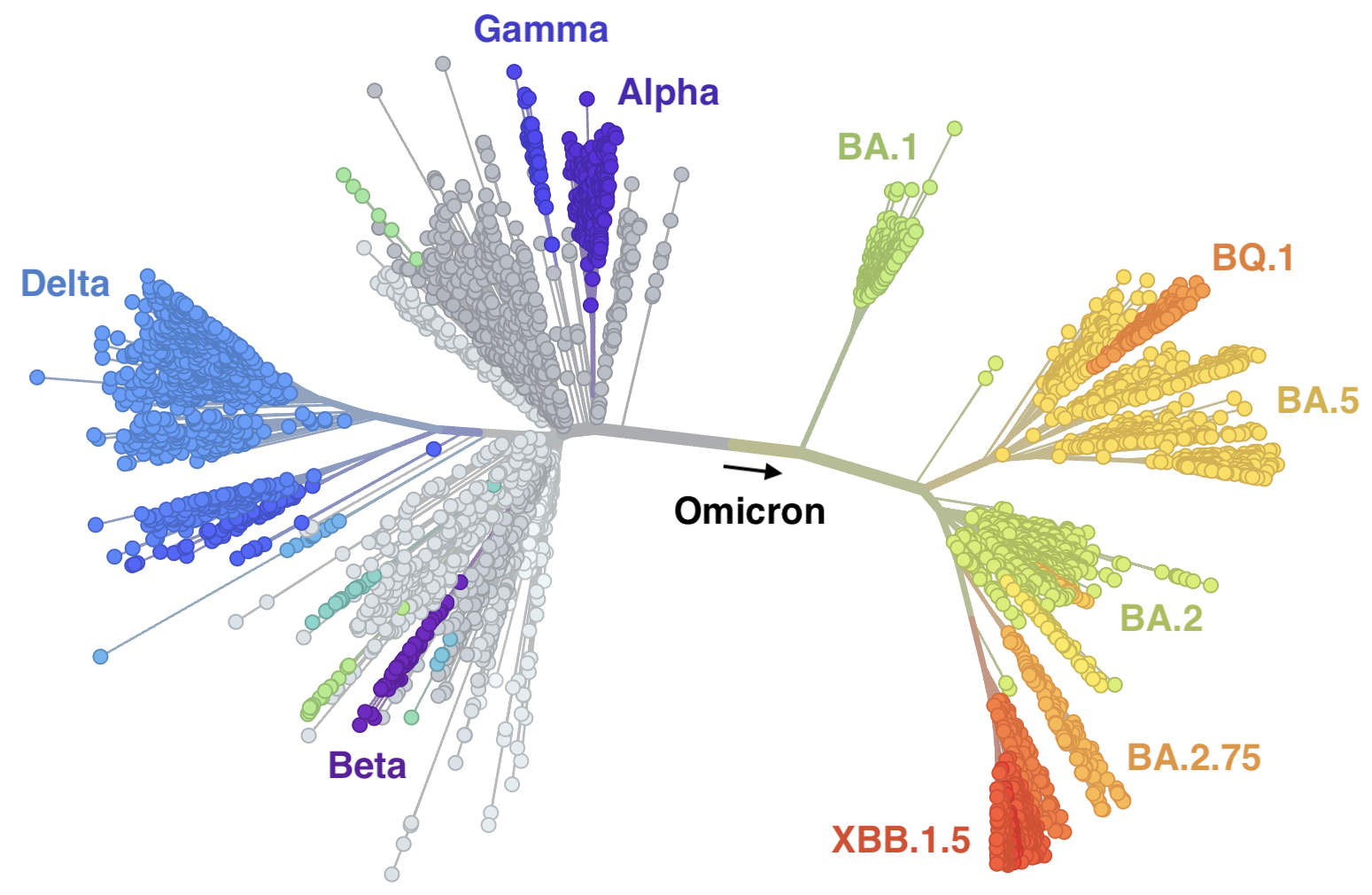
Mutations at spike S1 propel escape from population immunity
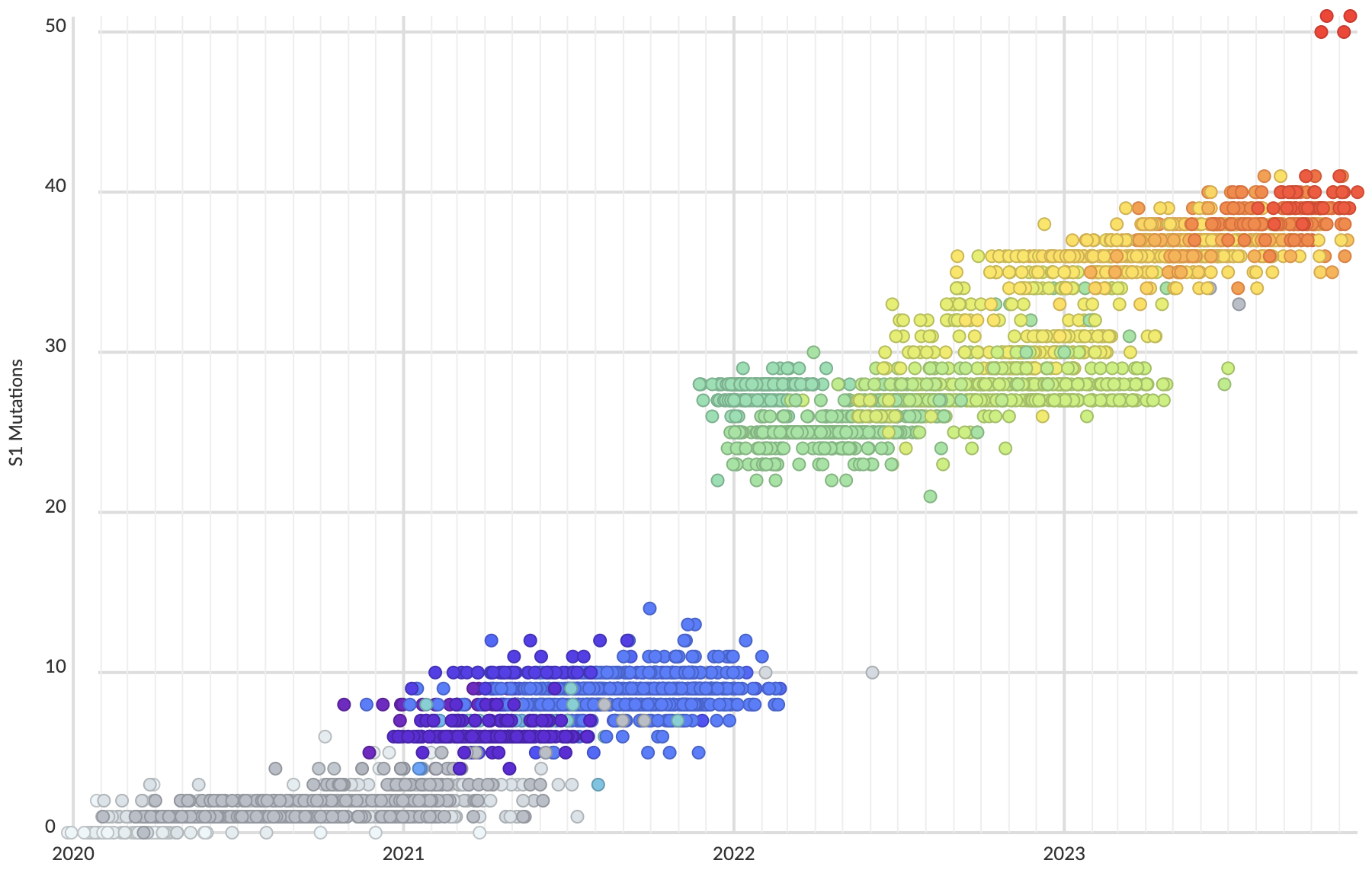
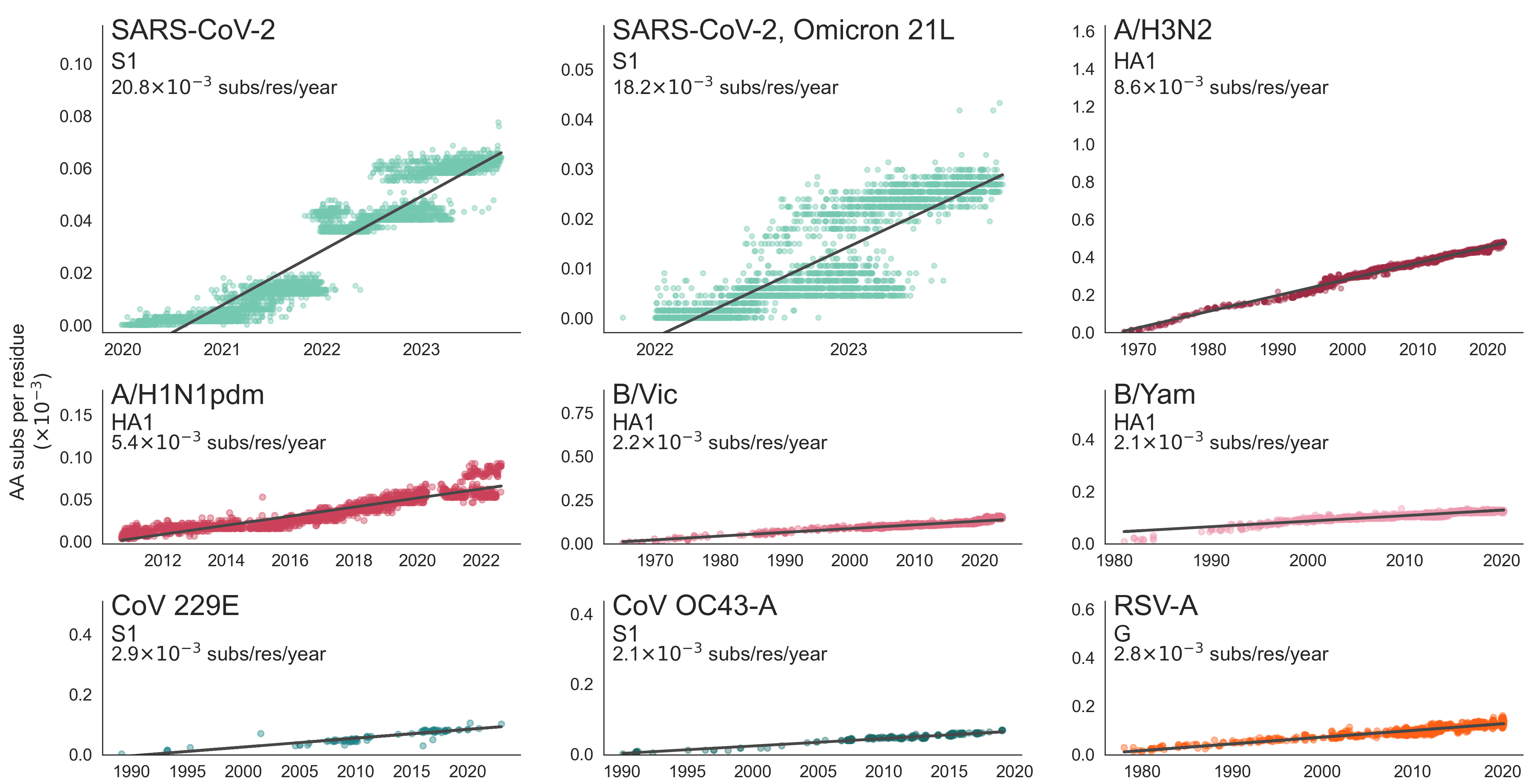
These mutations are accruing much more rapidly than other endemic viruses
New variants emerge that escape from existing population immunity and spread rapidly
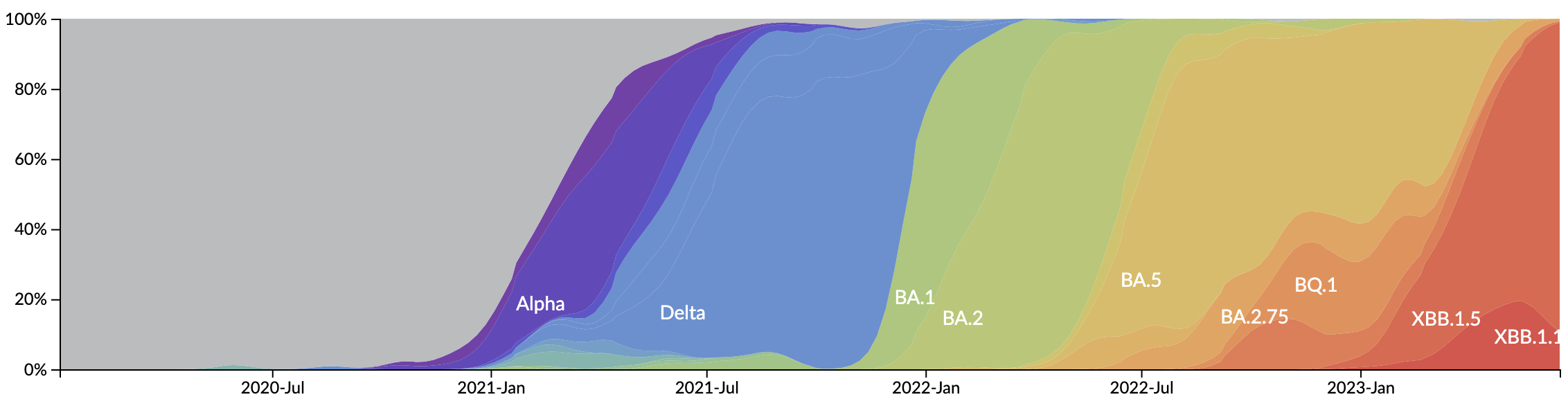
Novel variants sweep globally in months rather than years
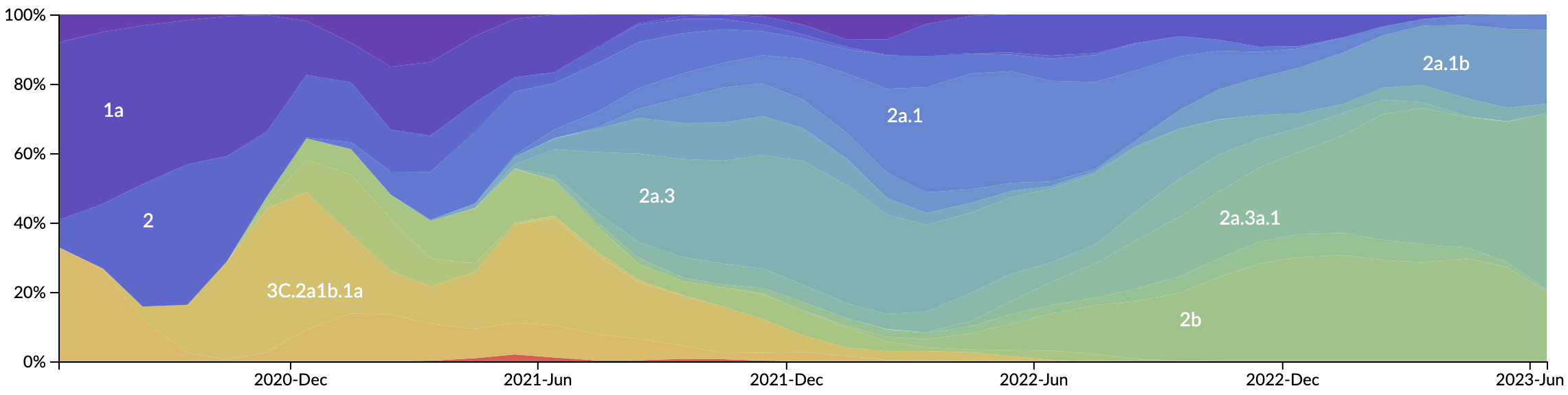
Variant frequency dynamics
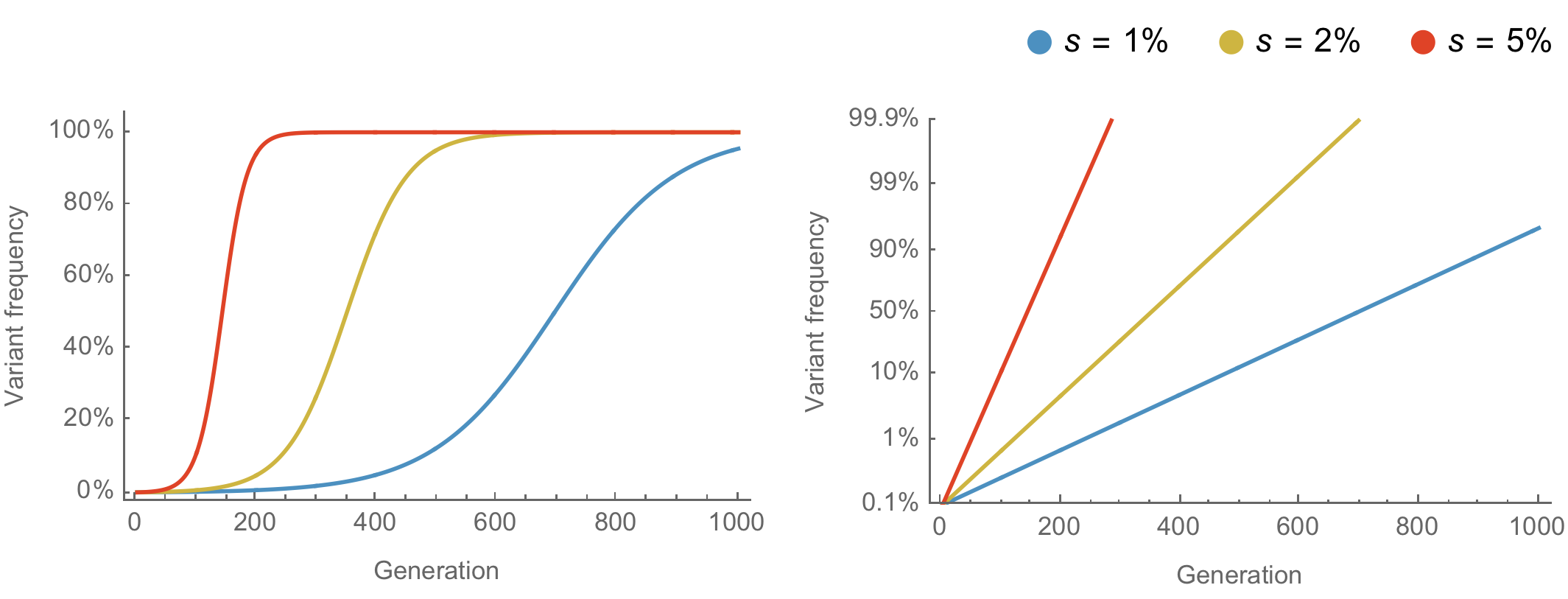
Population genetic expectation of variant frequency under selection
$x' = \frac{x \, (1+s)}{x \, (1+s) + (1-x)}$ for frequency $x$ over one generation with selective advantage $s$
$x(t) = \frac{x_0 \, (1+s)^t}{x_0 \, (1+s)^t + (1-x_0)}$ for initial frequency $x_0$ over $t$ generations
