Real-time tracking of virus evolution
Trevor Bedford (@trvrb)
24 Jun 2016
Federation Meeting of Korean Basic Medical Scientists
Incheon, Republic of Korea
Slides at bedford.io/talks/
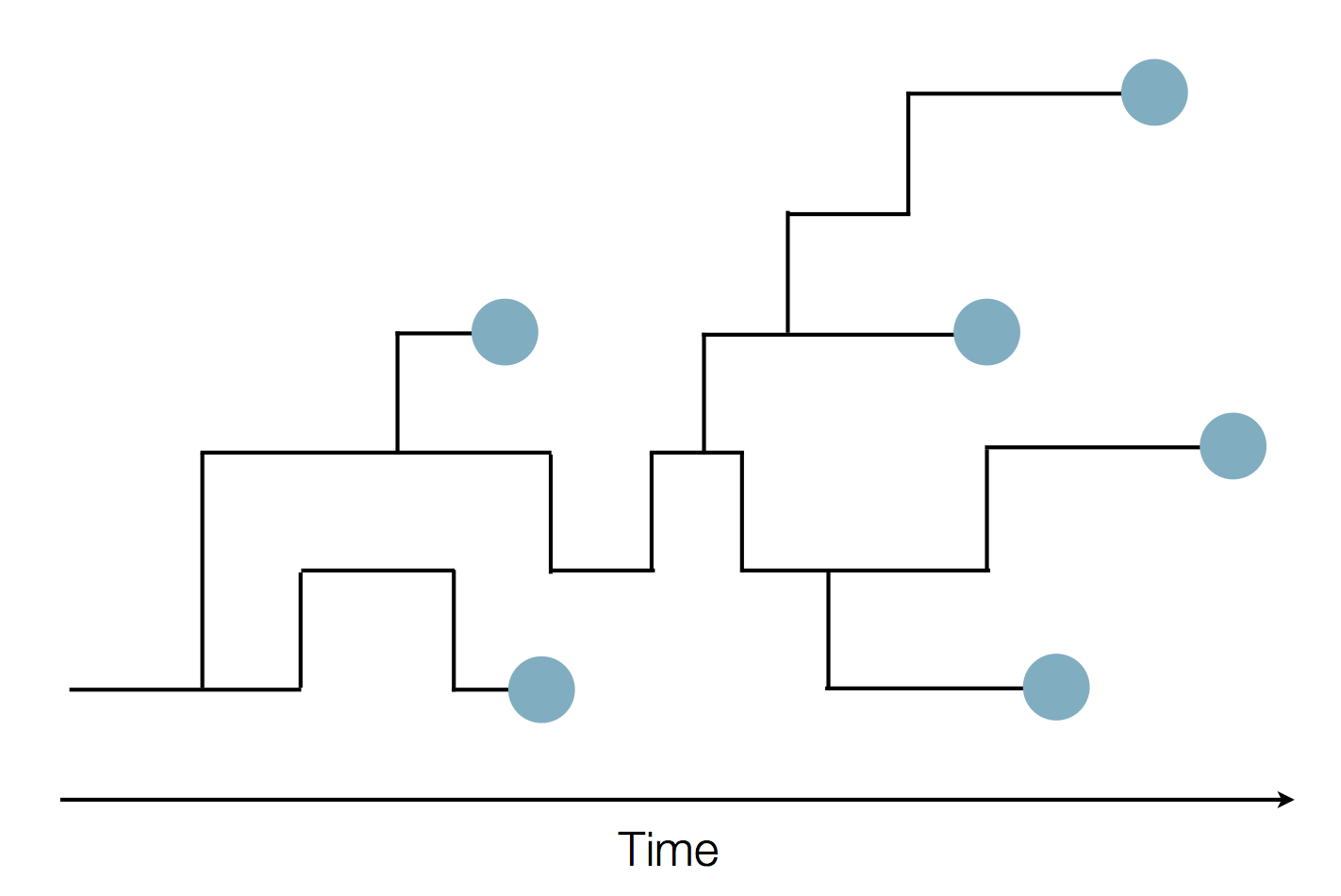
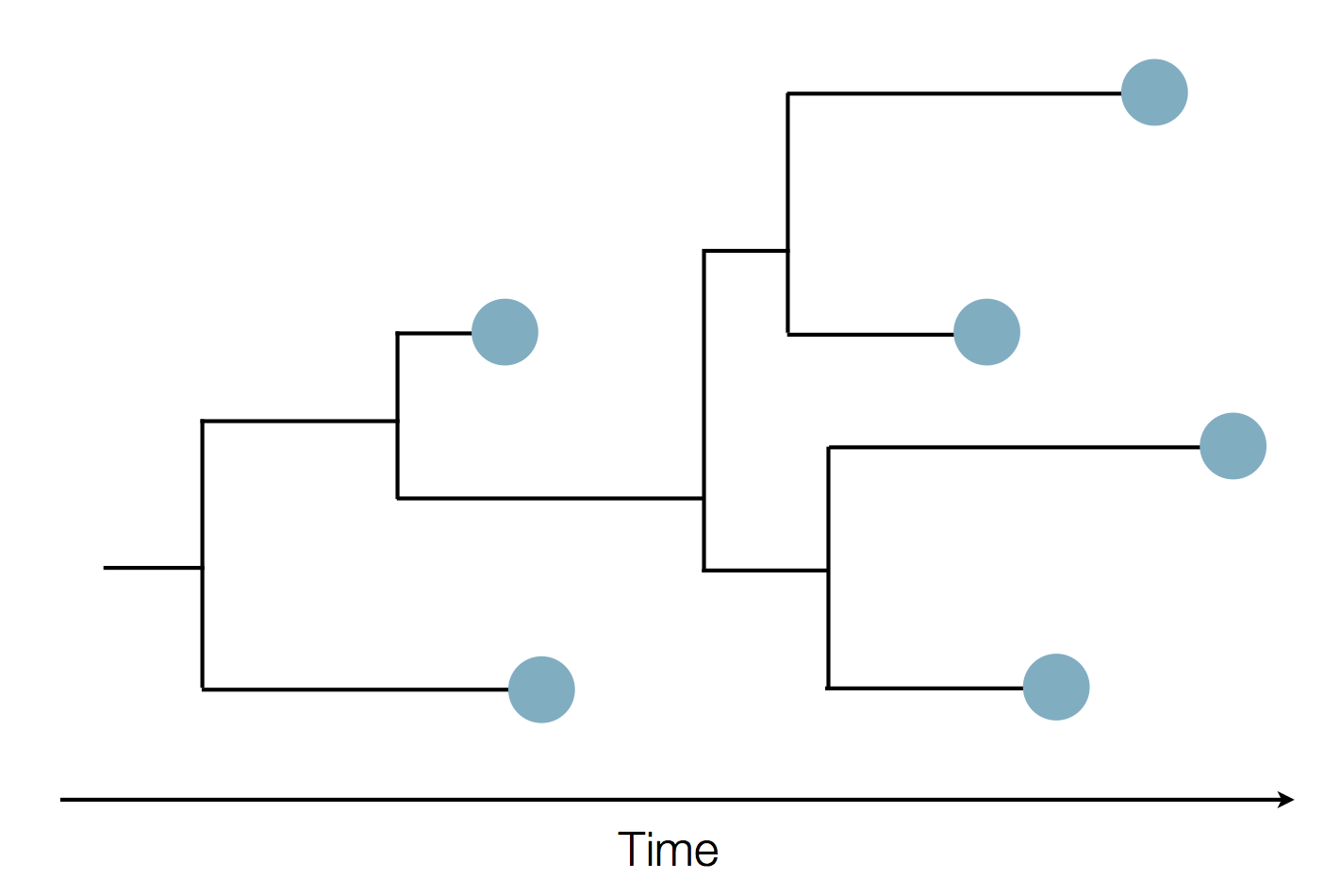
Phylogenies
Phylogenies describe history
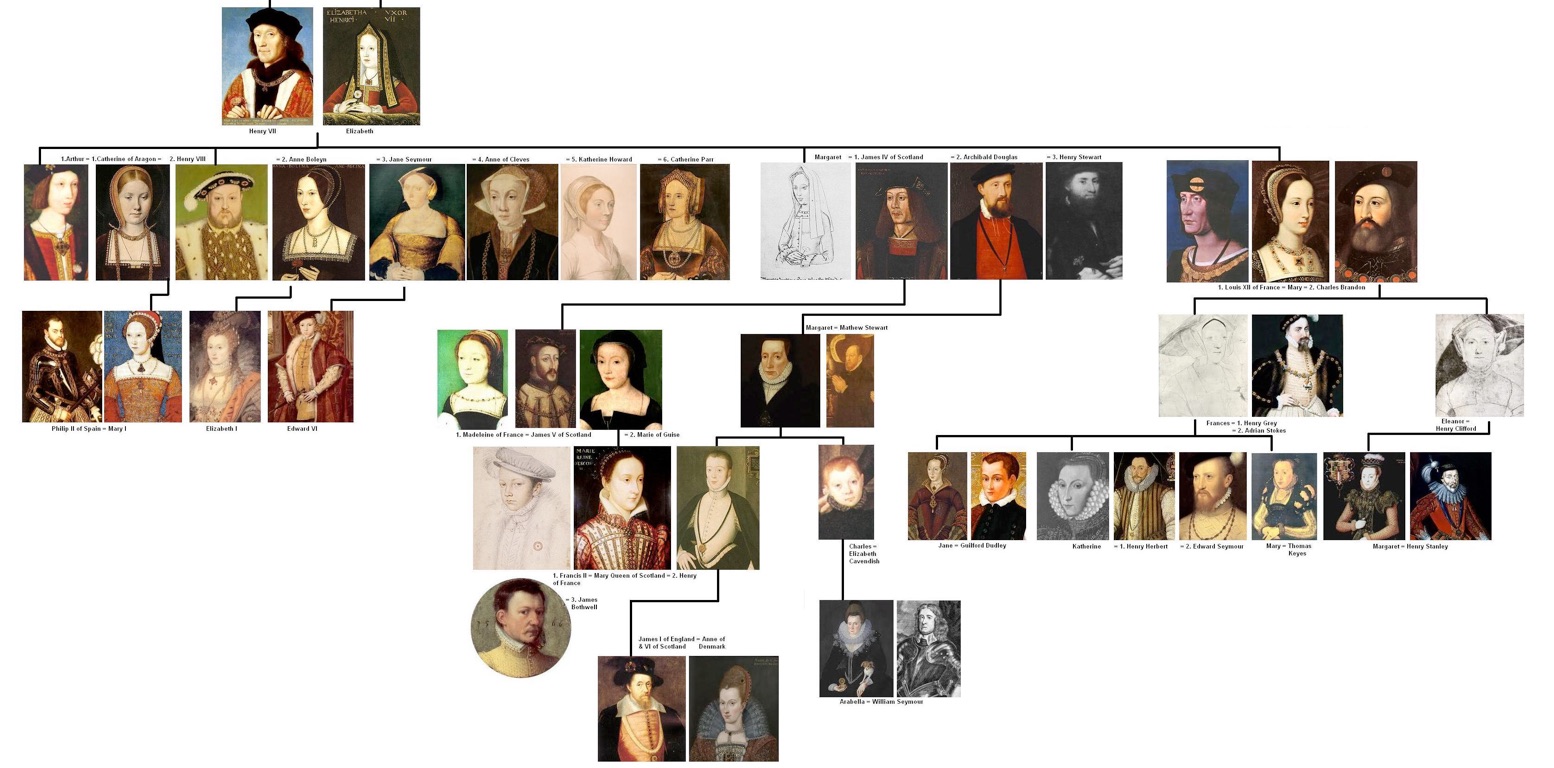
Phylogenies describe history
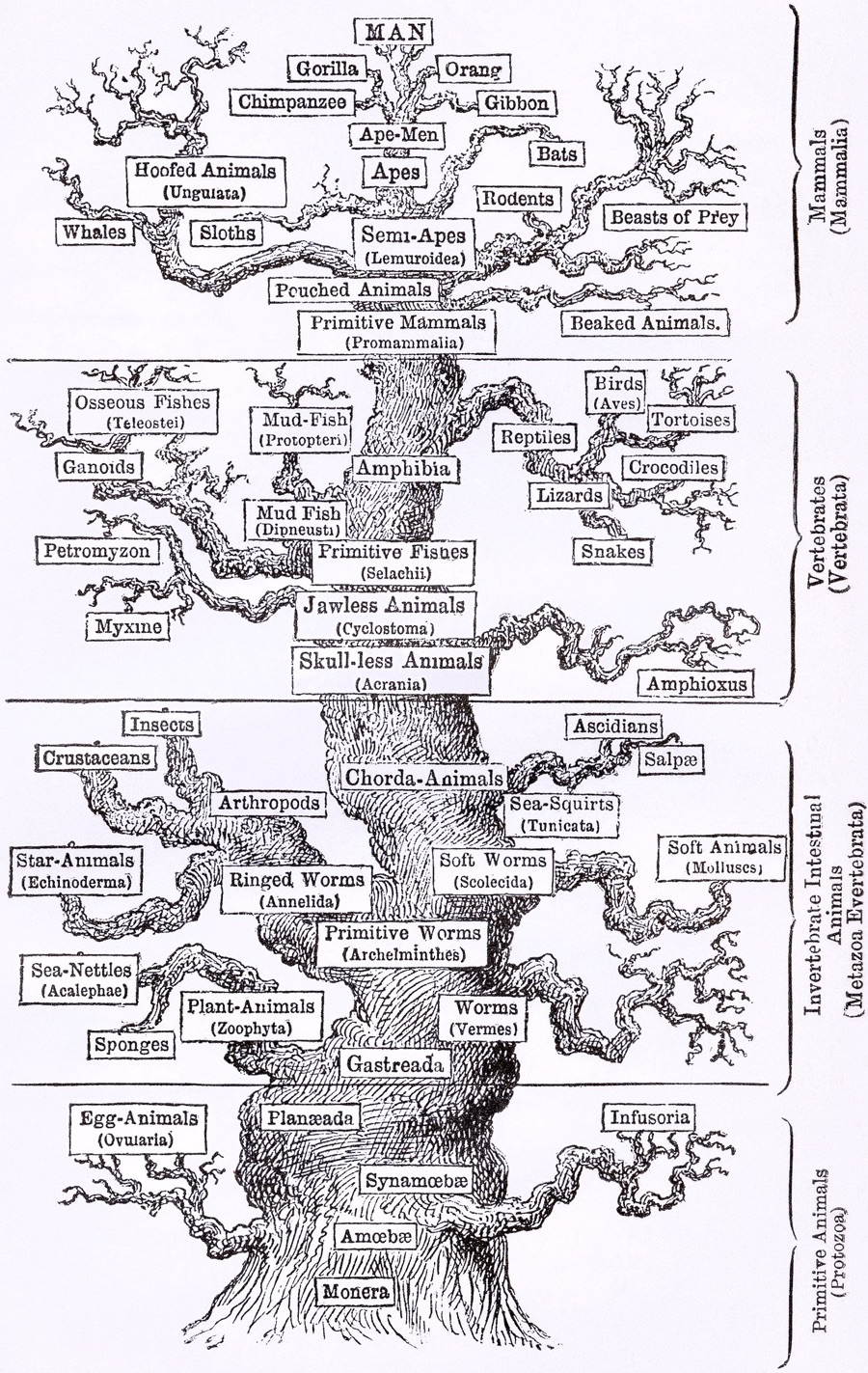
Phylogenies describe history
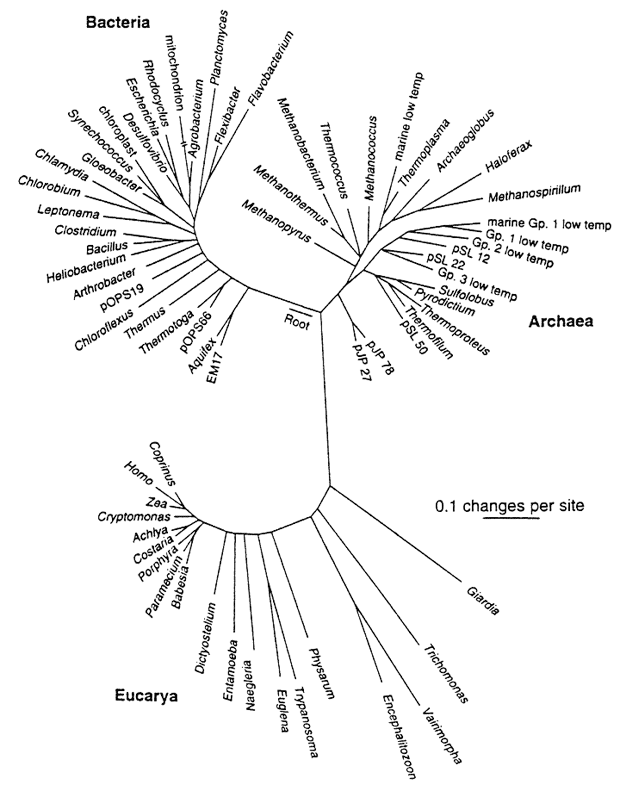
Phylogenies reveal process
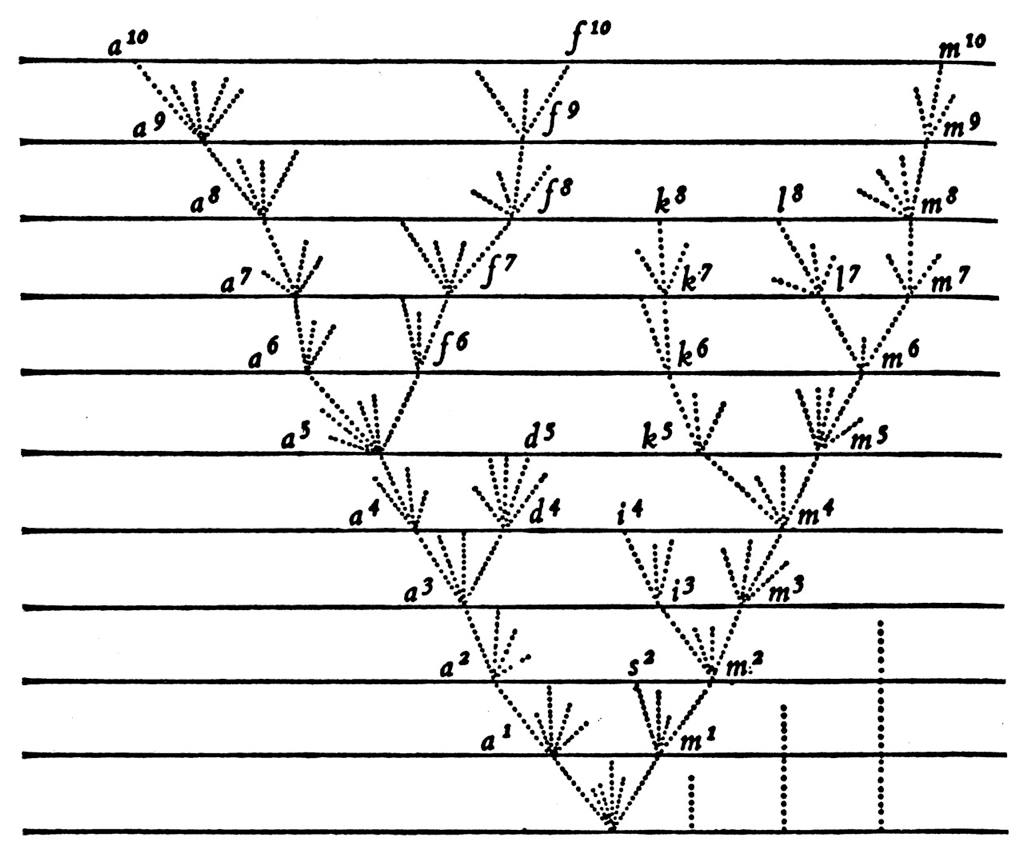
Epidemics
Epidemic process
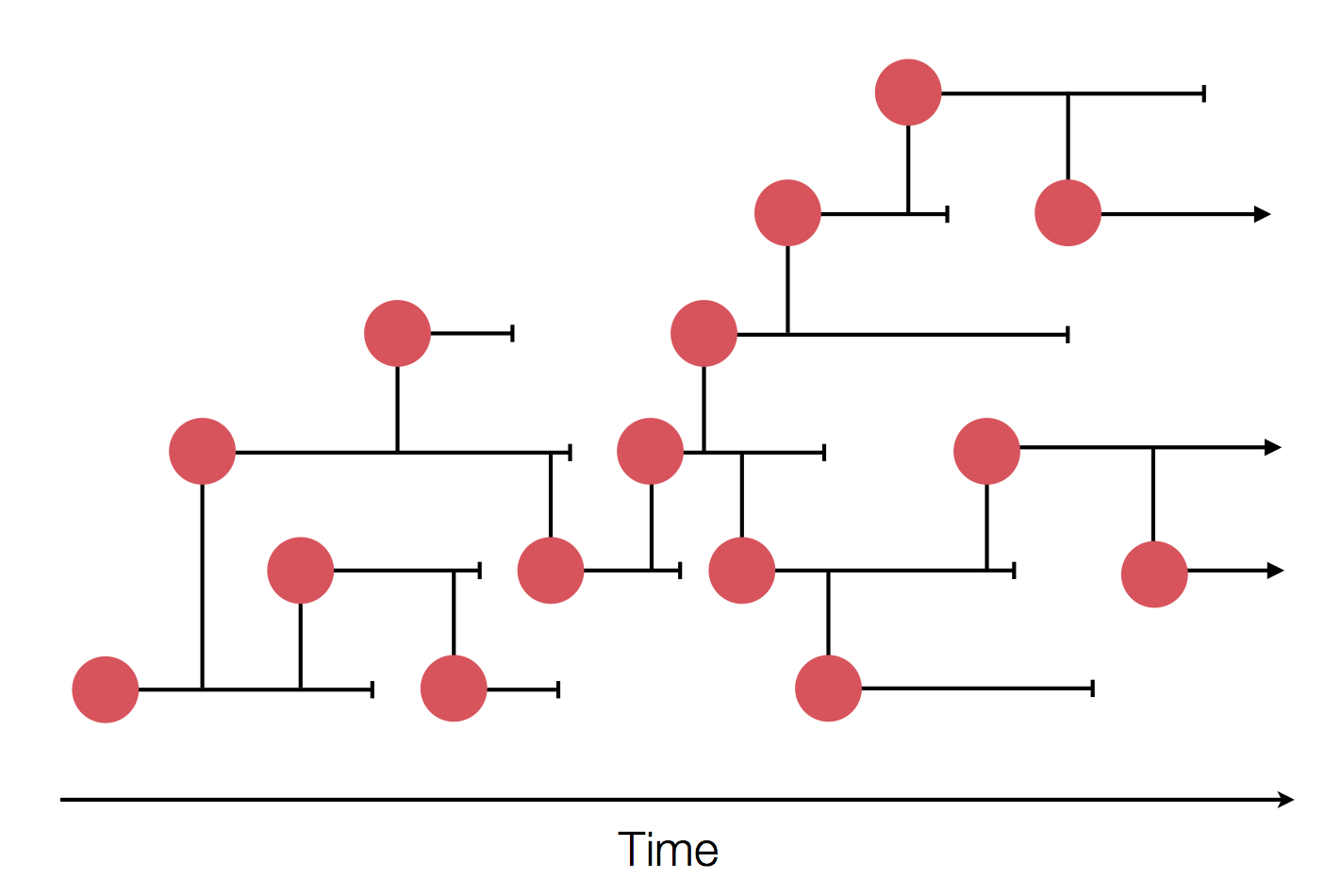
Sample some individuals
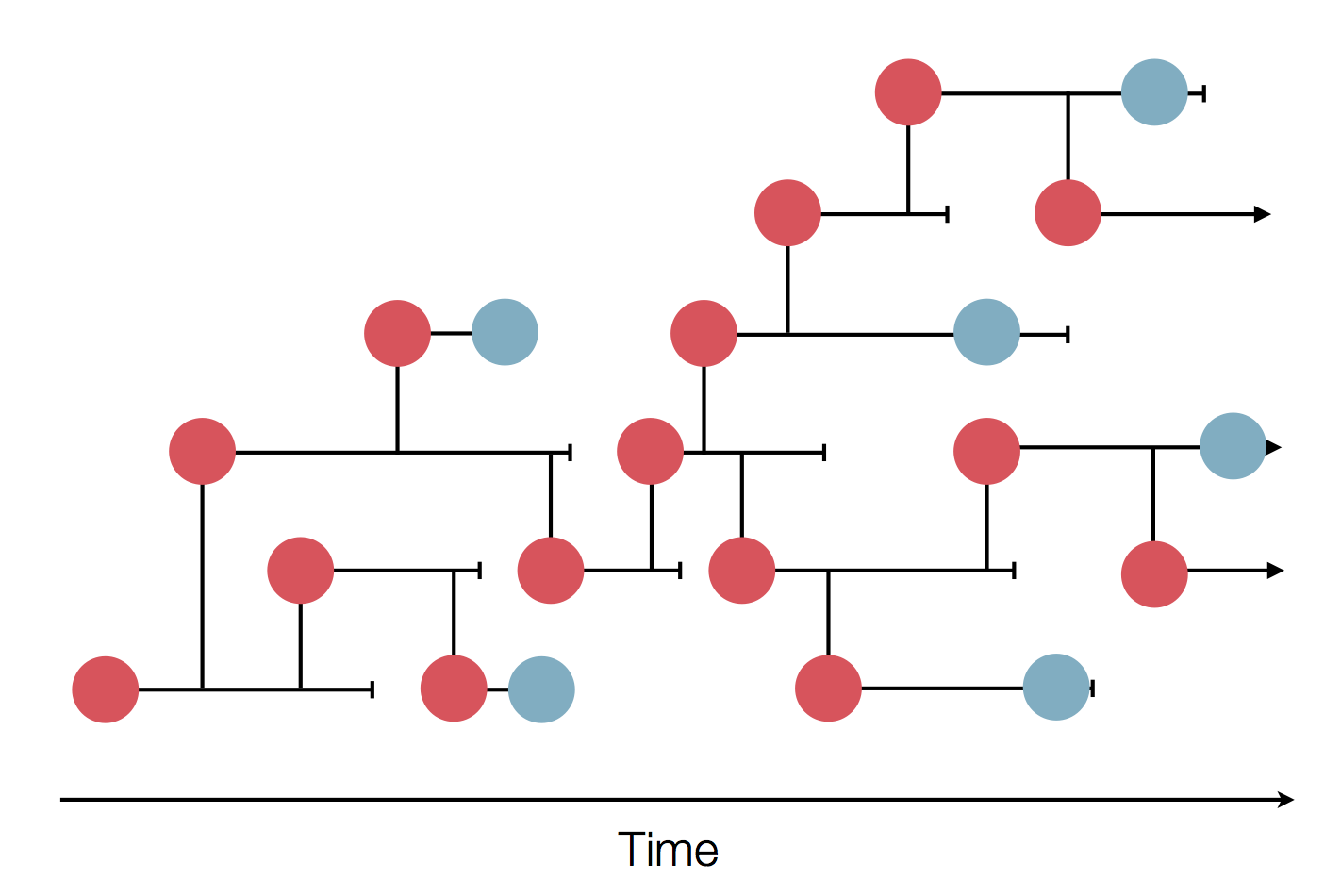