Trajectories are linear once logit transformed via $\mathrm{log}(\frac{x}{1 - x})$
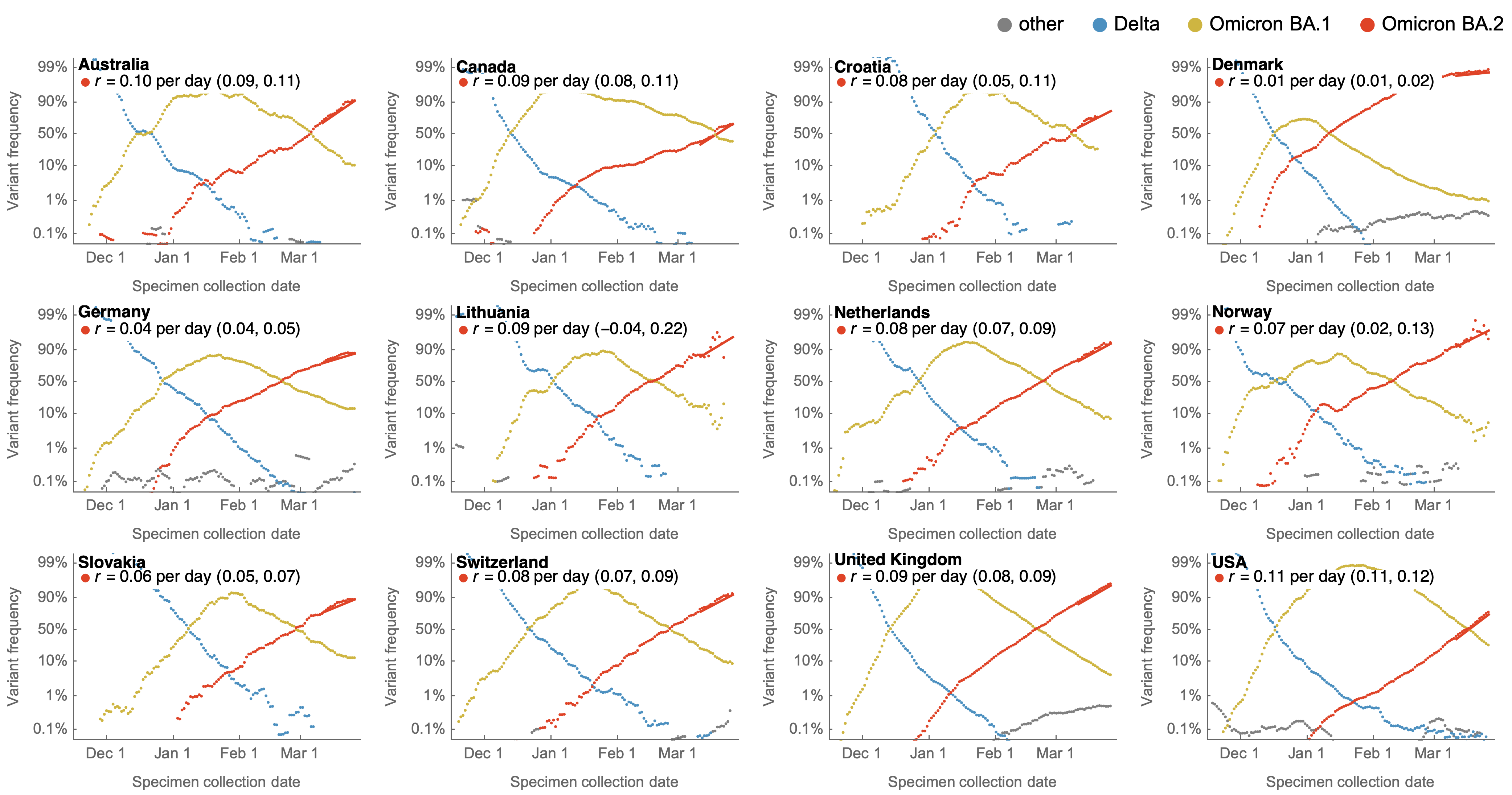
Consistent frequency dynamics in logit space (BA.2 Mar 2022)
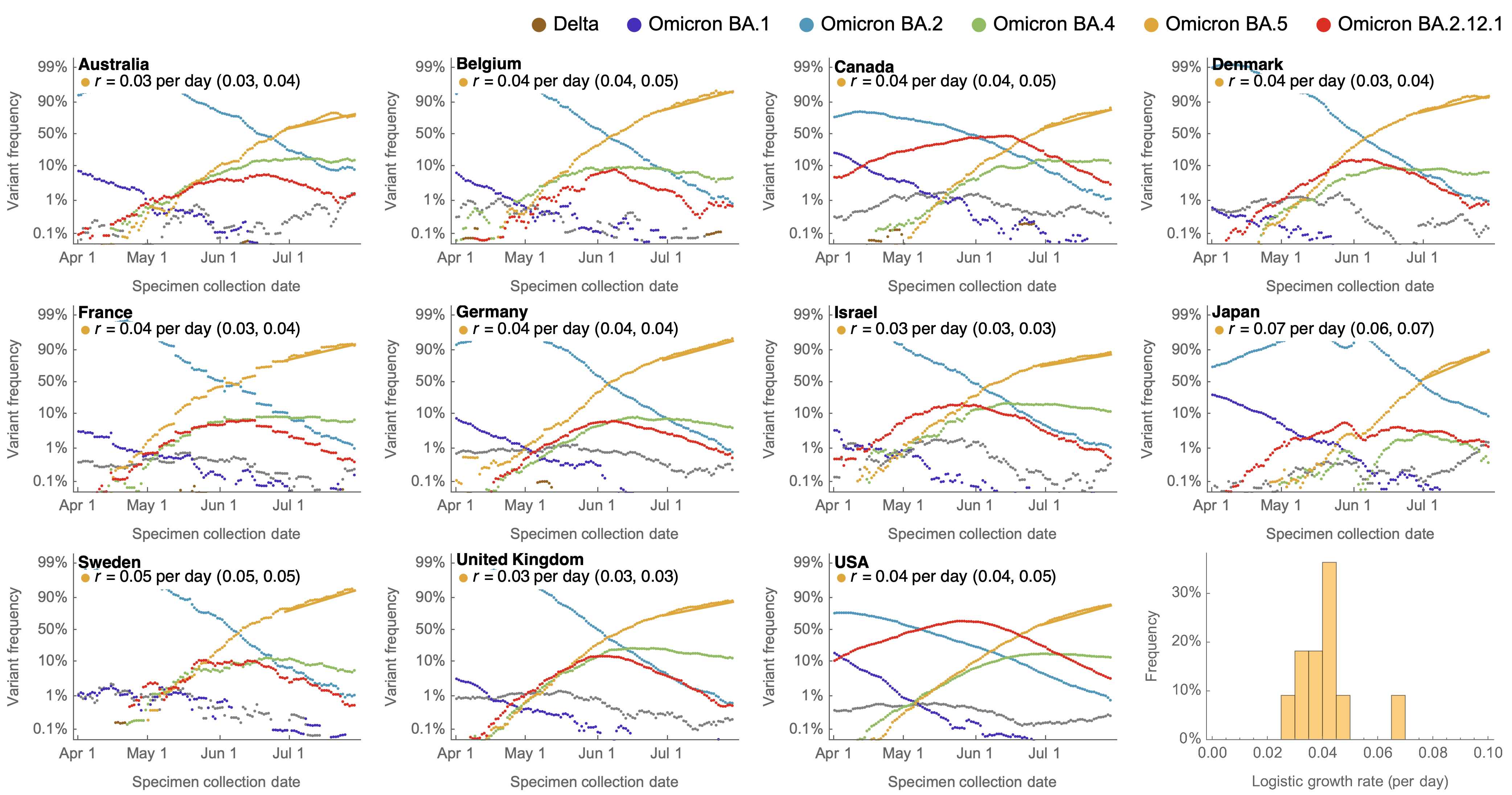
Consistent frequency dynamics in logit space (BA.5 Jul 2022)
Consistent frequency dynamics in logit space (JN.1 Dec 2023)
Multinomial logistic regression
Multinomial logistic regression across $n$ variants models the probability of a virus sampled at time $t$ belonging to variant $i$ as
$$\mathrm{Pr}(X = i) = x_i(t) = \frac{p_i \, \mathrm{exp}(f_i \, t)}{\sum_{1 \le j \le n} p_j \, \mathrm{exp}(f_j \, t) }$$
with $2n$ parameters consisting of $p_i$ the frequency of variant $i$ at initial timepoint and $f_i$ the growth rate or fitness of variant $i$.