Sequence and determine phylogeny
Sequence and determine phylogeny
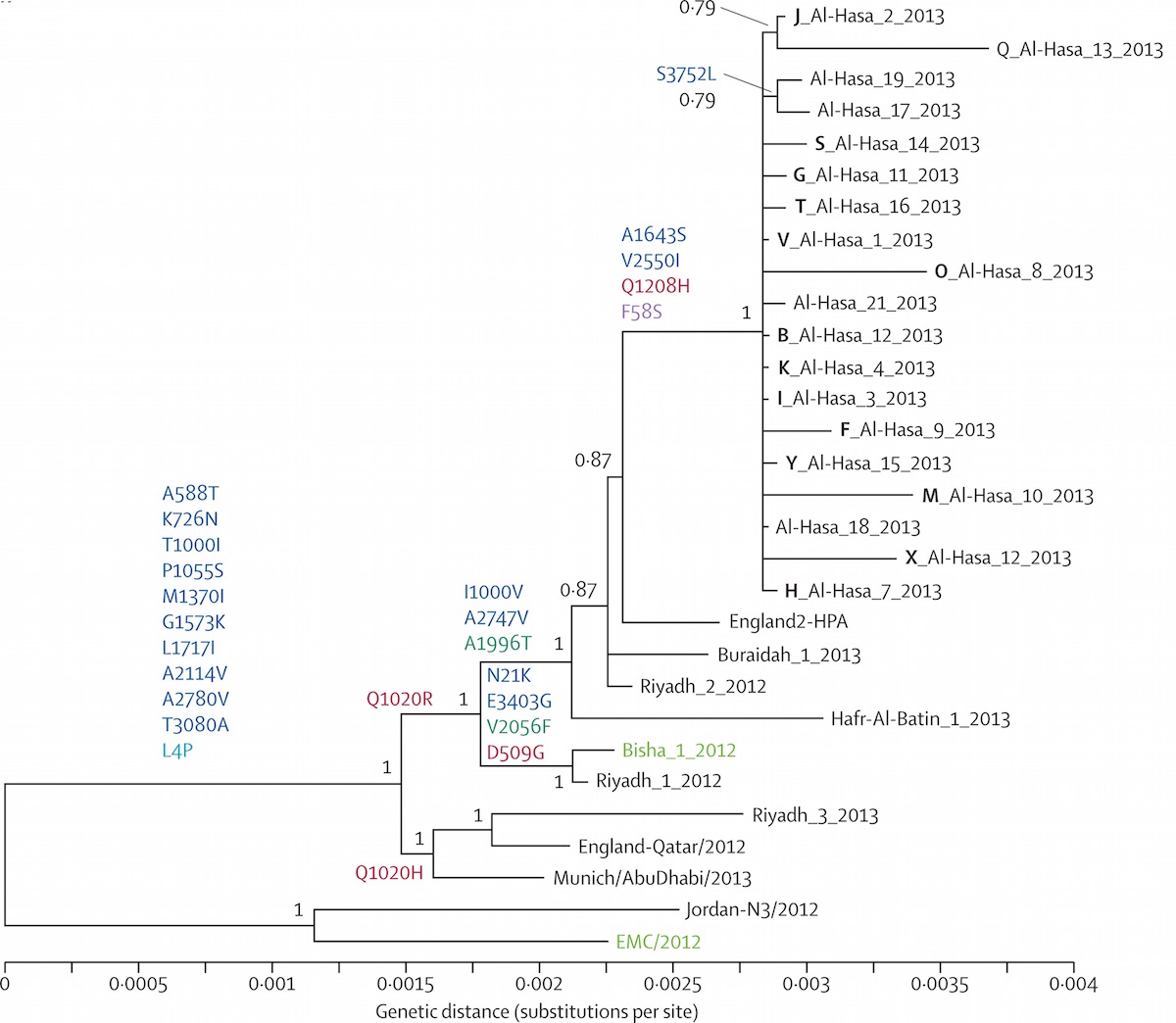
Localized Middle Eastern MERS-CoV phylogeny
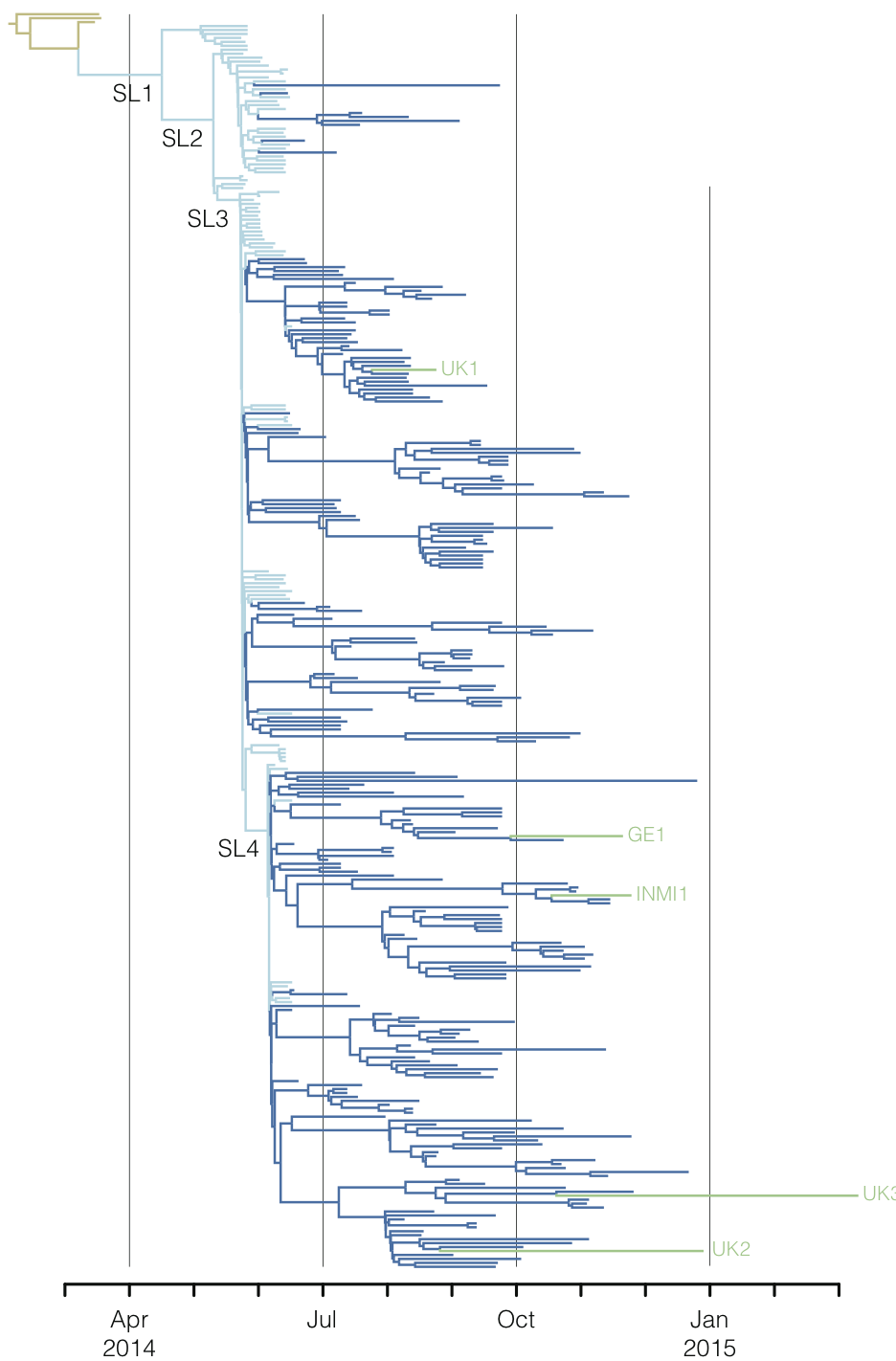
Regional West African Ebola phylogeny
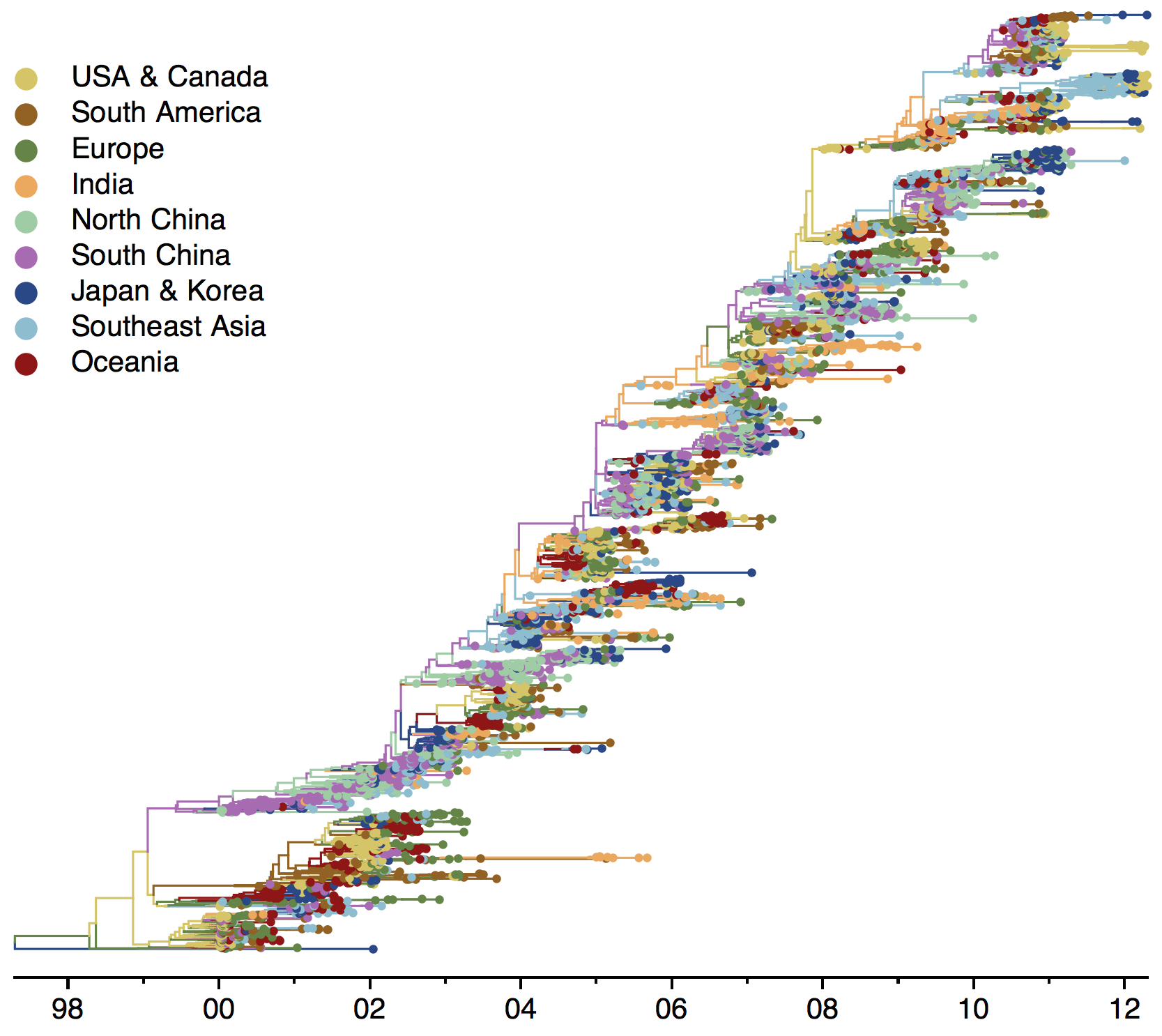
Global influenza phylogeny
Applications of evolutionary analysis for vaccine strain selection and charting outbreak spread
Influenza
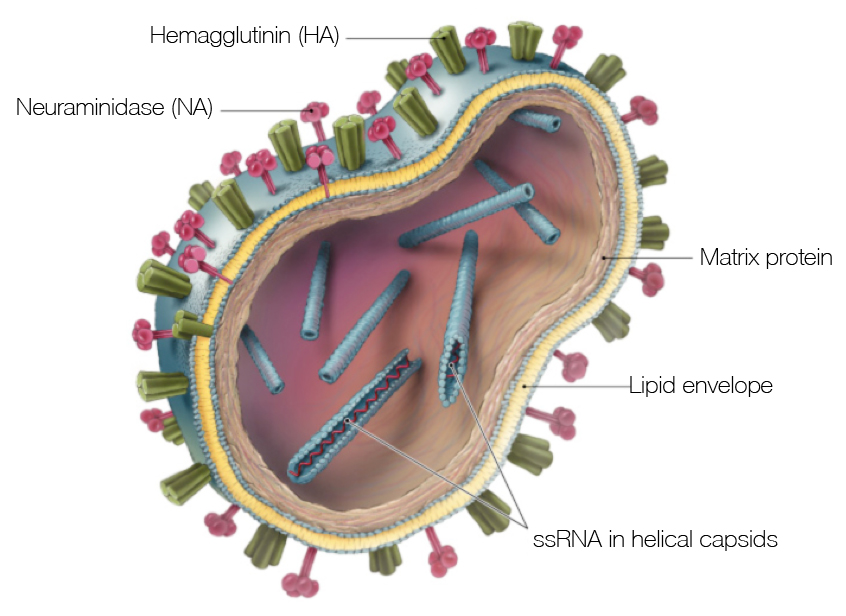
Influenza virion
Influenza H3N2 vaccine updates
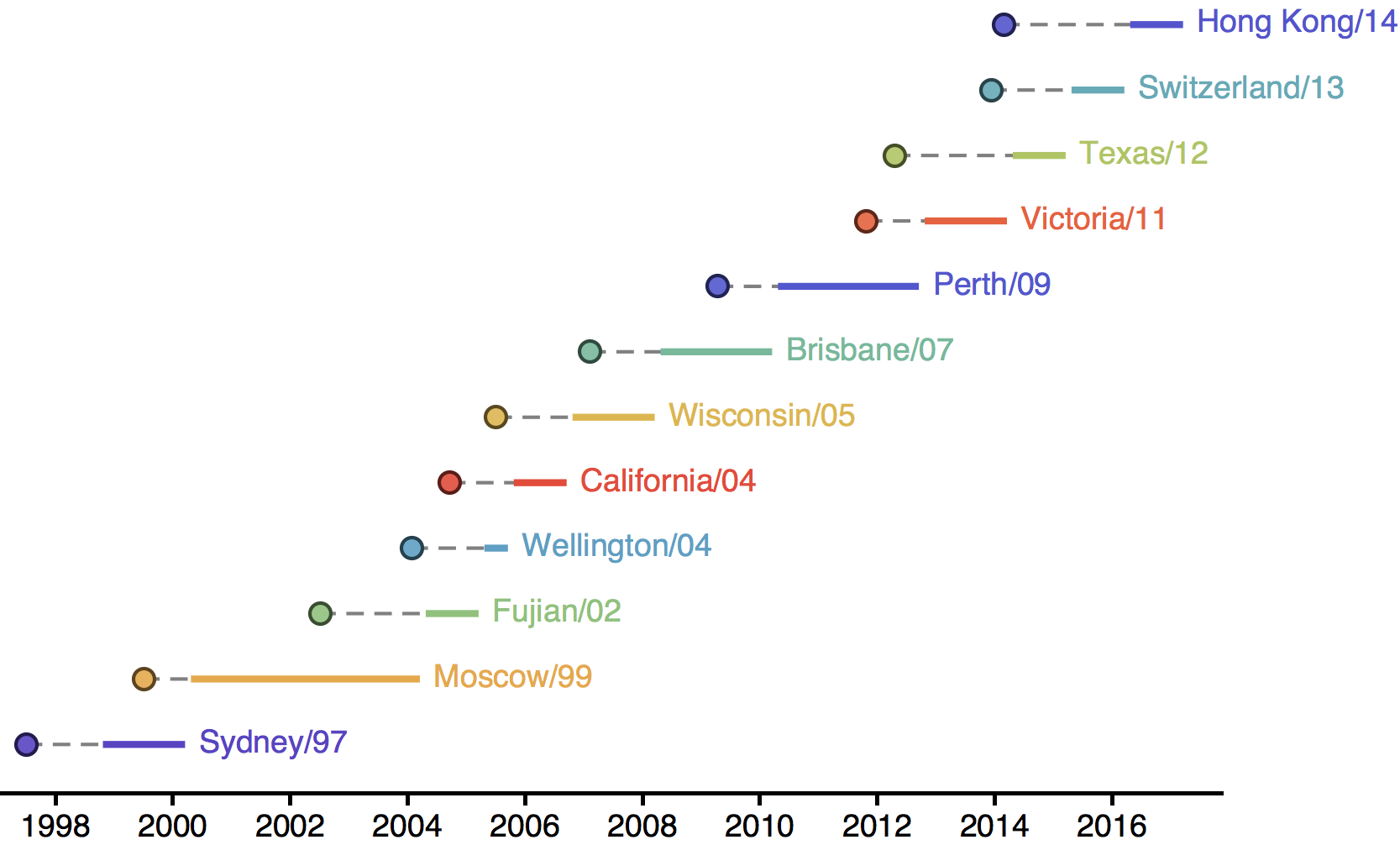
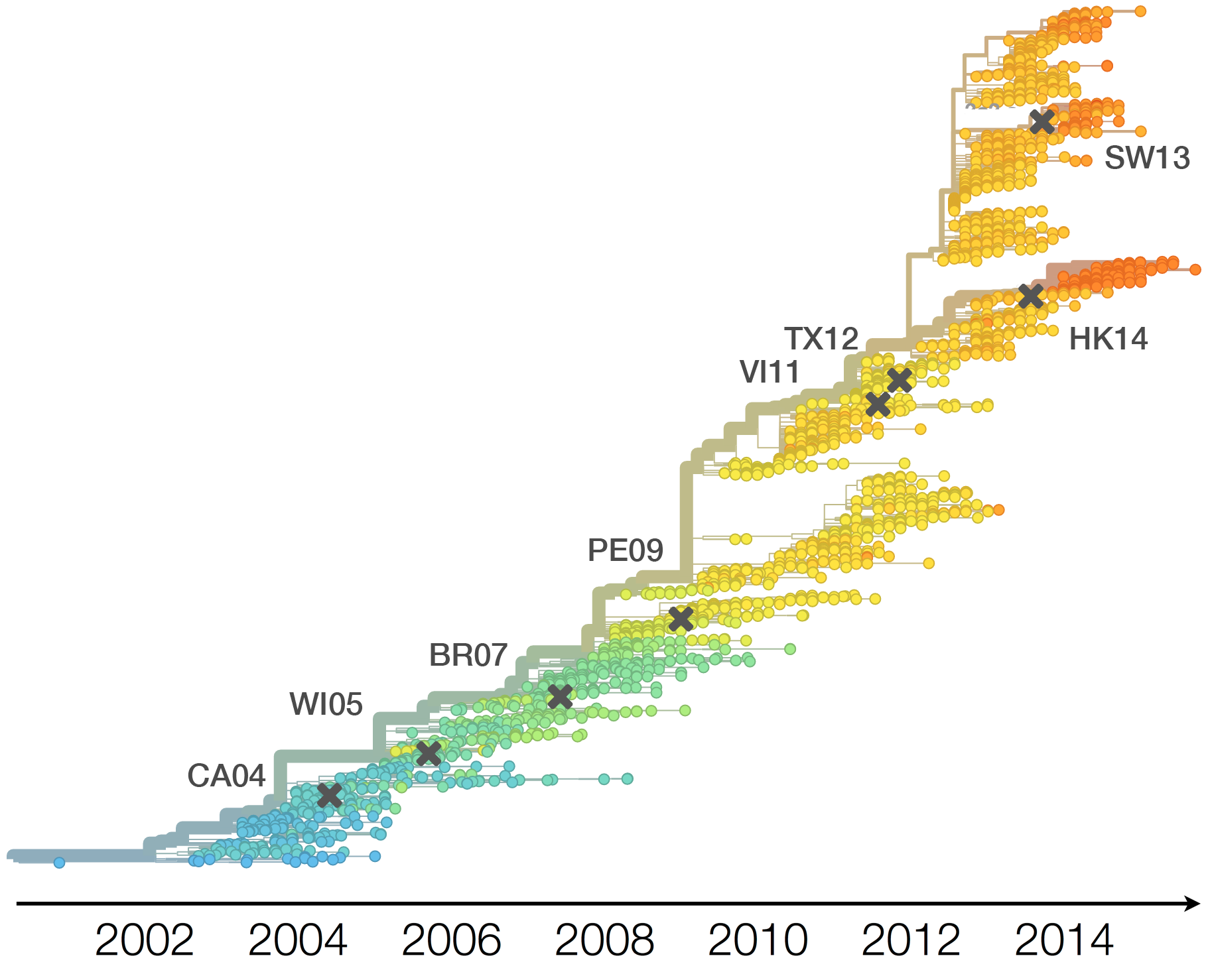
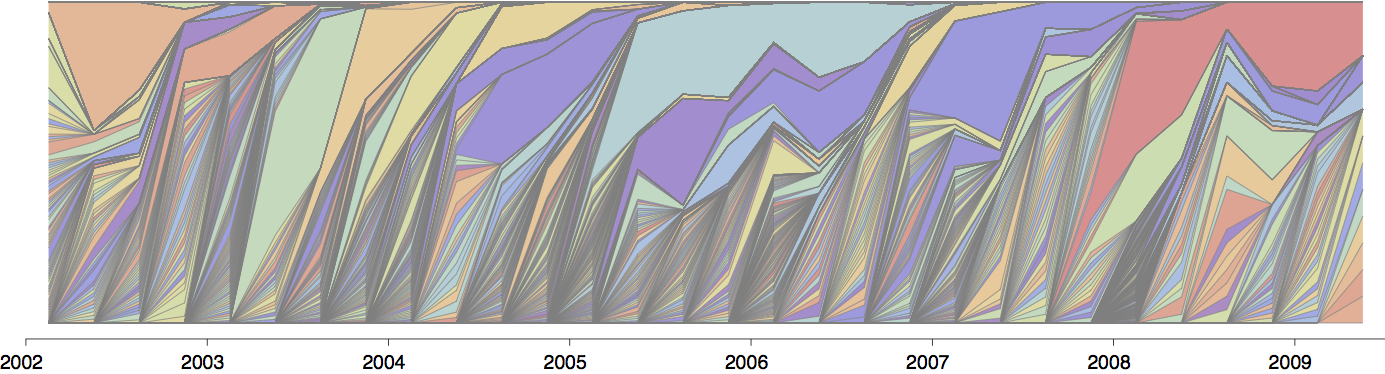
H3N2 phylogeny showing antigenic drift
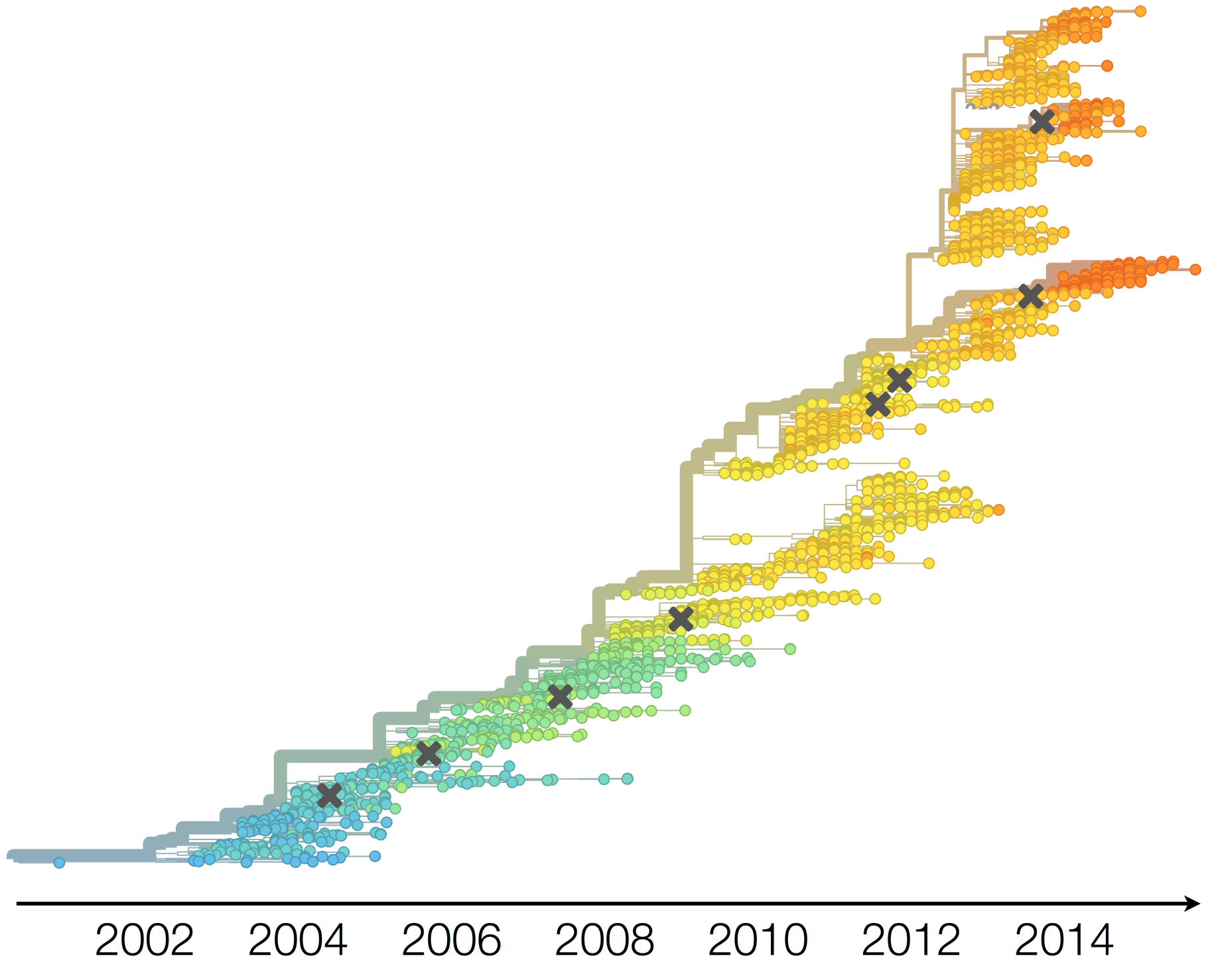
H3N2 phylogeny showing antigenic drift
Drift variants rapidly take over the virus population
Timely surveillance and rapid analysis essential to understand ongoing influenza evolution
nextflu
Project to provide a real-time view of the evolving influenza population
All in collaboration with Richard Neher

nextflu pipeline
- Download all recent HA sequences from GISAID
- Filter to remove outliers
- Subsample across time and space
- Align sequences
- Build tree
- Estimate frequencies
- Export for visualization
Up-to-date analysis publicly available at:
nextflu.org
Forecasting
The future is here, it's just not evenly distributed yet
— William Gibson
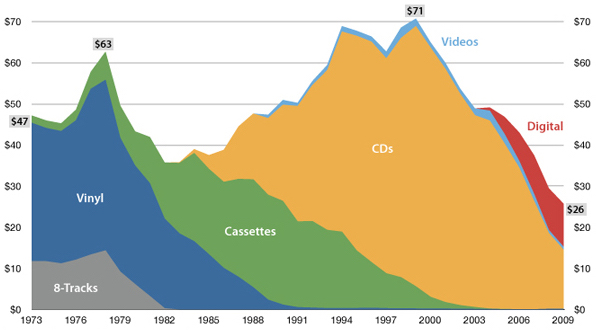
USA music industry, 2011 dollars per capita
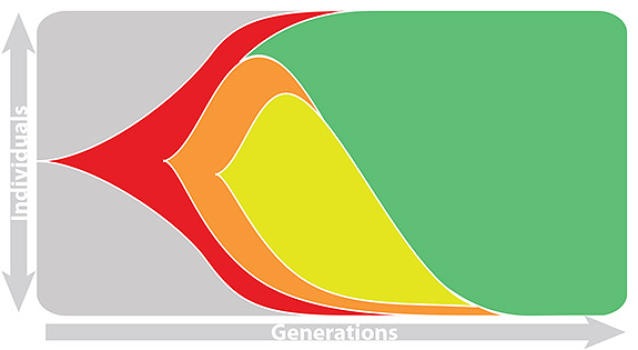
Influenza population turnover
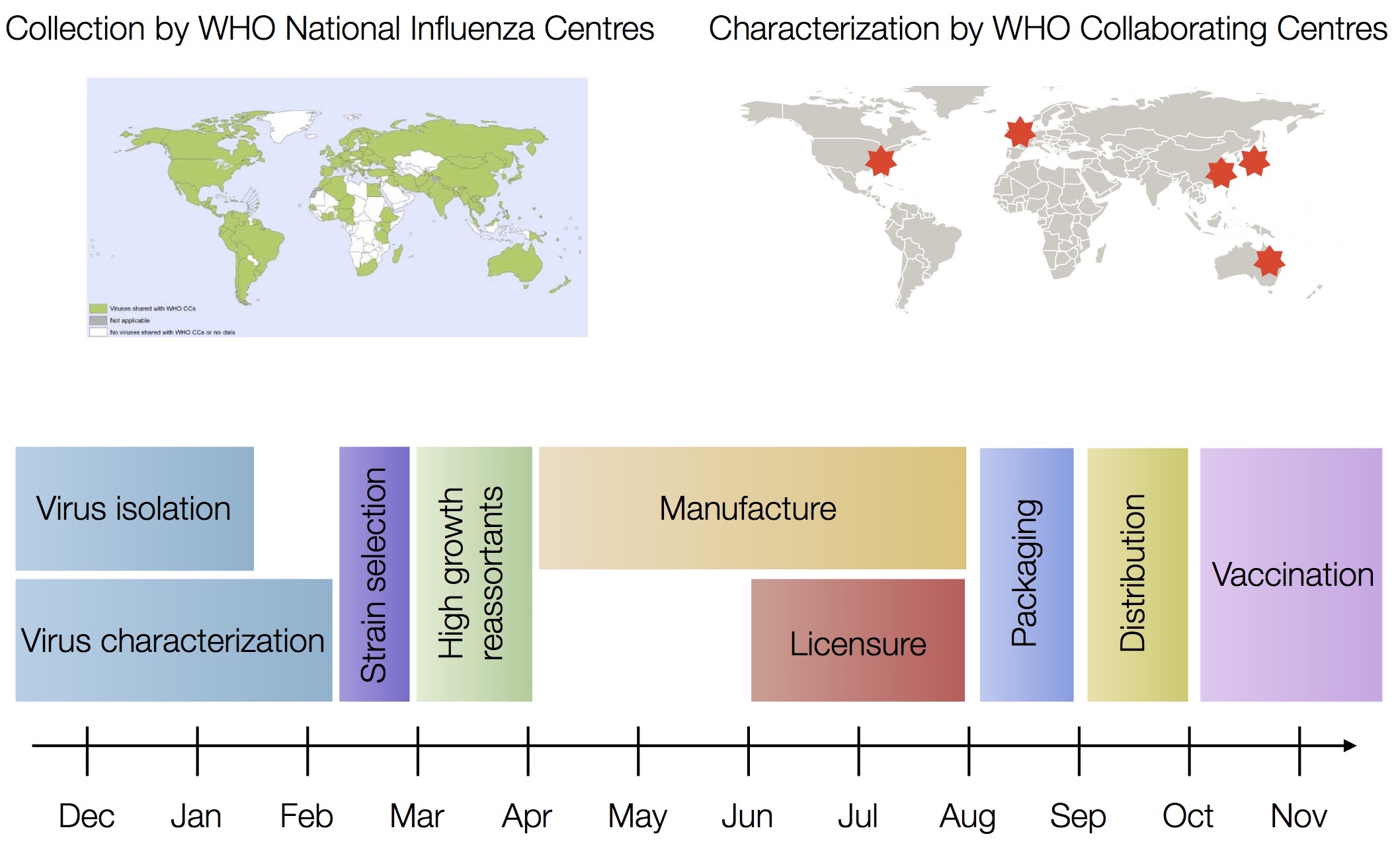
Vaccine strain selection timeline
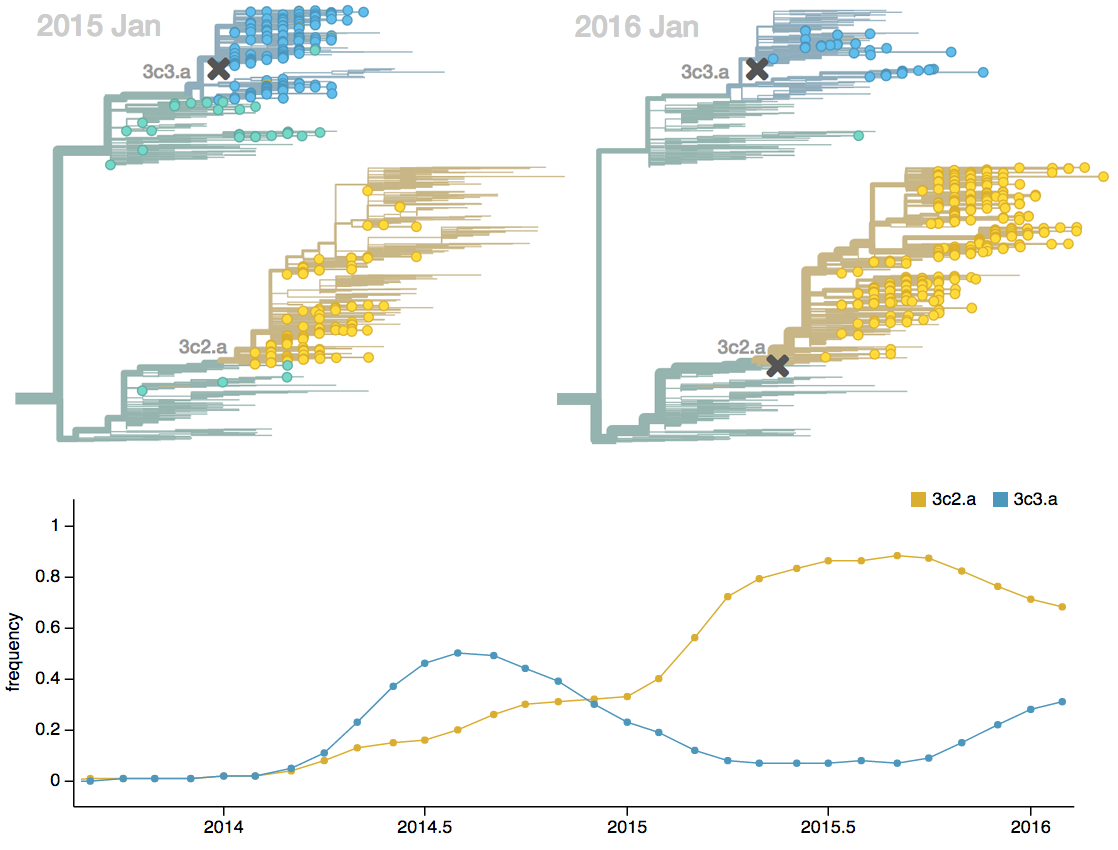
Seek to explain change in clade frequencies over 1 year
Fitness models can project clade frequencies
Clade frequencies $X$ derive from the fitnesses $f$ and frequencies $x$ of constituent viruses, such that
$$\hat{X}_v(t+\Delta t) = \sum_{i:v} x_i(t) \, \mathrm{exp}(f_i \, \Delta t)$$
This captures clonal interference between competing lineages
Predictive fitness models
A simple predictive model estimates the fitness $f$ of virus $i$ as