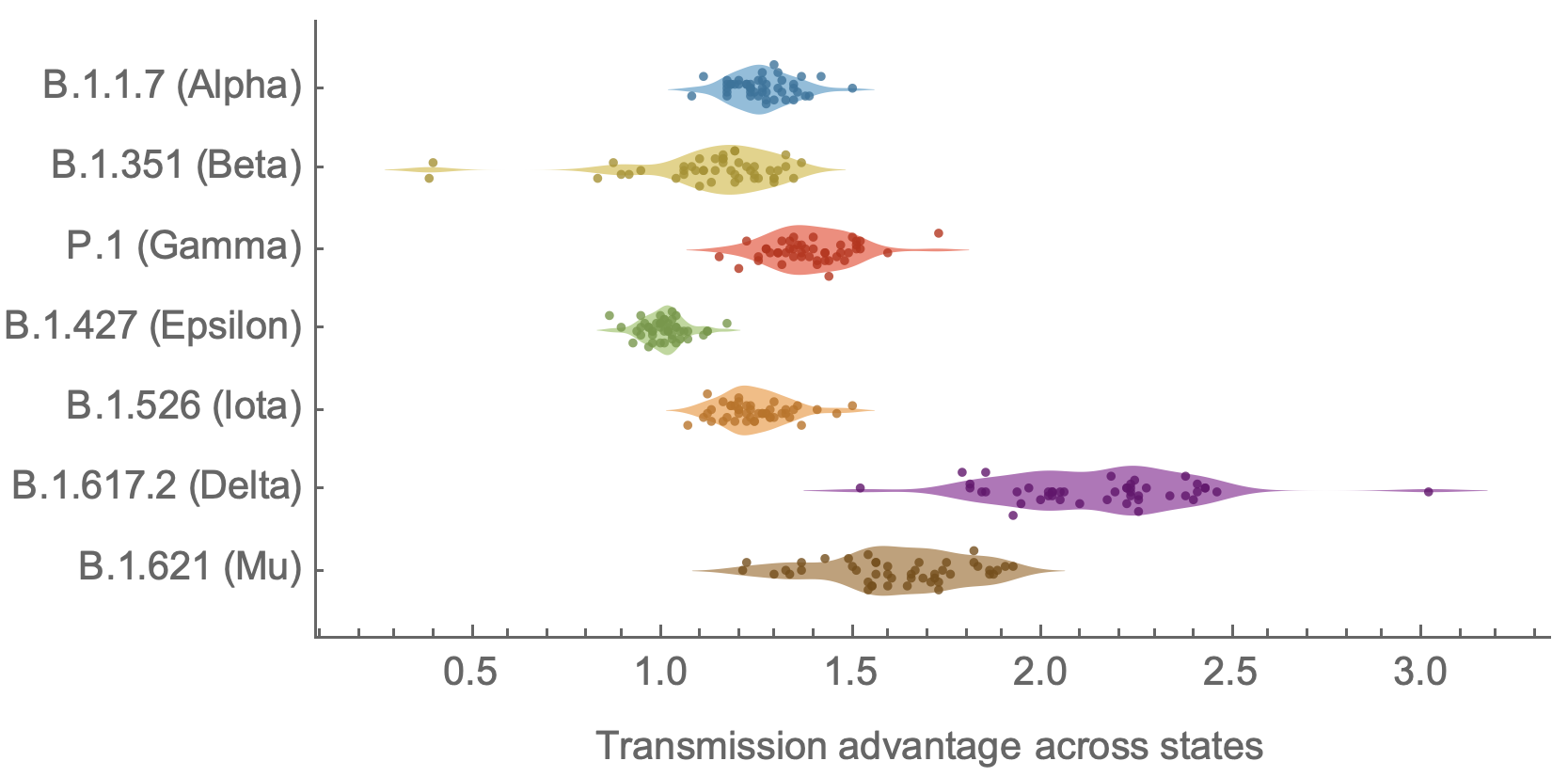
Original VOC viruses had substantially increased transmissibility
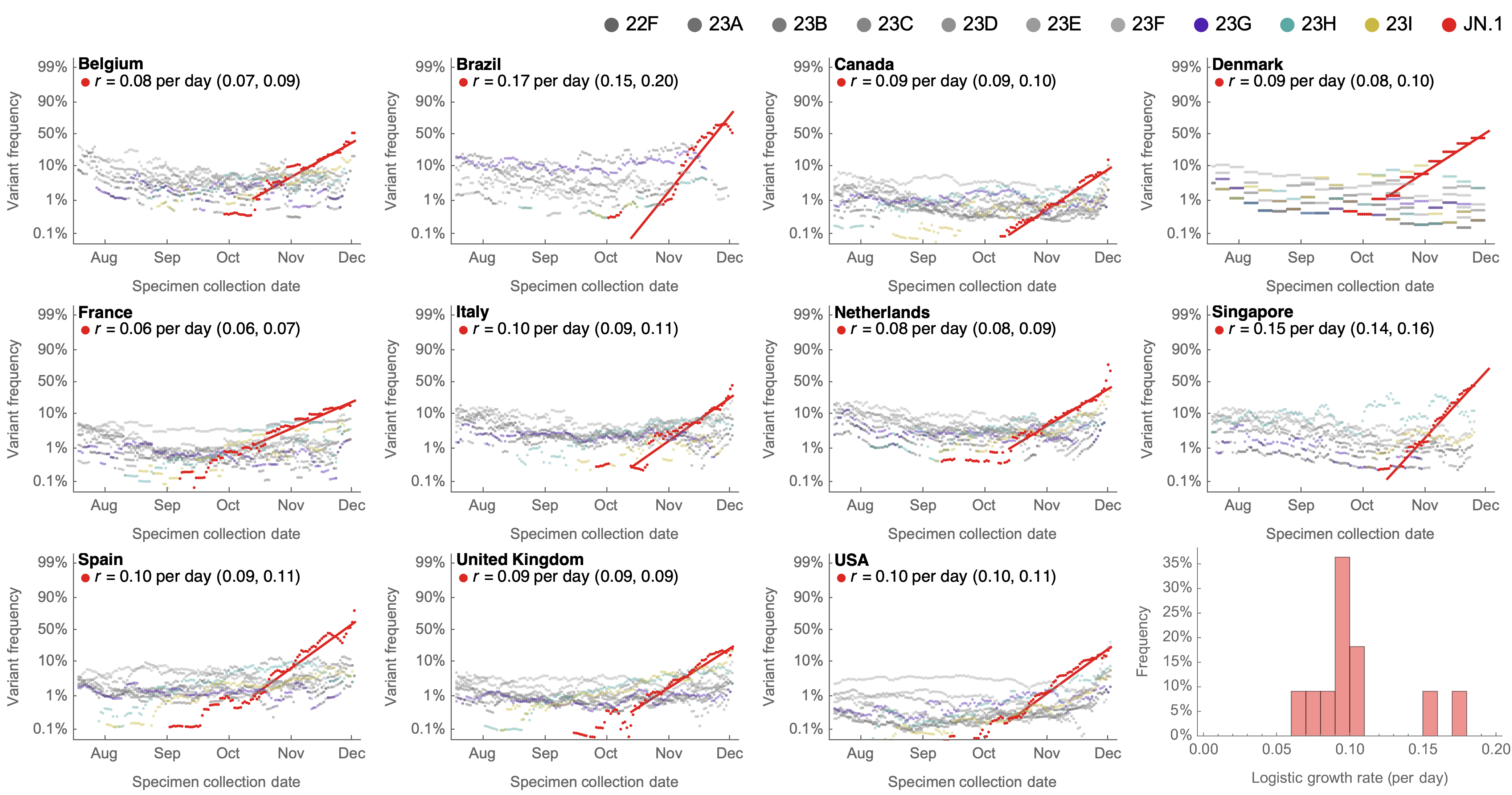
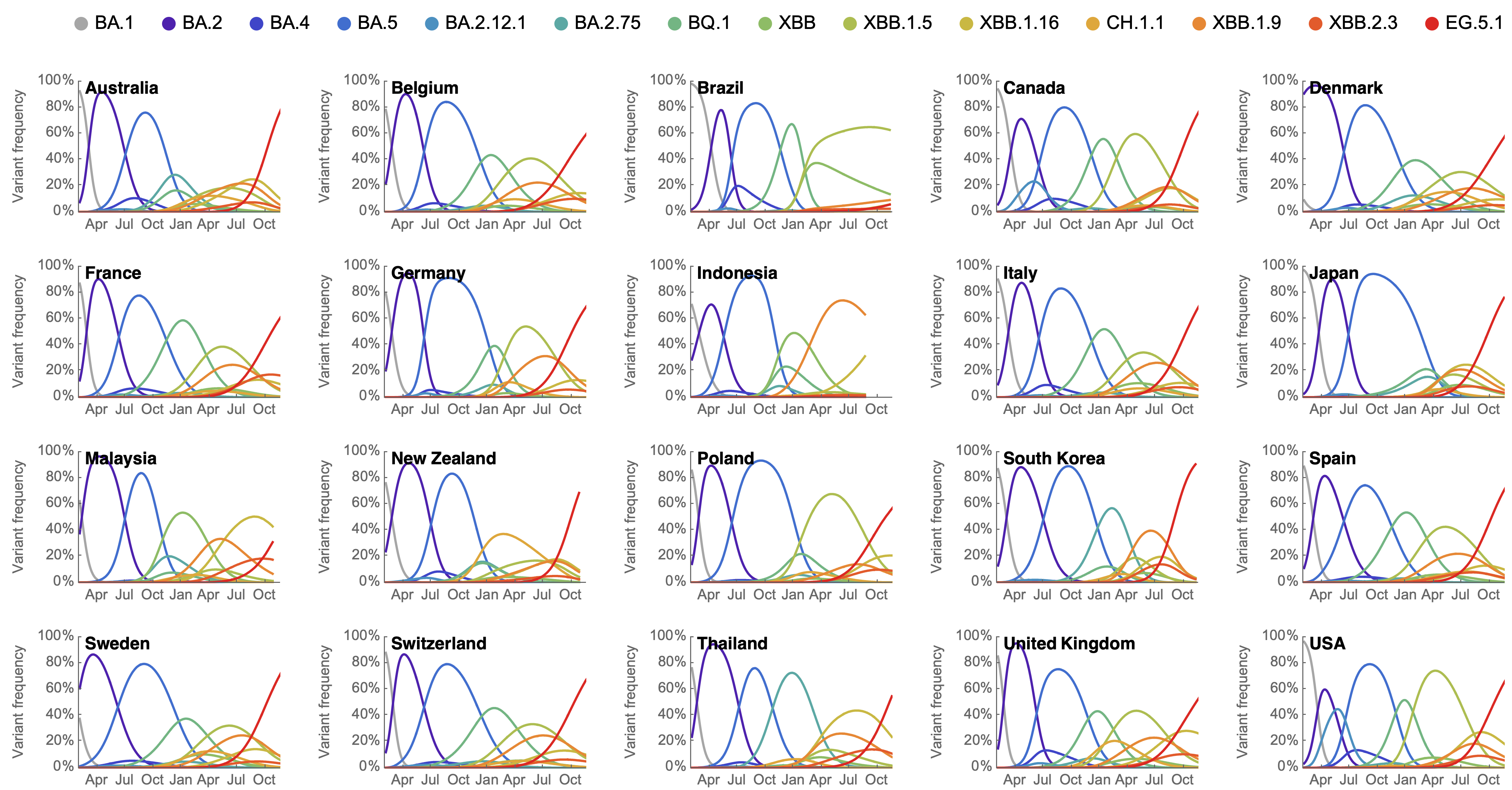
Variant frequencies across countries from Feb 2022 to present
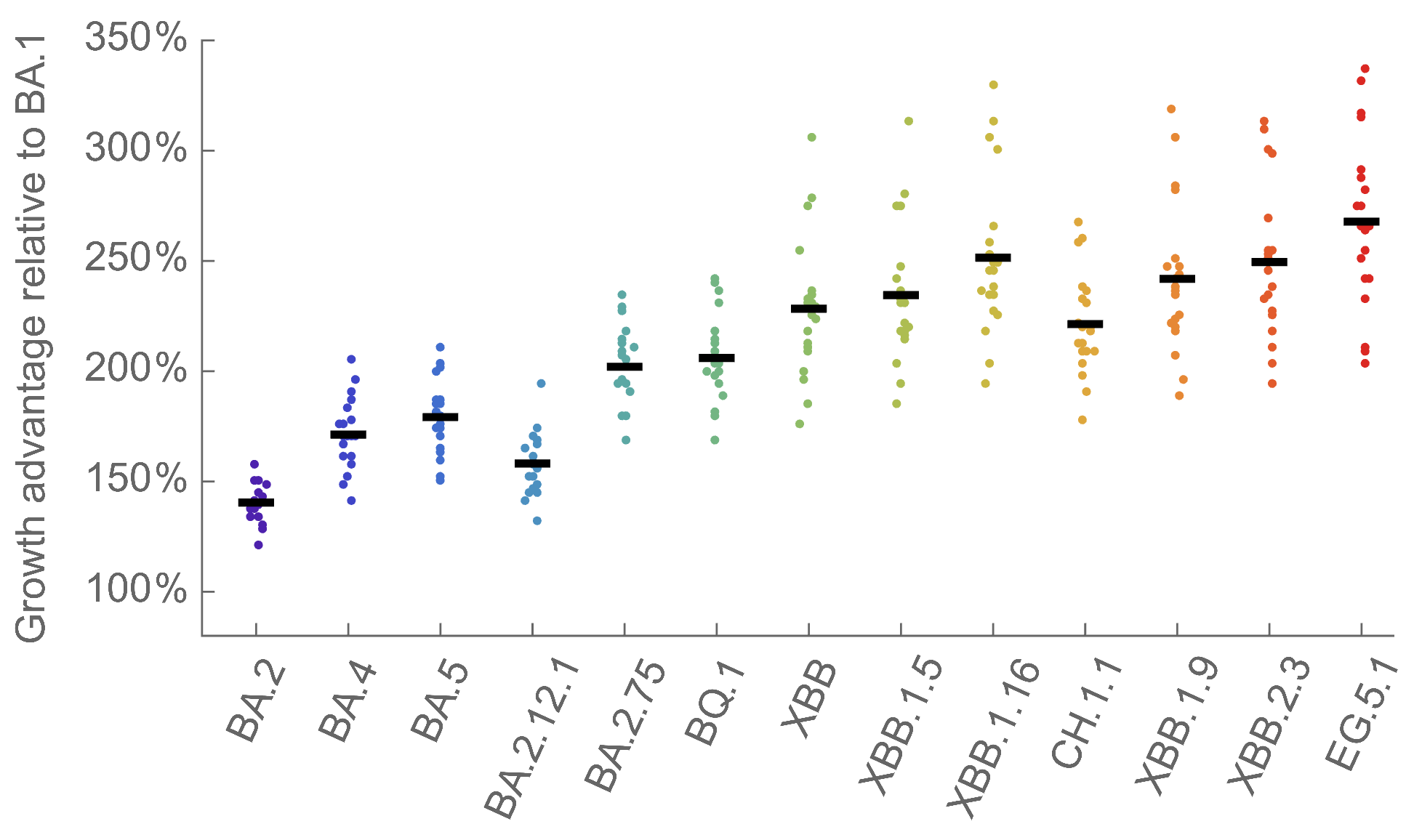
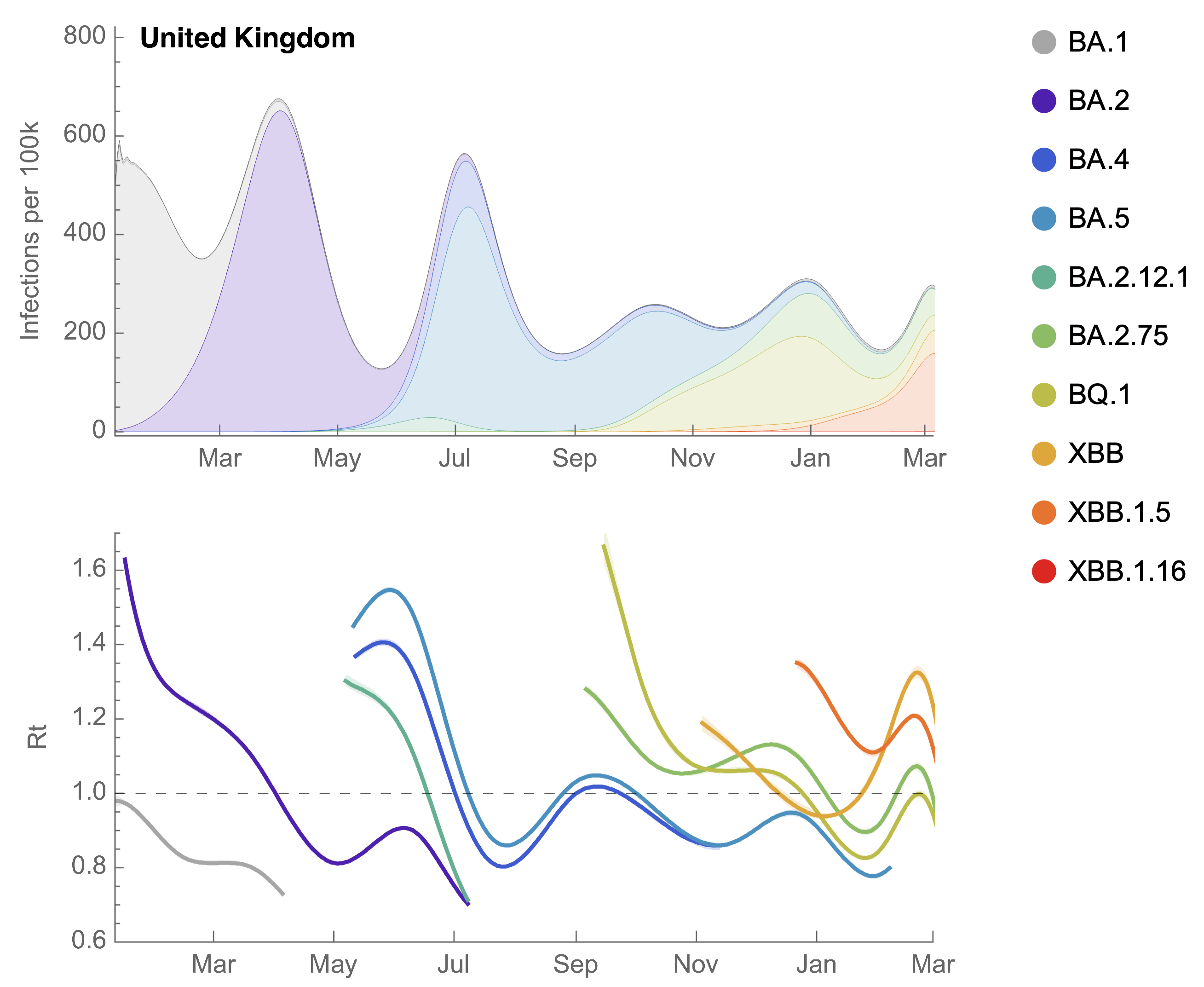
We find that recent variants like EG.5.1 are ~250% fitter than original Omicron BA.1
Evolution driving epidemics
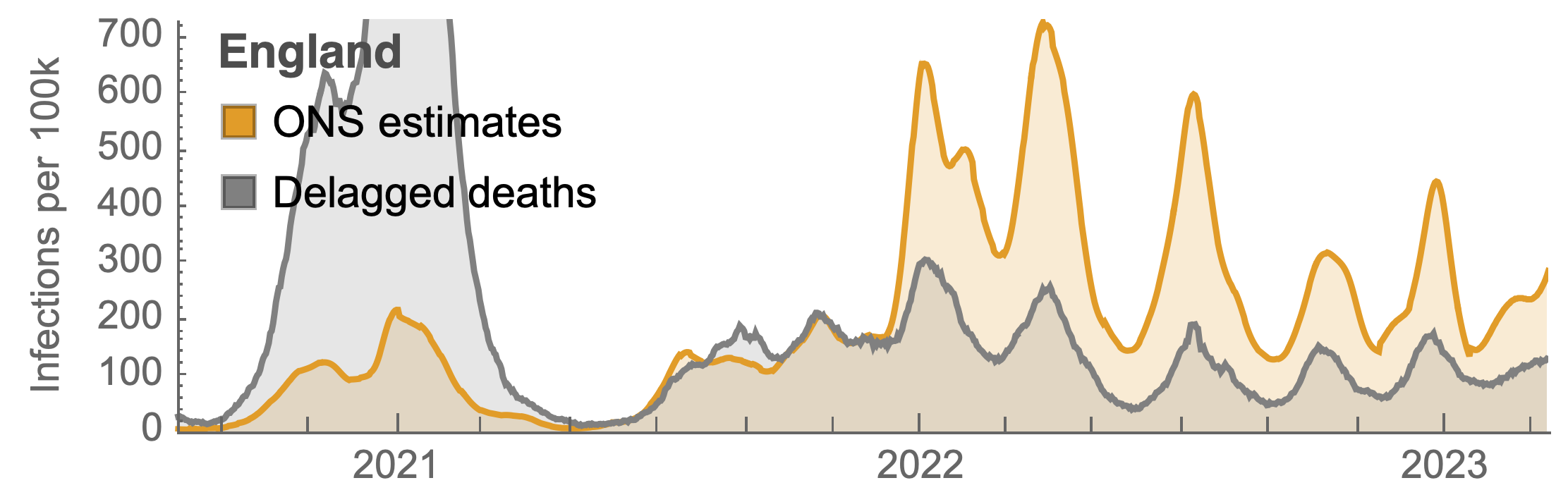
Many fewer reported cases in England post-Omicron
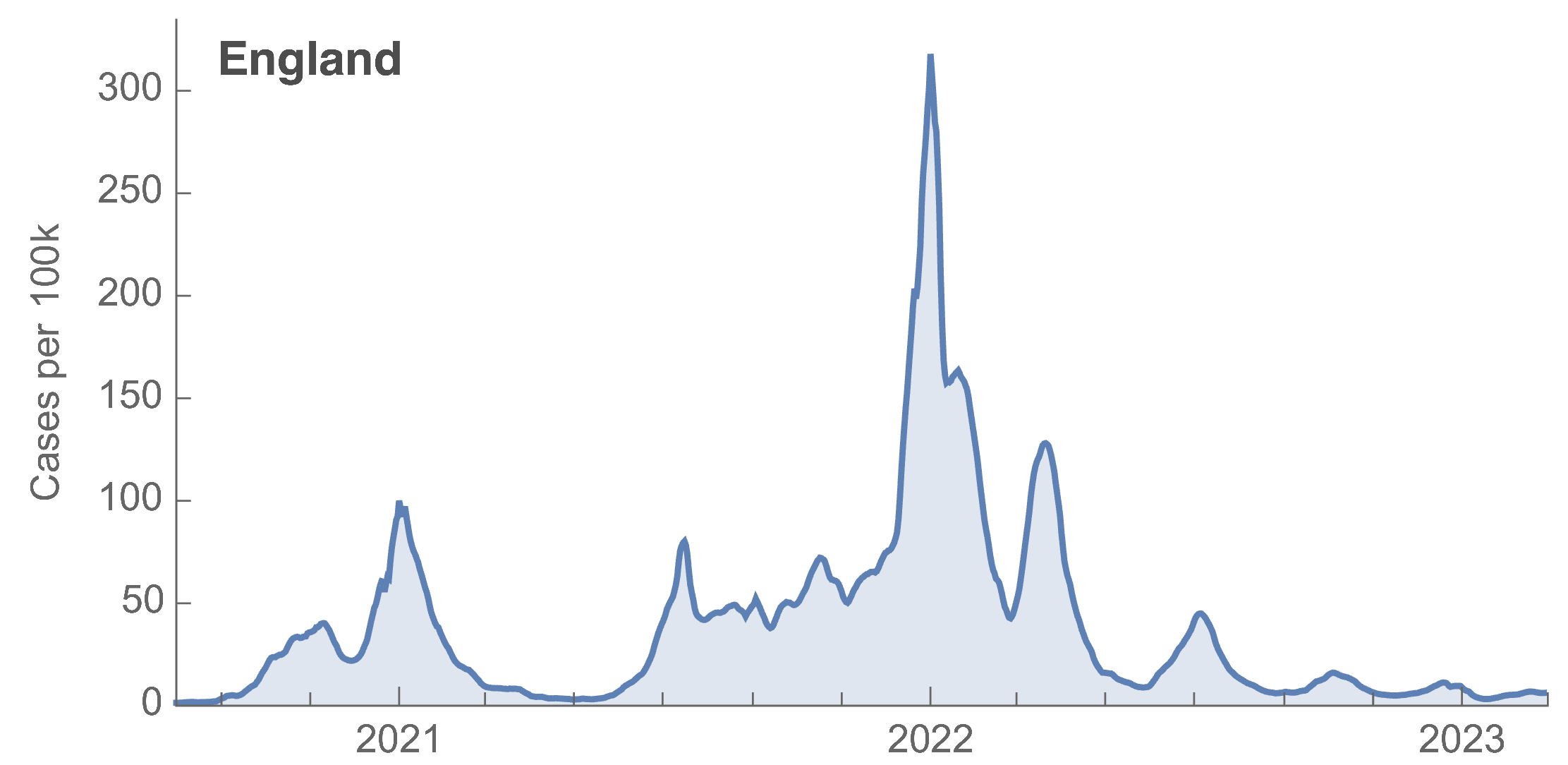
ONS Infection Survey provides rare source of ground truth