$$\hat{f}_i = \beta^\mathrm{ep} \, f_i^\mathrm{ep} + \beta^\mathrm{ne} \, f_i^\mathrm{ne}$$
where $f_i^\mathrm{ep}$ measures cross-immunity via substitutions at epitope sites and $f_i^\mathrm{ep}$ measures mutational load via substitutions at non-epitope sites
We implement a similar model based on two predictors
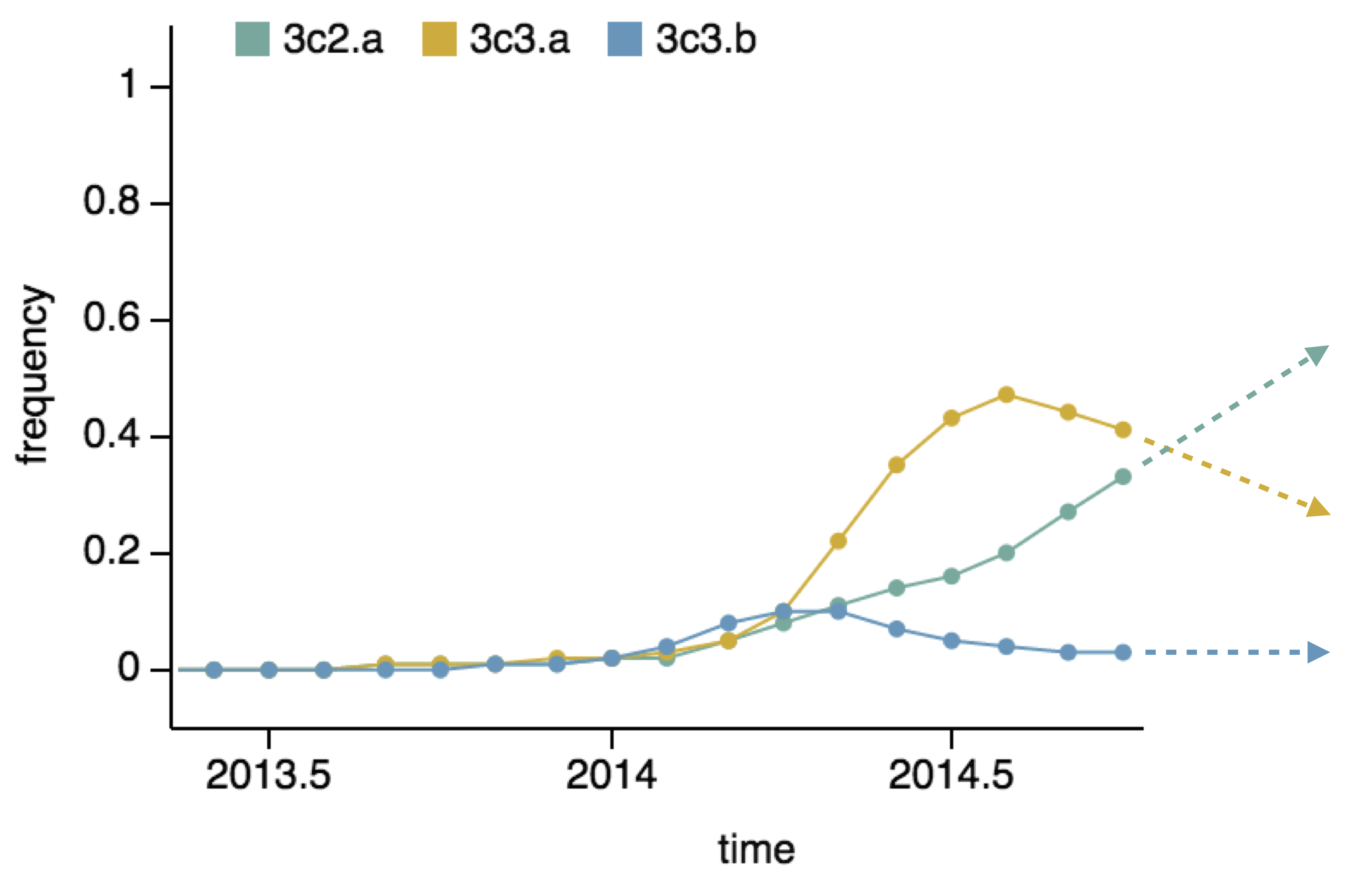
- Clade frequency change
- Antigenic advancement
Project frequencies forward,
growing clades have high fitness
Calculate HI drop from ancestor,
drifted clades have high fitness
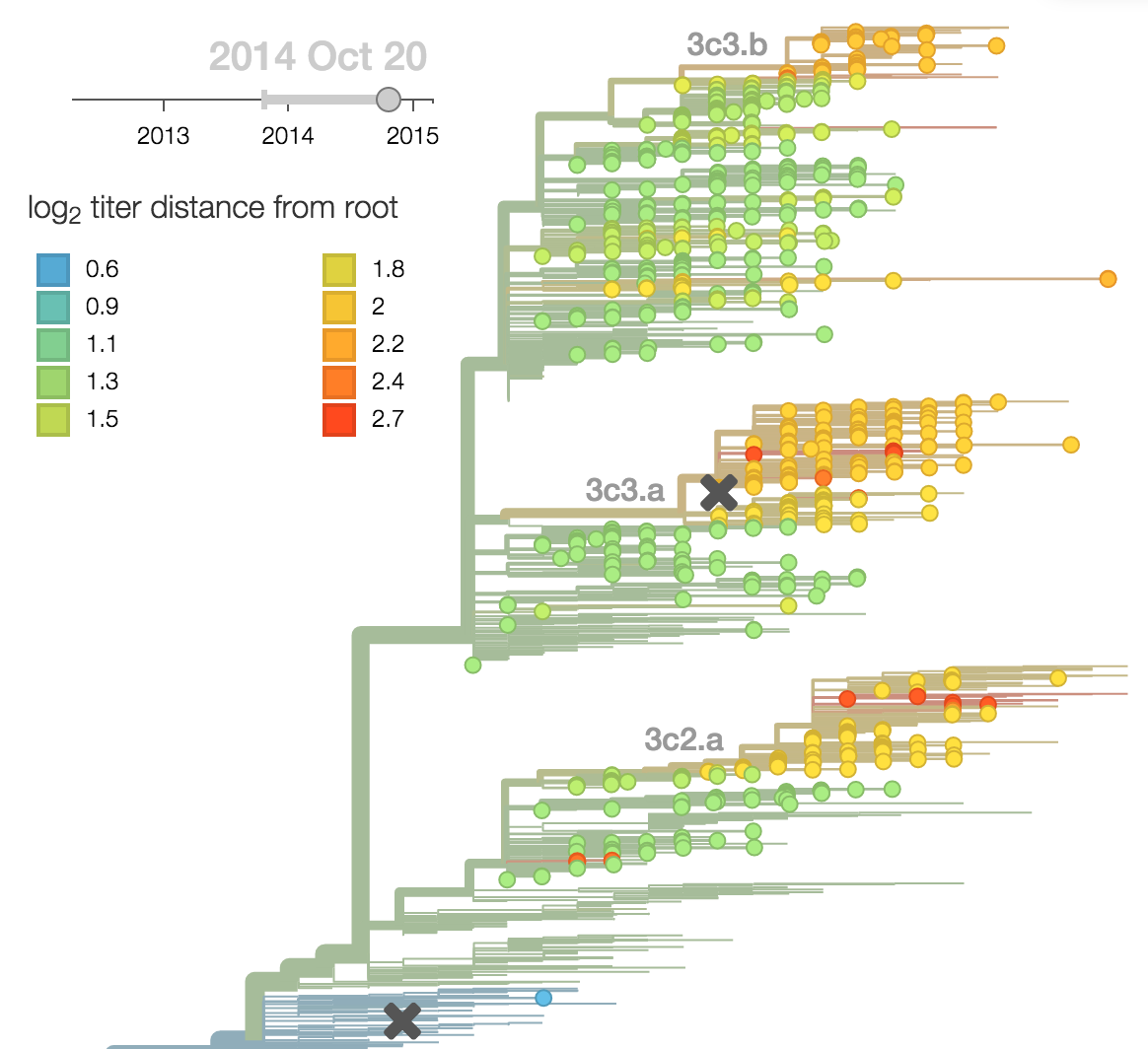
Fitness model parameterization
Our predictive model estimates the fitness $f$ of virus $i$ as