Roughly 1 in 3 infections detected in 2021, while 1 in 40 in 2023
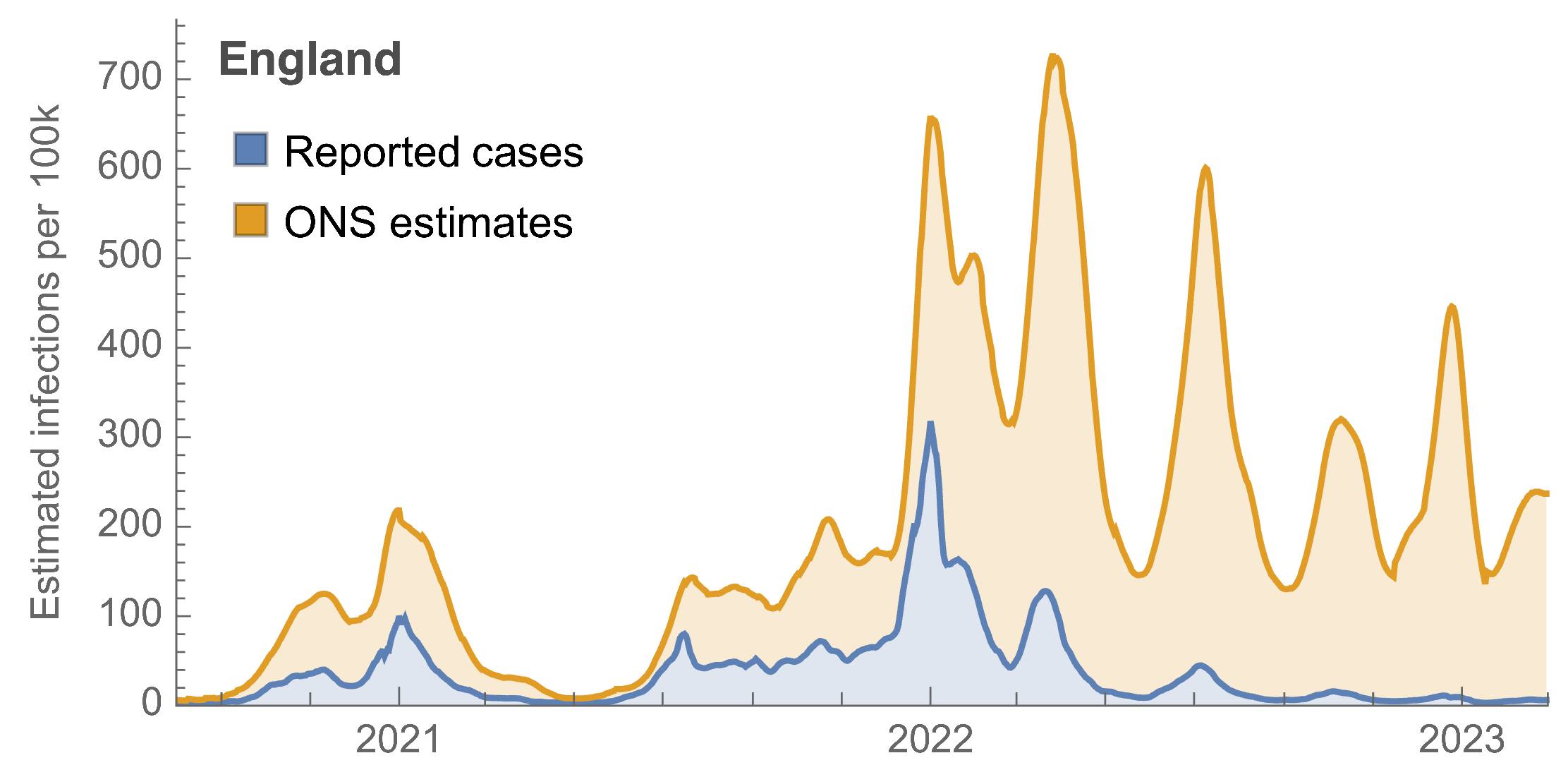
Partitioning ONS incidence based on sequencing data shows variant-driven epidemics
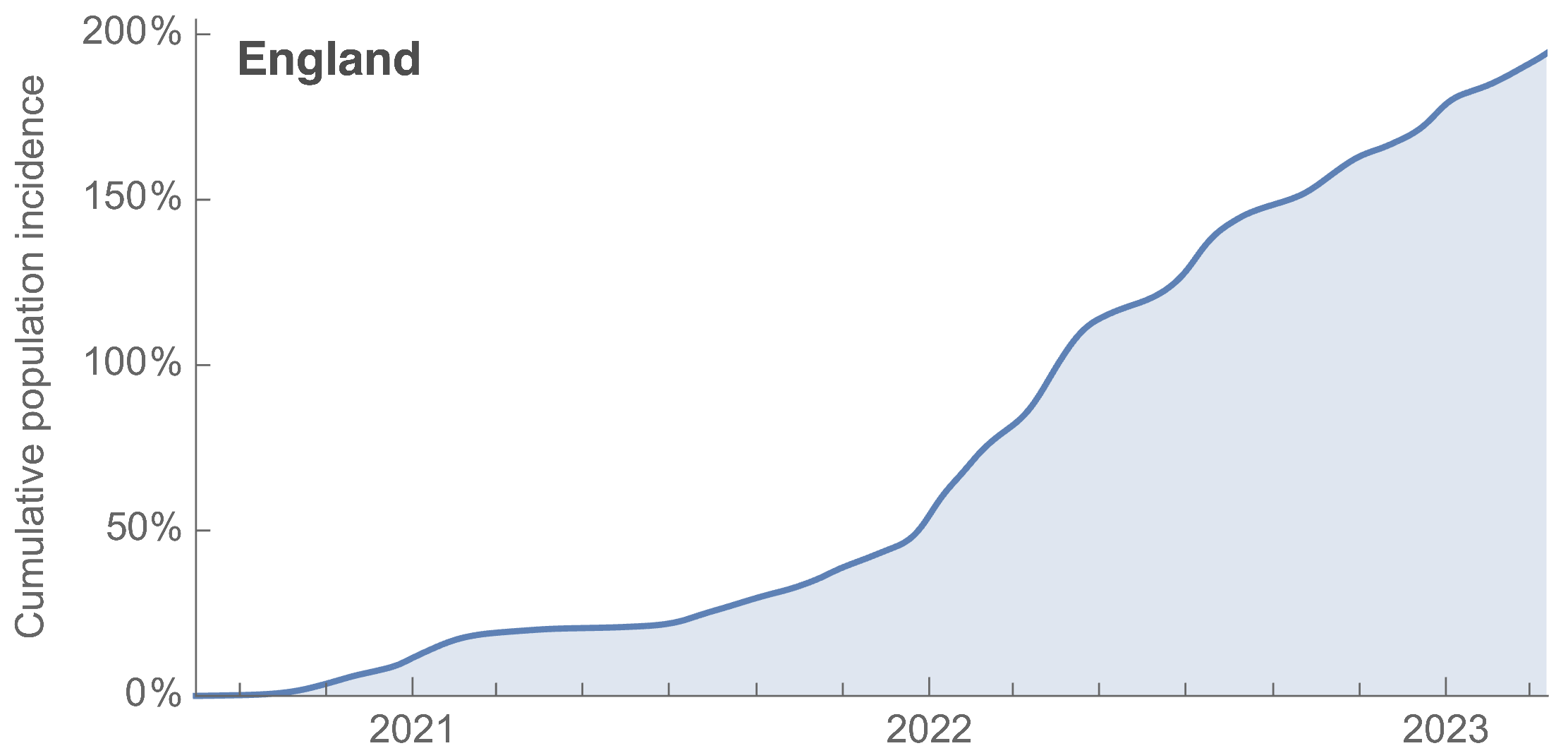
~110% population attack rate from March 2022 to March 2023
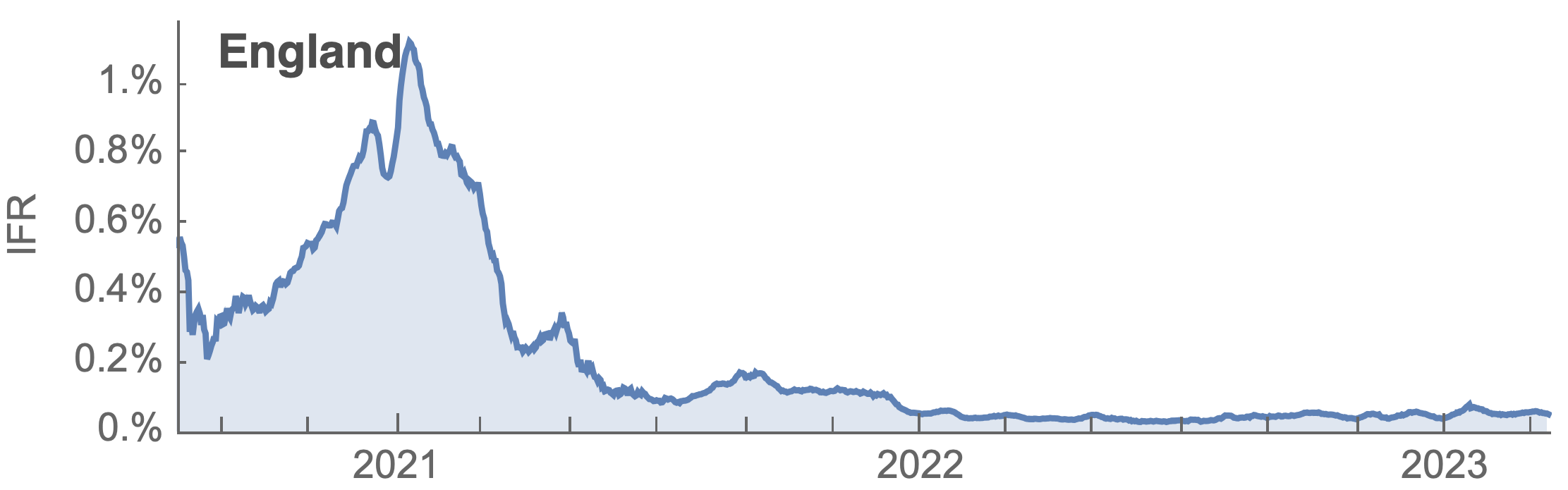
Post-Omicron period shows consistent IFR of 0.04%
Forecasting
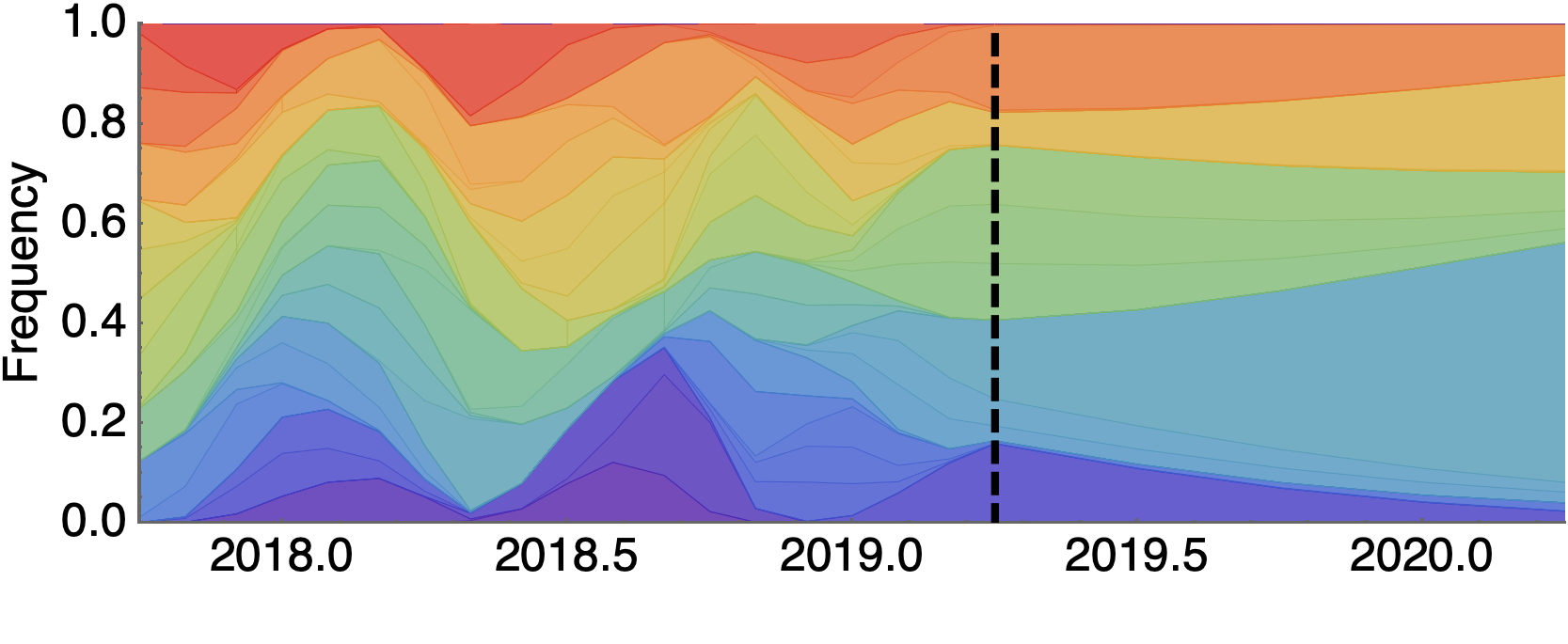
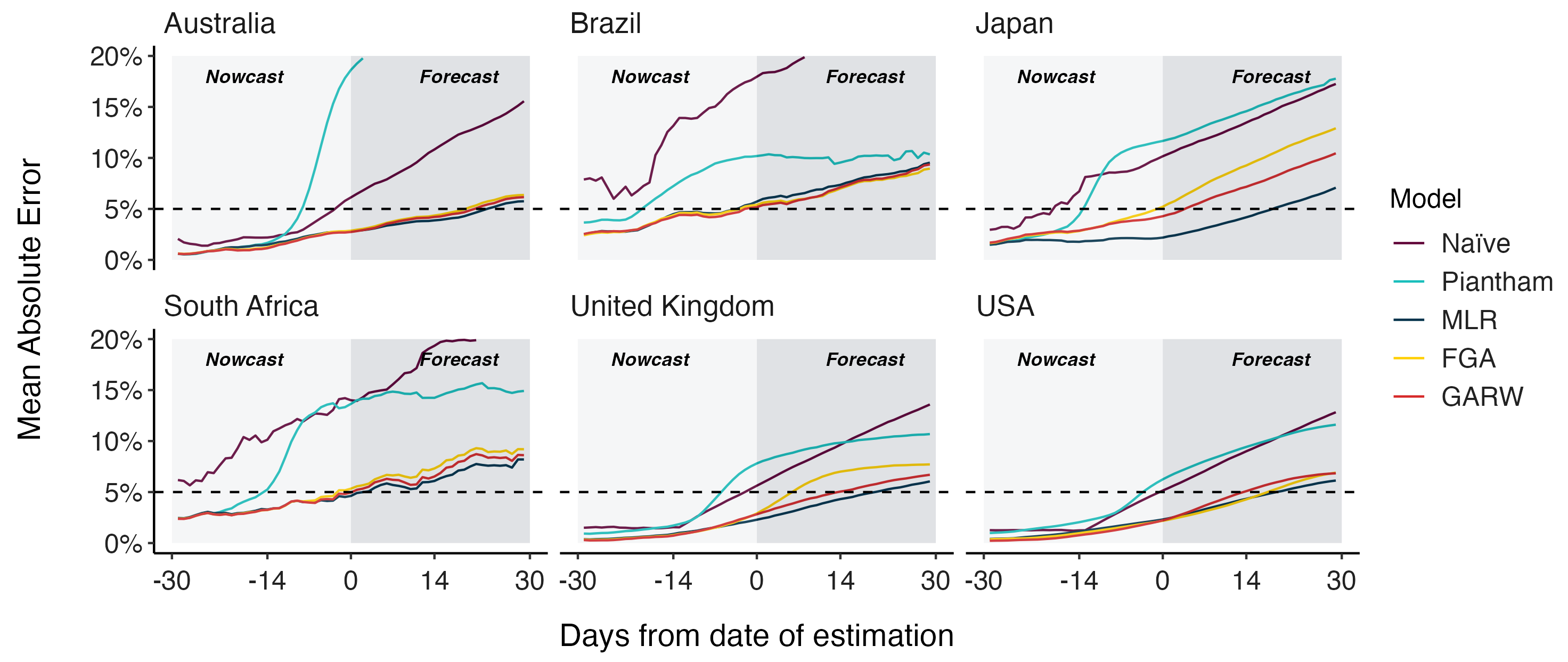
Assessing MLR models for short-term frequency forecasting
Retrospective projections twice monthly during 2022
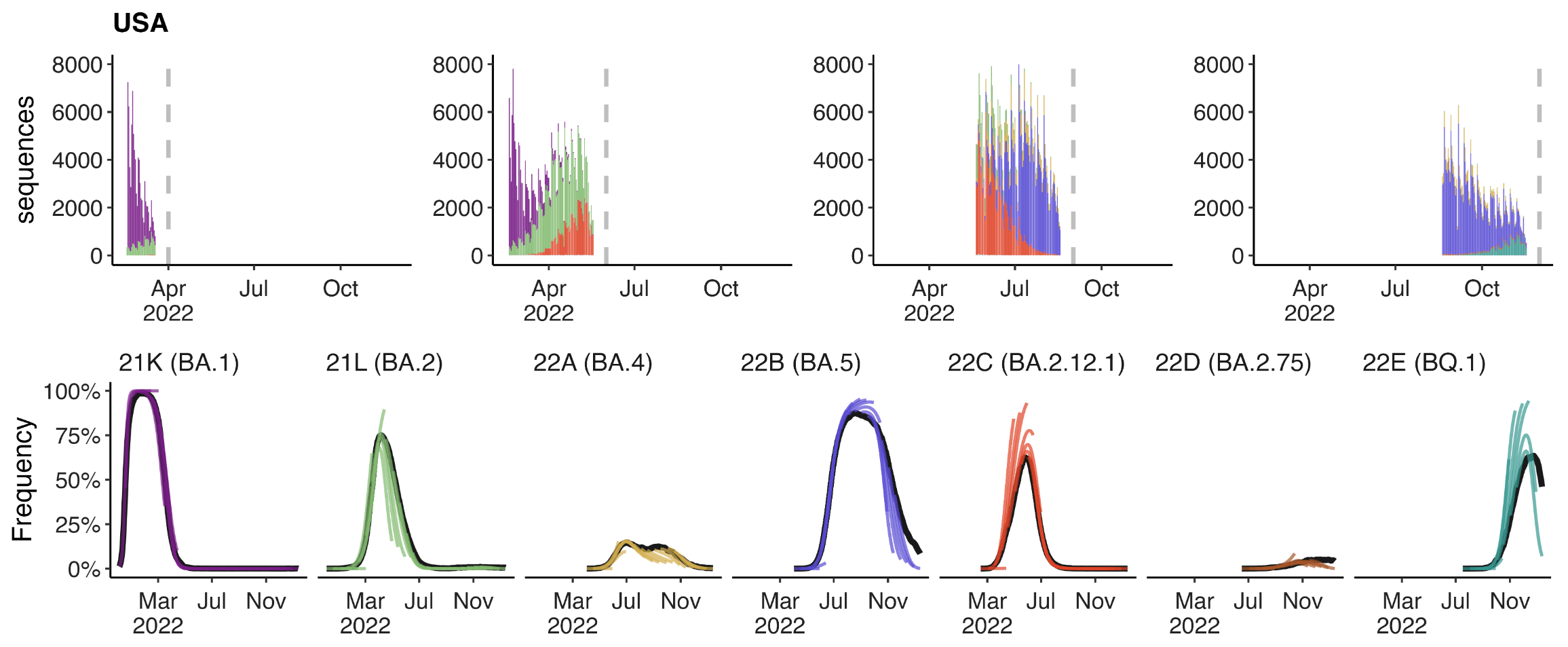
+30 day short-term forecasts across different countries
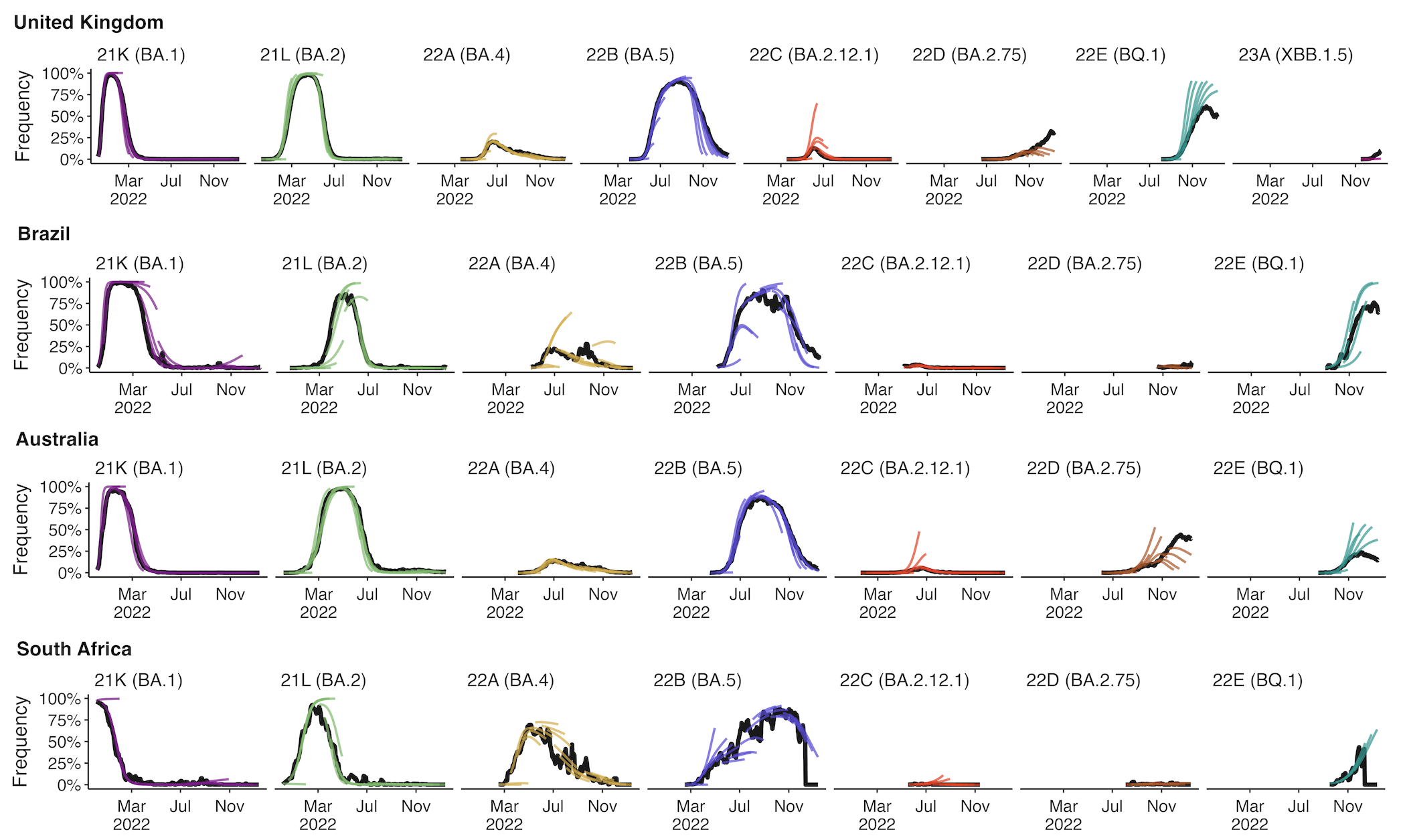
MLR models generate accurate short-term forecasts
30 days out, countries range from 6 to 10% mean absolute error
Clade and lineage forecasts continuously updated at nextstrain.org
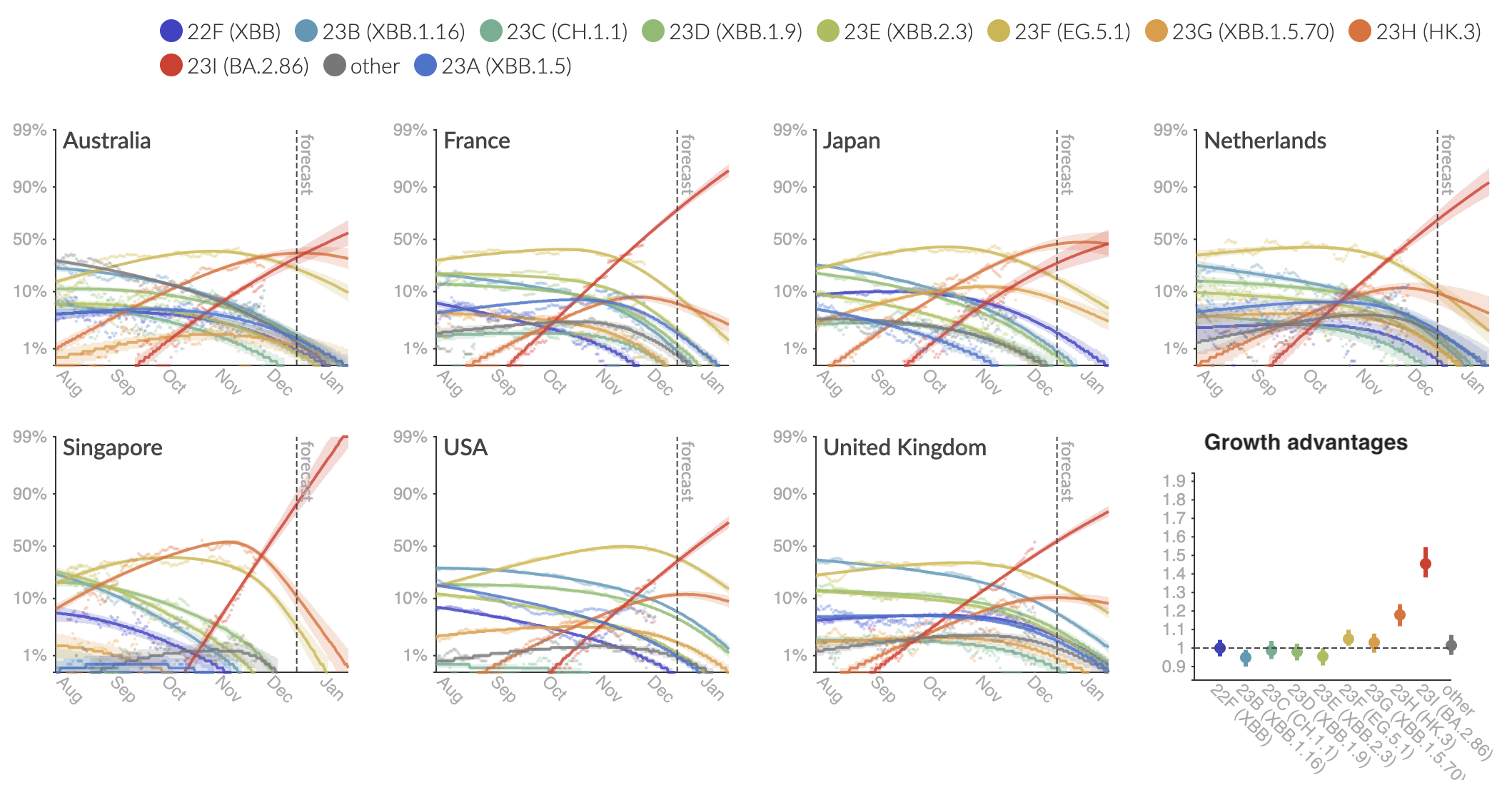
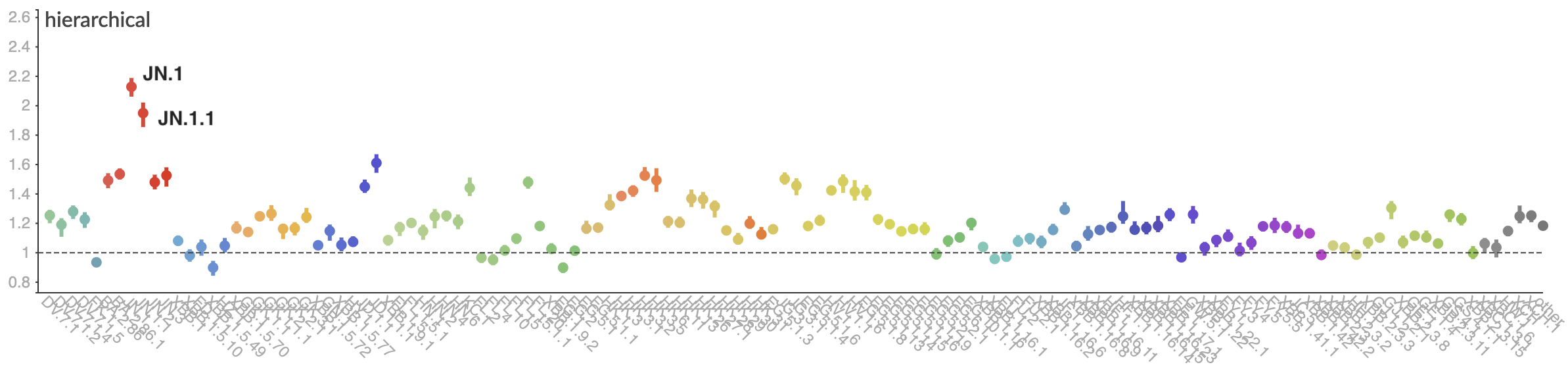
Pango-level growth advantages place JN.1 far ahead of the curve
Multinomial logistic regression should work well for SARS-CoV-2 prediction, except new variants have been emerging fast enough that the prediction horizon is really quite short
Could we predict the spread of new mutations using DMS data?