$$\hat{f}_i = \beta^\mathrm{freq} \, f_i^\mathrm{freq} + \beta^\mathrm{HI} \, f_i^\mathrm{HI}$$
We learn coefficients and validate model based on previous 15 H3N2 seasons
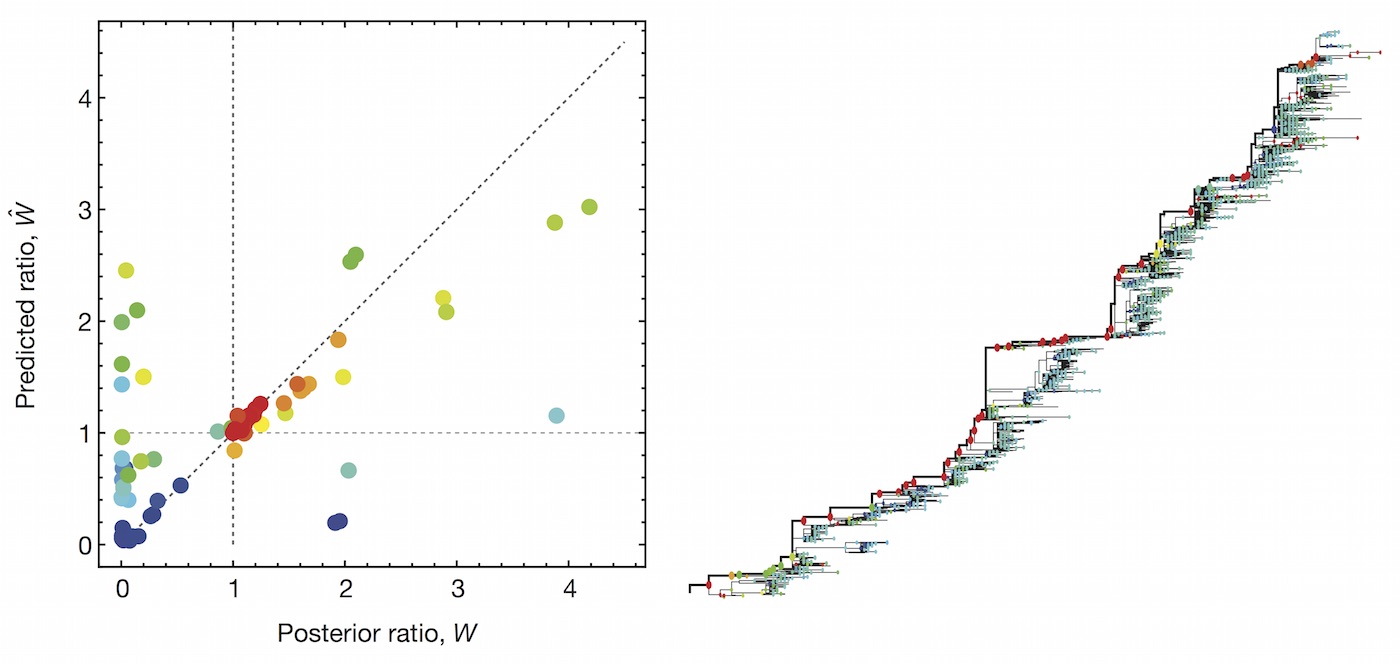
Clade growth rate is well correlated (ρ = 0.66)
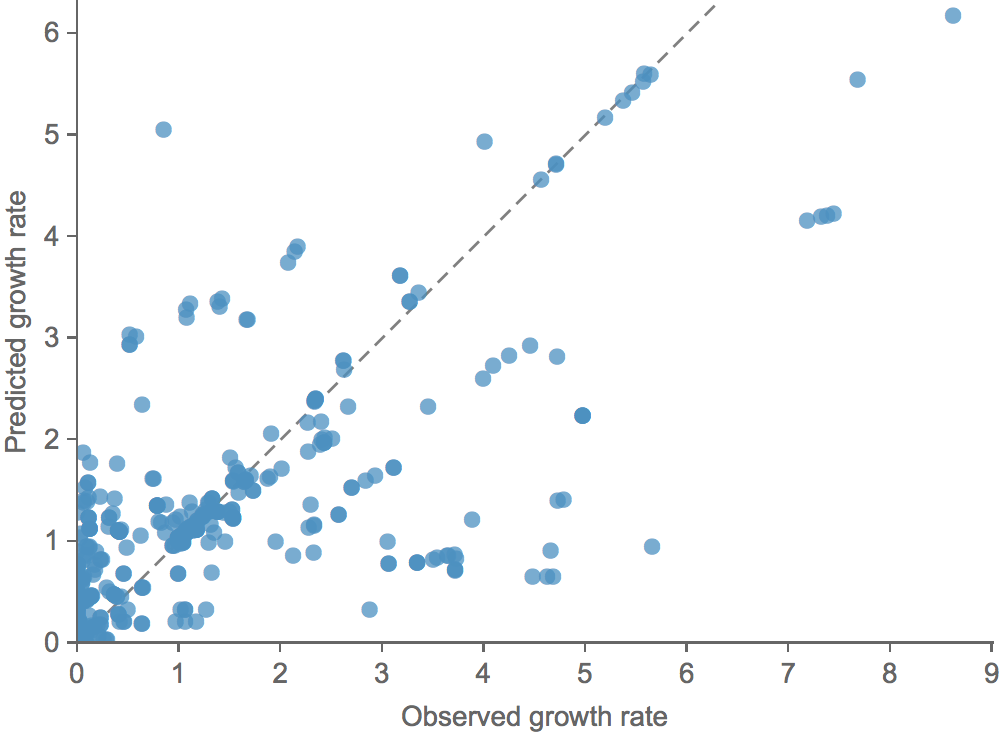
Growth vs decline correct in 84% of cases
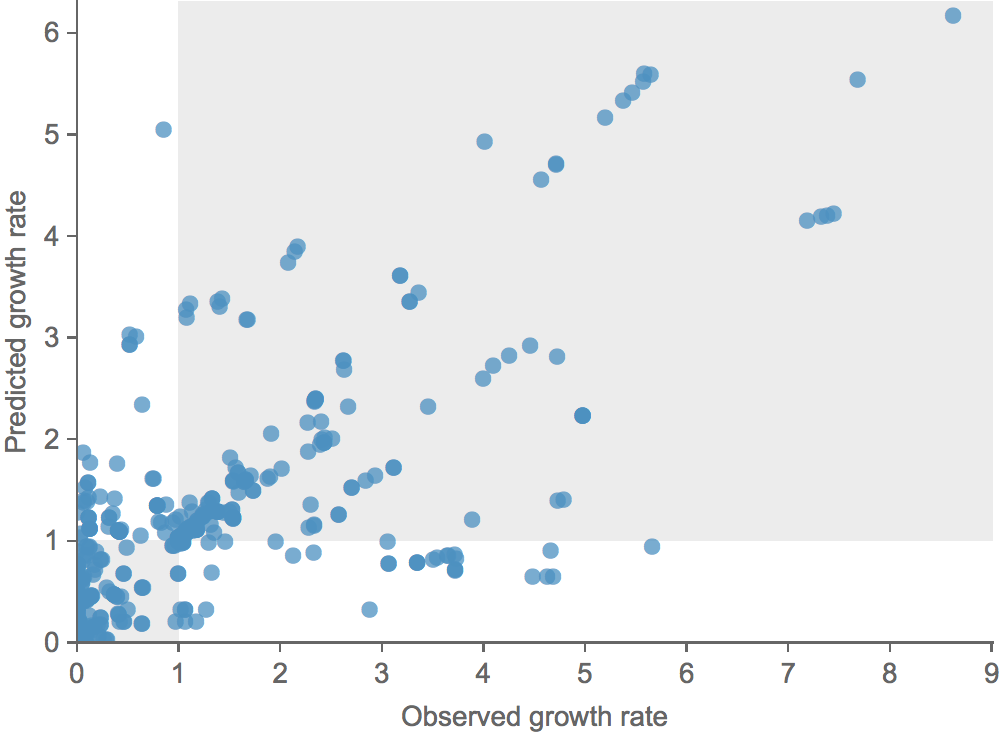
Trajectories show more detailed congruence
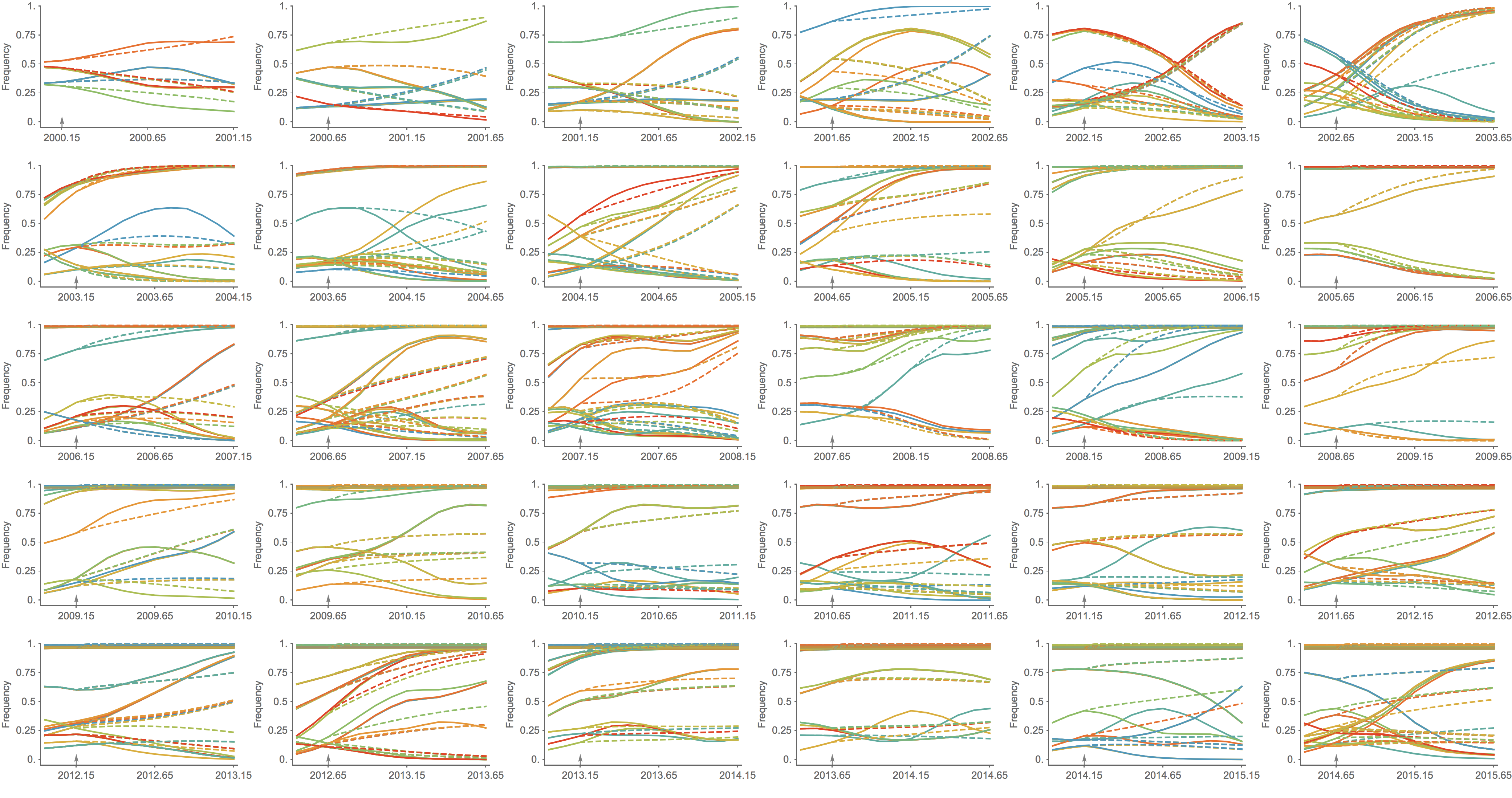
Further work on predictive modeling
- Integrate data predictors and data sources, e.g. geography
- Possible to build predictive models for H1N1 and B and to forecast NA evolution
Real-time analyses are actionable and thus, may inform influenza vaccine strain selection
Outbreak analysis
Ebola
Epidemic basically contained, but resulted in >28,000 confirmed cases and >11,000 deaths
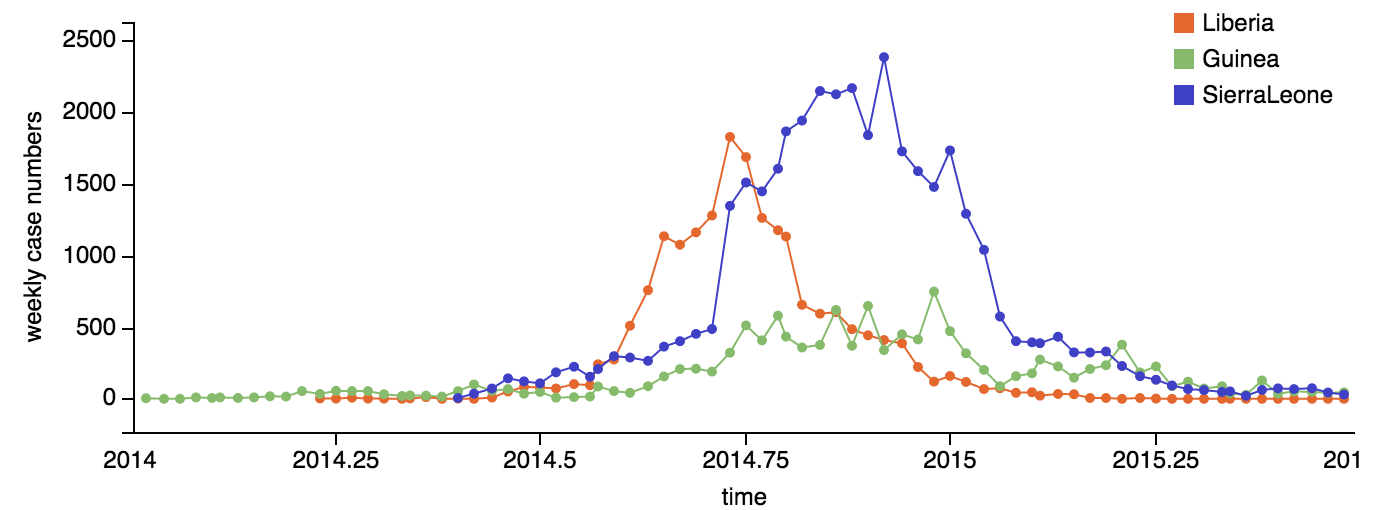
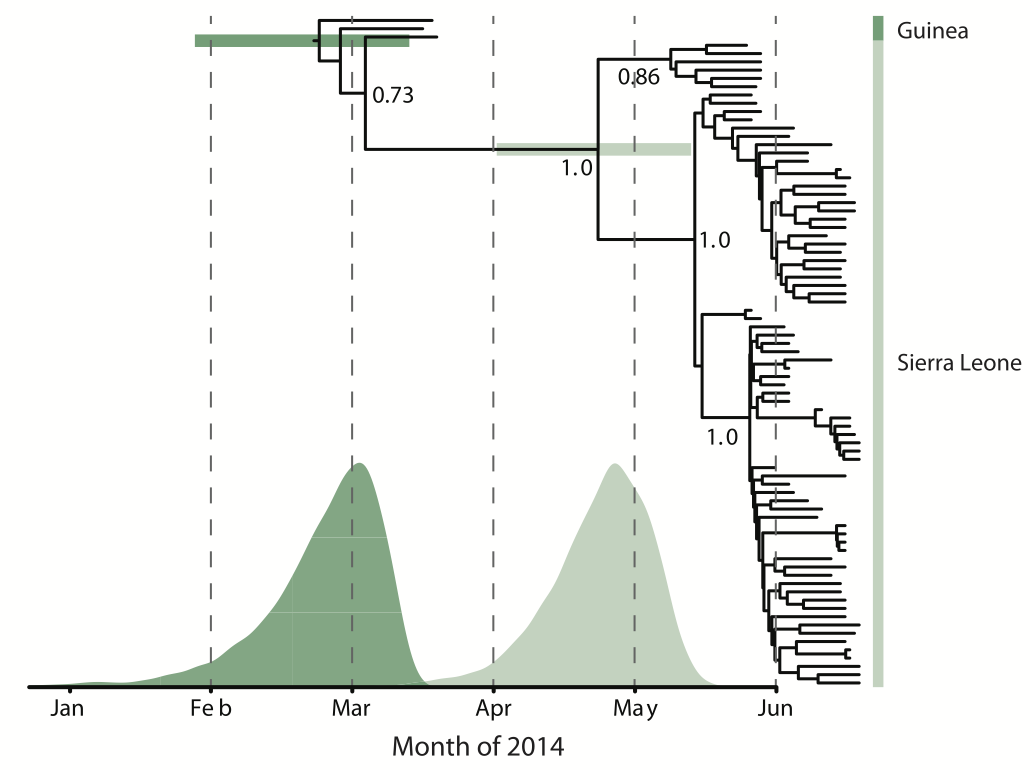
Early sequencing showed single origin of epidemic
Continued spread through Dec 2014
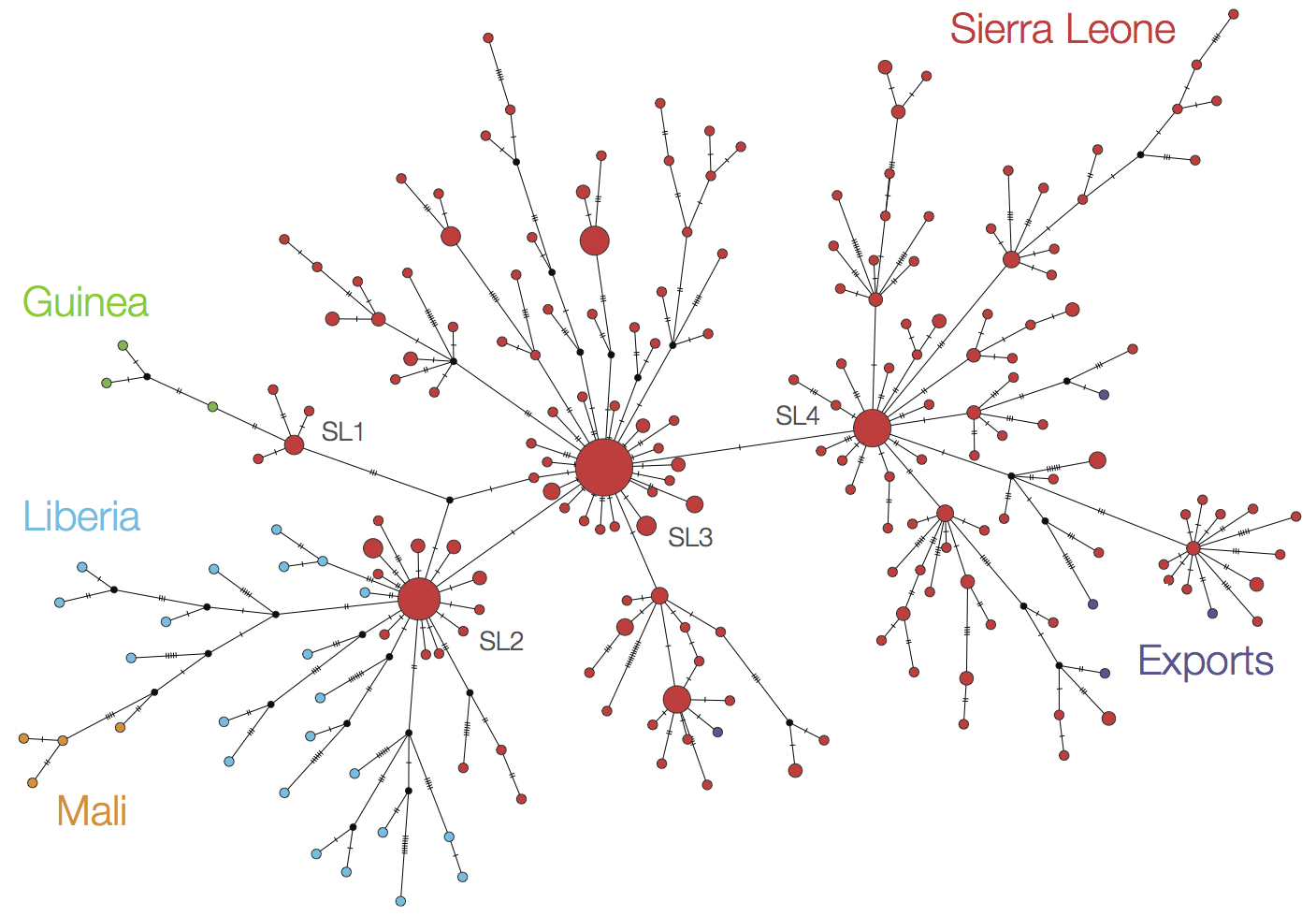
At epidemic height, geographic spread of particular interest
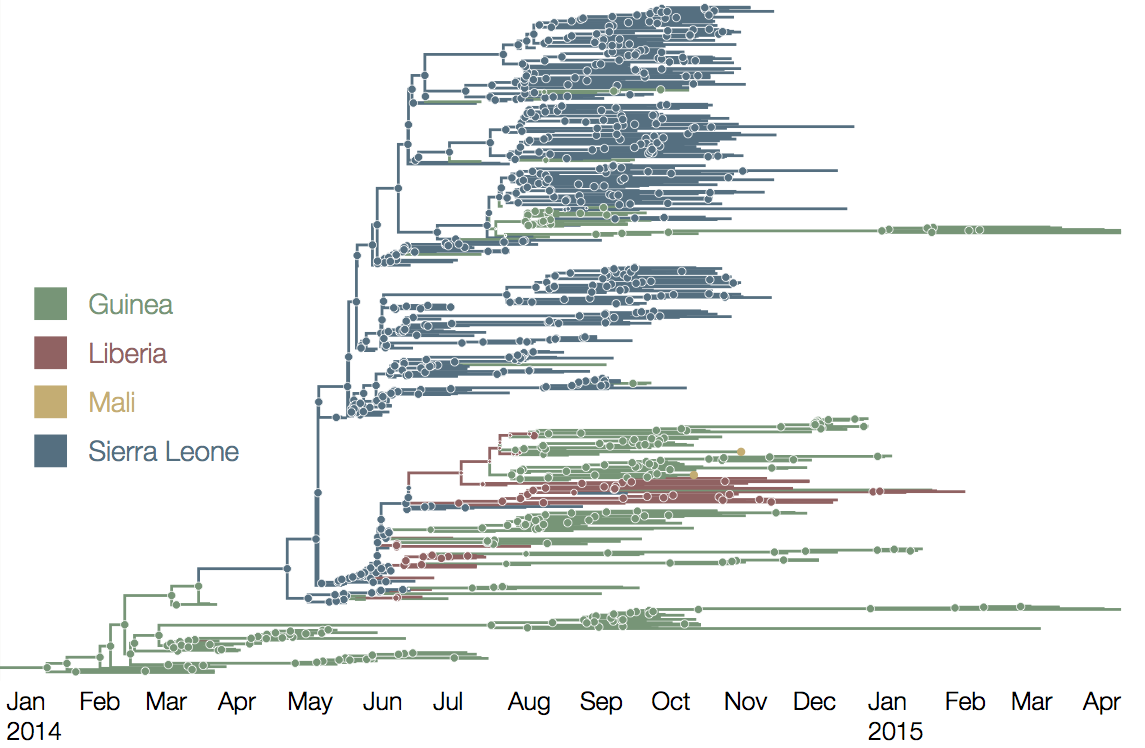
Later on, tracking transmission clusters of primary importance
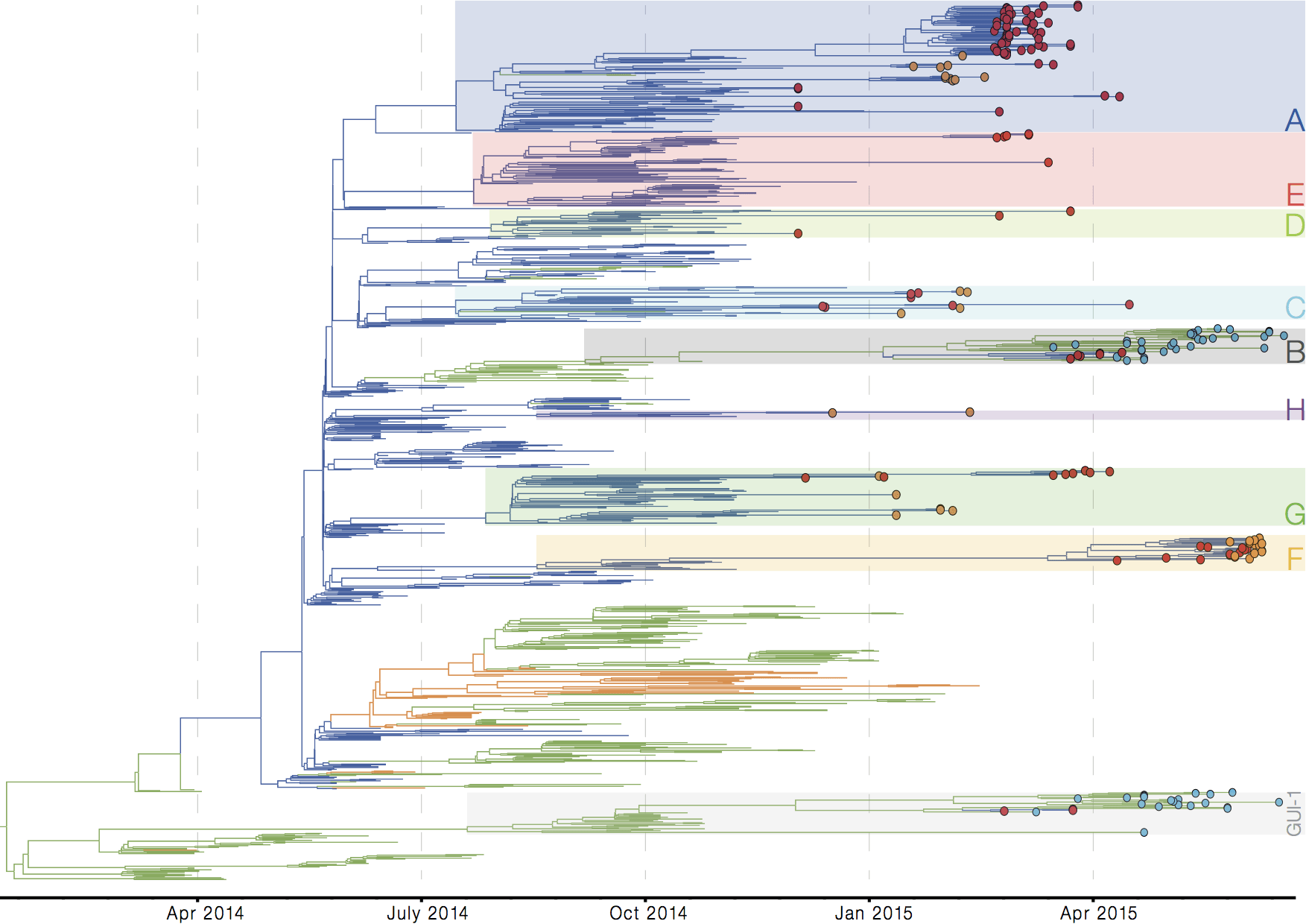
Evolutionary analyses helped to establish the degree of adaptive evolution occurring
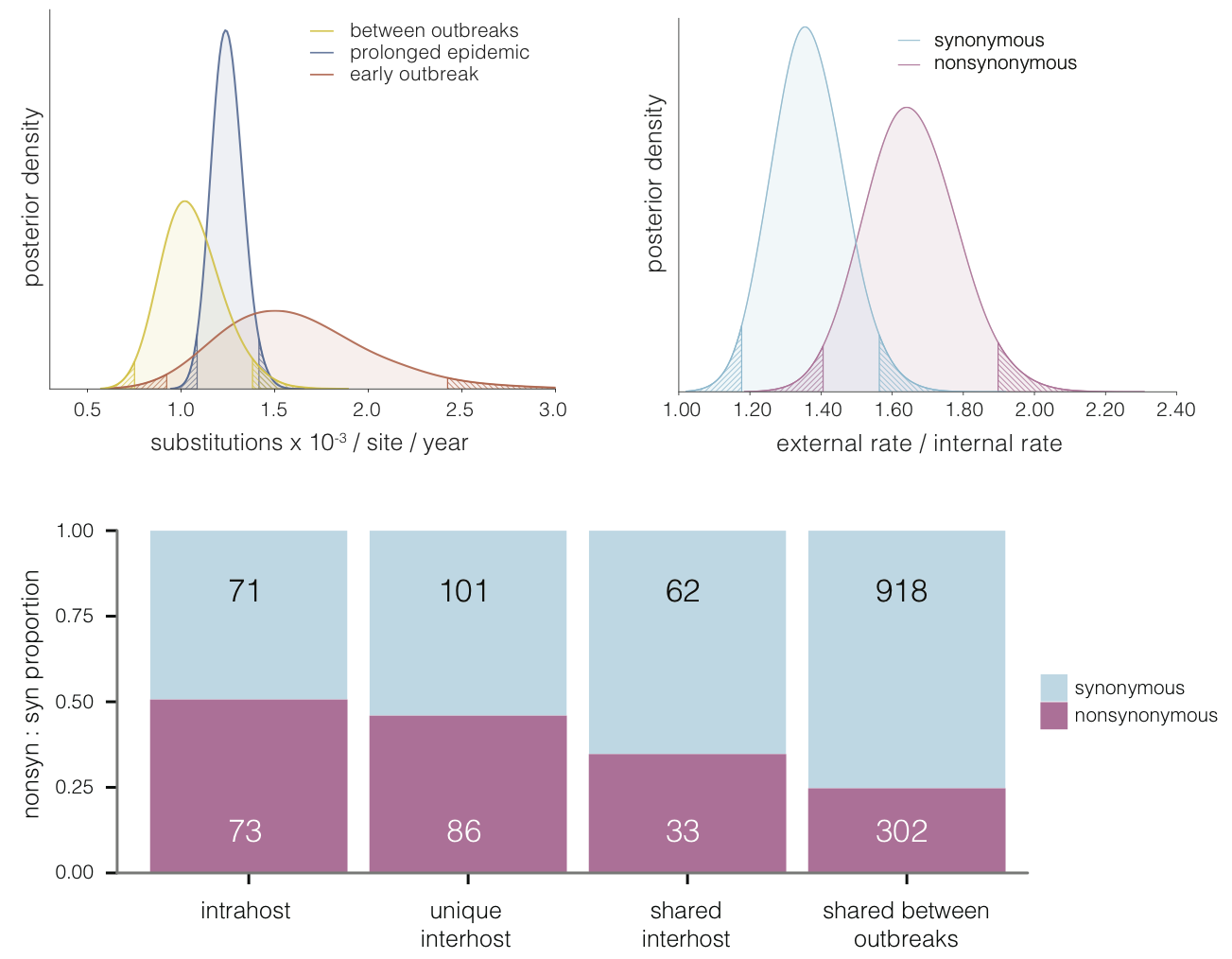