Escape from antibodies that potently neutralize BA.2
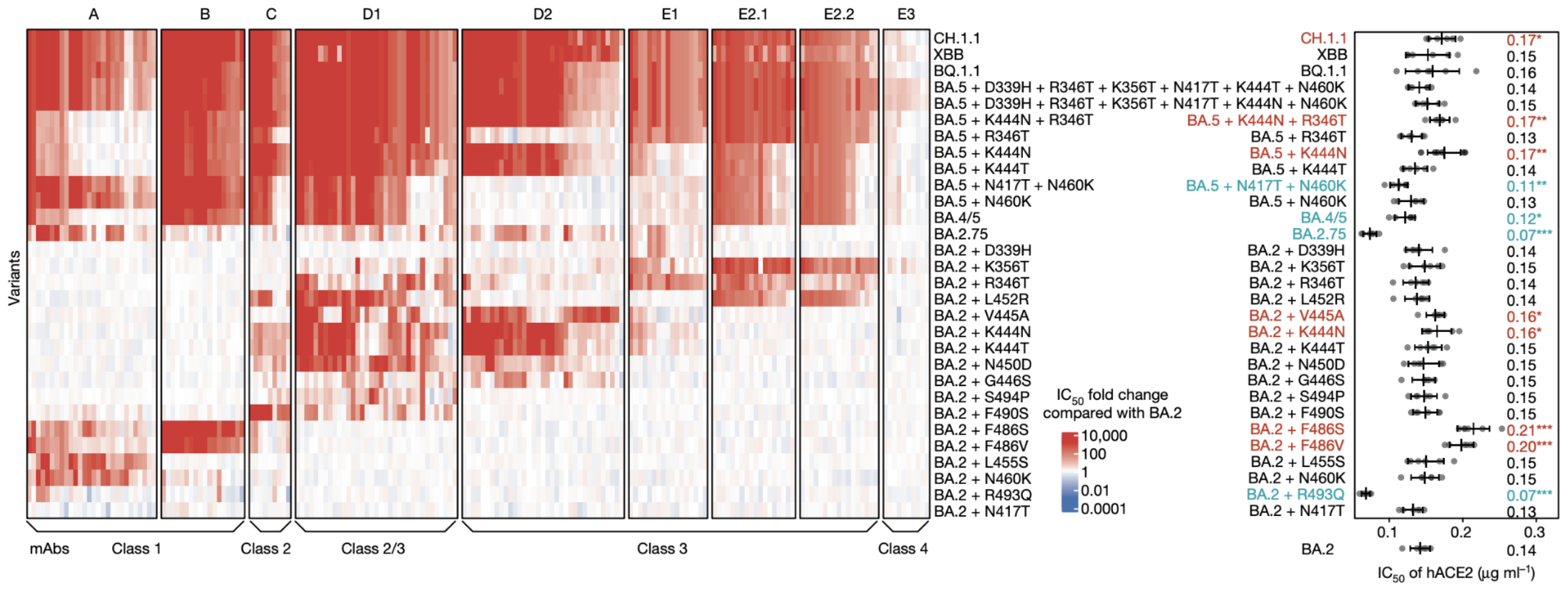
Can calculate escape of arbitrary RBD against antibodies known to neutralize BA.2
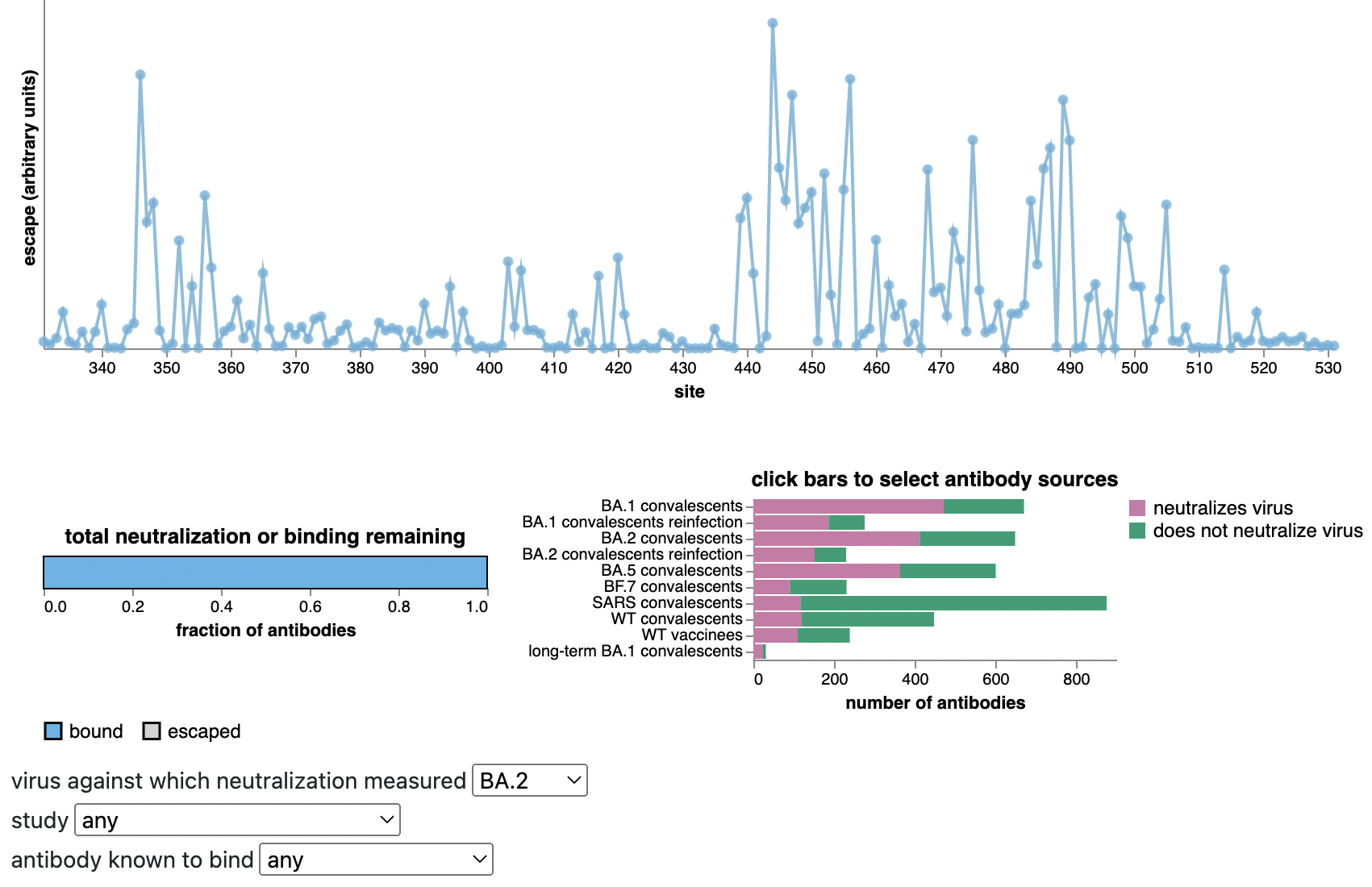
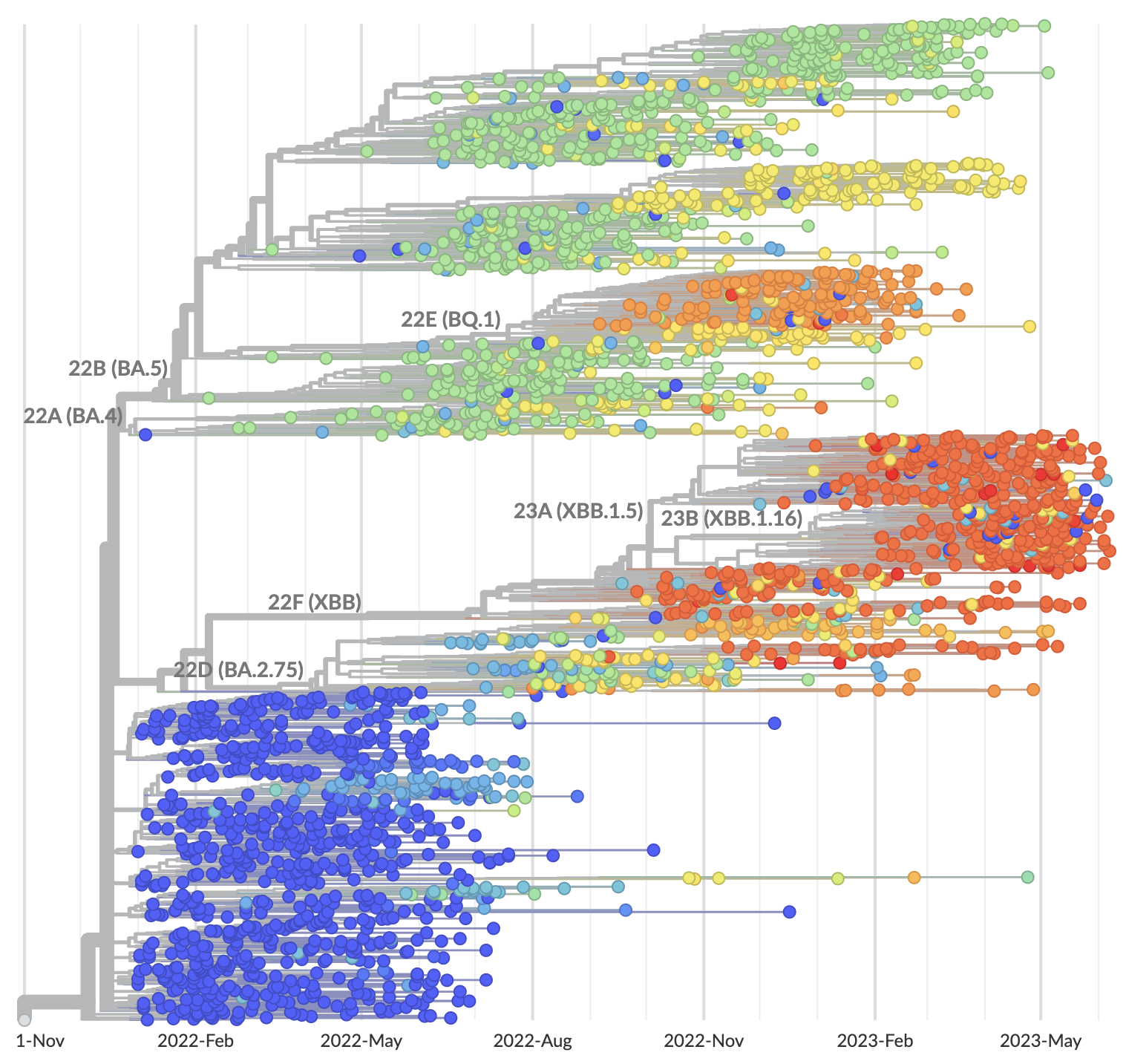
Strong correlation between DMS immune escape and lineage-level MLR growth advantage
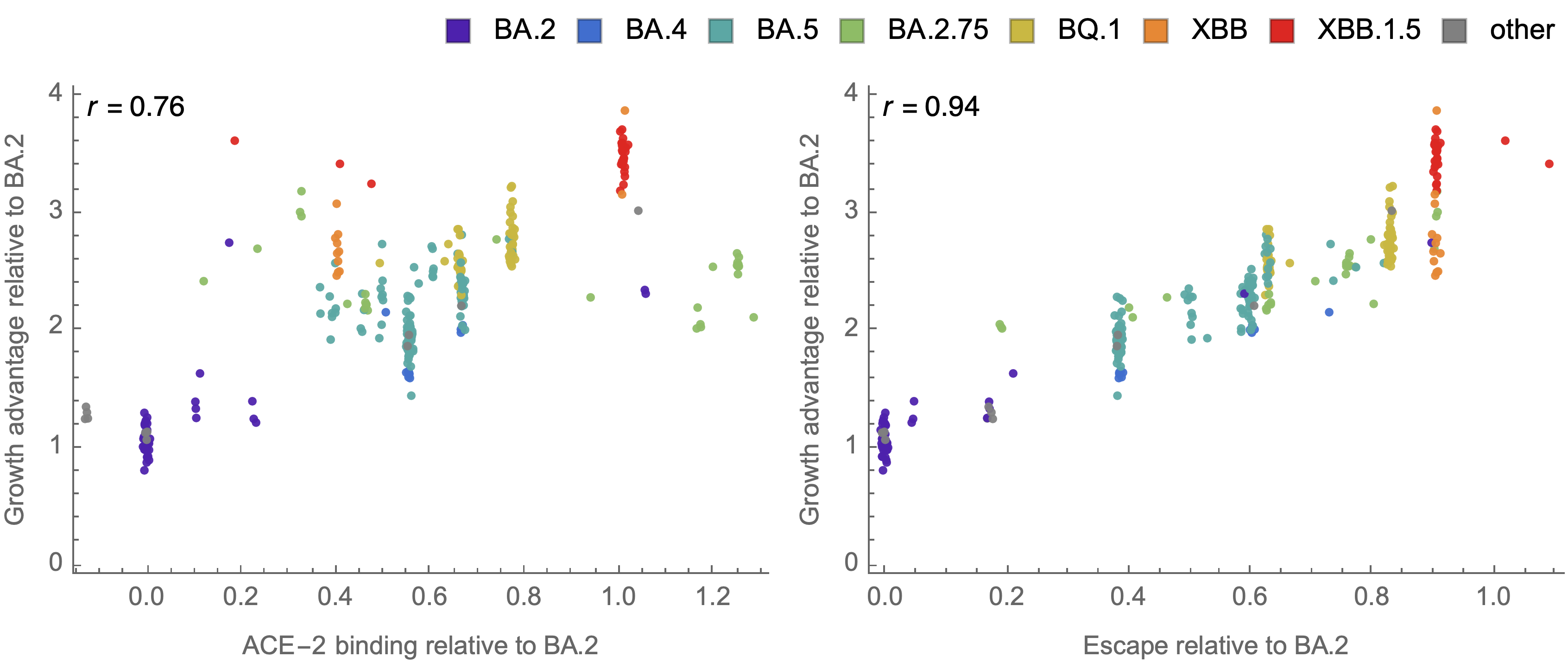
Similar results for new DMS platform measuring cell entry vs ACE2 binding vs escape from serum panel
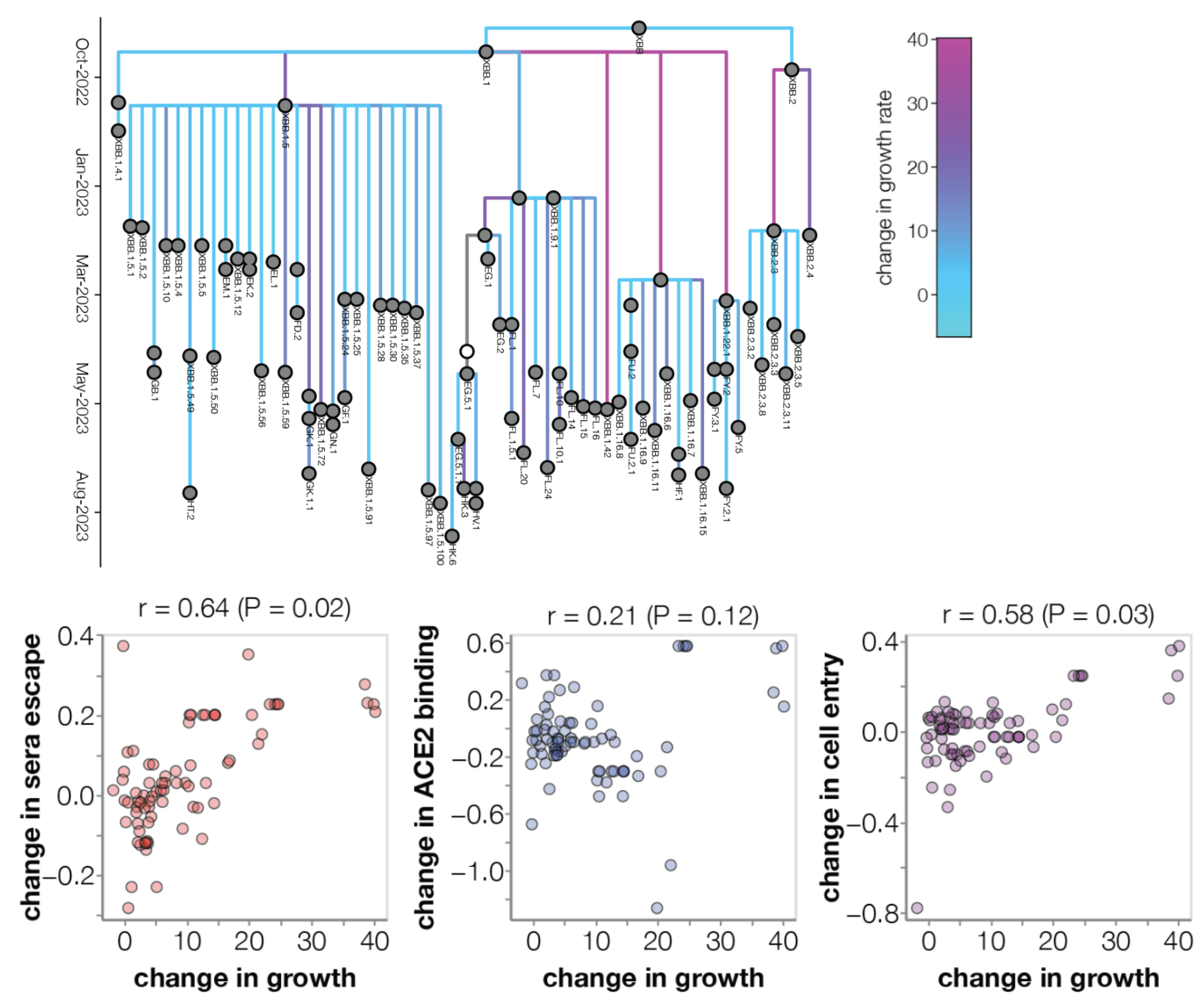
Continued research
- Application of MLR models to seasonal influenza and other pathogens
- Assessing and improving accuracy of "live" models at nextstrain.org/sars-cov-2/forecasts/
- Implementing DMS priors to predict fitness of emerging and yet-to-emerge lineages
Acknowledgements
Flu: WHO Global Influenza Surveillance and Response System, other data producers, GISAID, John Huddleston, Richard Neher, Jennifer Chang, Jover Lee
SARS-CoV-2: Data producers from all over the world, GISAID, the Nextstrain team, Katie Kistler, Marlin Figgins, Eslam Abousamra, Jover Lee, James Hadfield
Bedford Lab:
![]() John Huddleston,
John Huddleston,
![]() James Hadfield,
James Hadfield,
![]() Katie Kistler,
Katie Kistler,
![]() Thomas Sibley,
Thomas Sibley,
![]() Jover Lee,
Jover Lee,
![]() Cassia Wagner,
Cassia Wagner,
![]() Miguel Paredes,
Miguel Paredes,
![]() Nicola Müller,
Nicola Müller,
![]() Marlin Figgins,
Marlin Figgins,
![]() Victor Lin,
Victor Lin,
![]() Jennifer Chang,
Jennifer Chang,
![]() Allison Li,
Allison Li,
![]() Eslam Abousamra,
Eslam Abousamra,
![]() Donna Modrell,
Donna Modrell,
![]() Nashwa Ahmed,
Nashwa Ahmed,
![]() Cécile Tran Kiem
Cécile Tran Kiem
Question
Which factors contributed to the observed high yearly attack rate of SARS-CoV-2 in 2022 and 2023?
- Intrinsic $R_0$ of SARS-CoV-2
- Rapid antigenic evolution of SARS-CoV-2 spike protein
- SARS-CoV-2 is a pandemic virus rather than epidemic virus