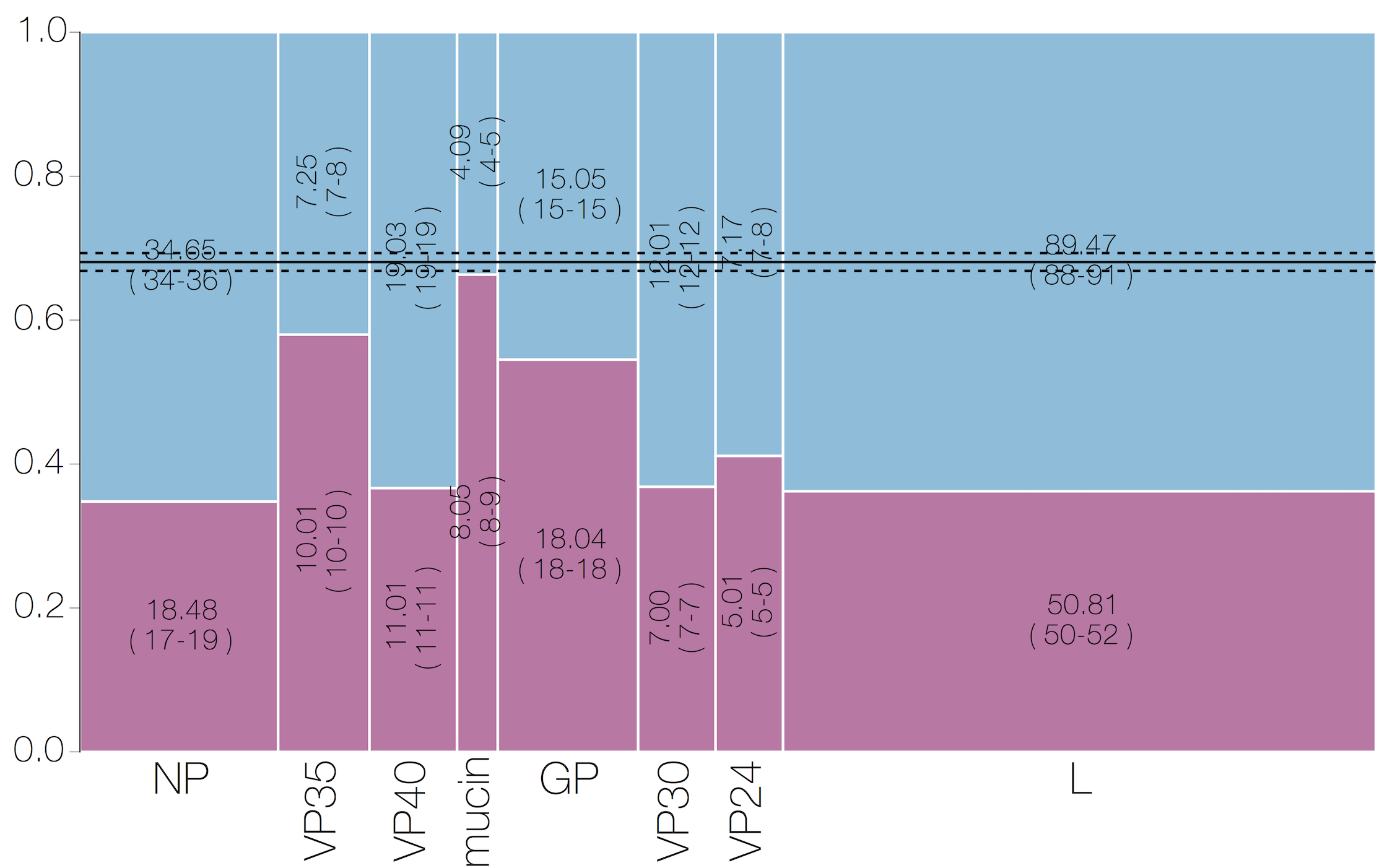
Selective patterns differ across genome
Phylogeographic analyses reveal detailed patterns of spatial movement
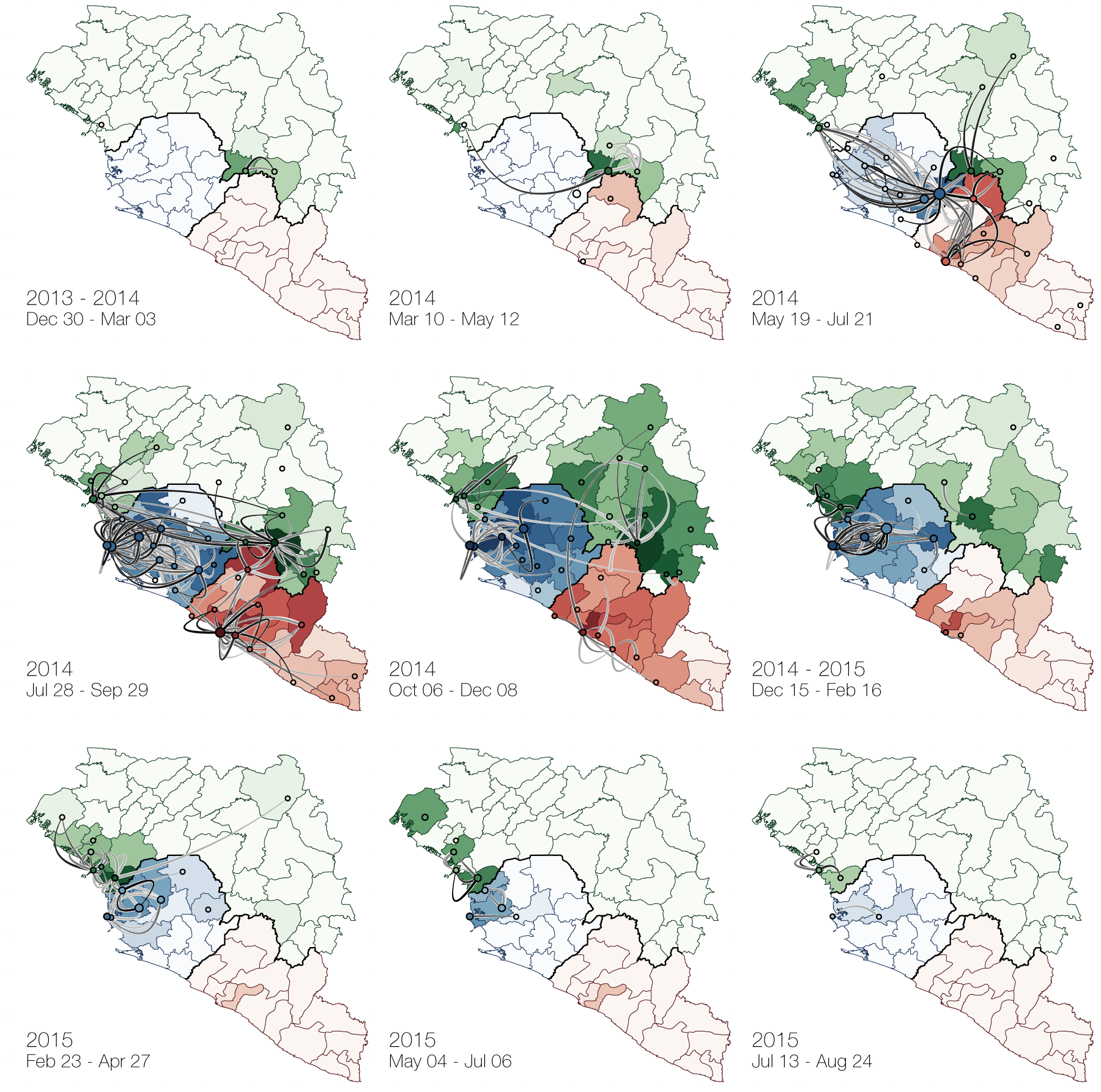
Animation by Gytis Dudas
Important analyses, let's make them more rapid and more automated
Tracking epidemic spread in real-time:
ebola.nextstrain.org
Rapid on-the-ground sequencing by Ian Goodfellow, Matt Cotten and colleagues
Deployment of MinION sequencing to Guinea by Nick Loman, Josh Quick, Lauren Cowley and colleagues

Zika
Virus endemic to Africa, emergence in Southeast Asia in the last century
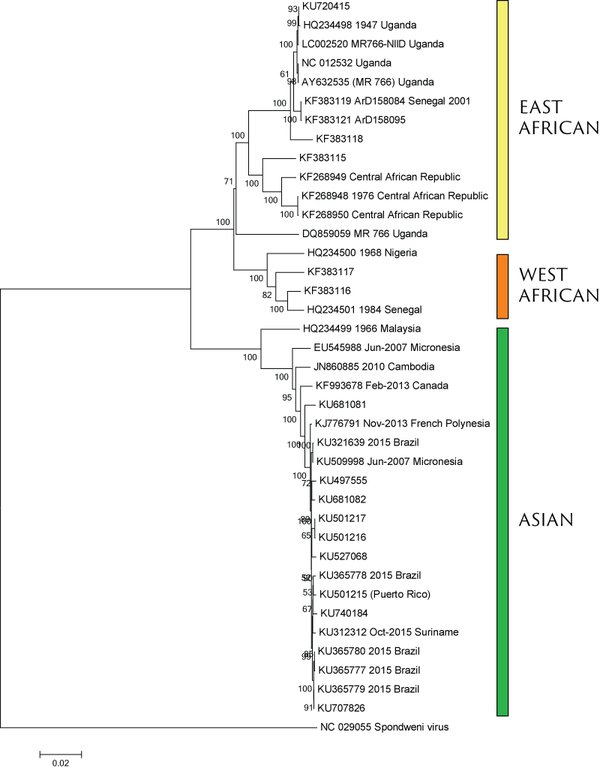
Spread eastward through the South Pacific
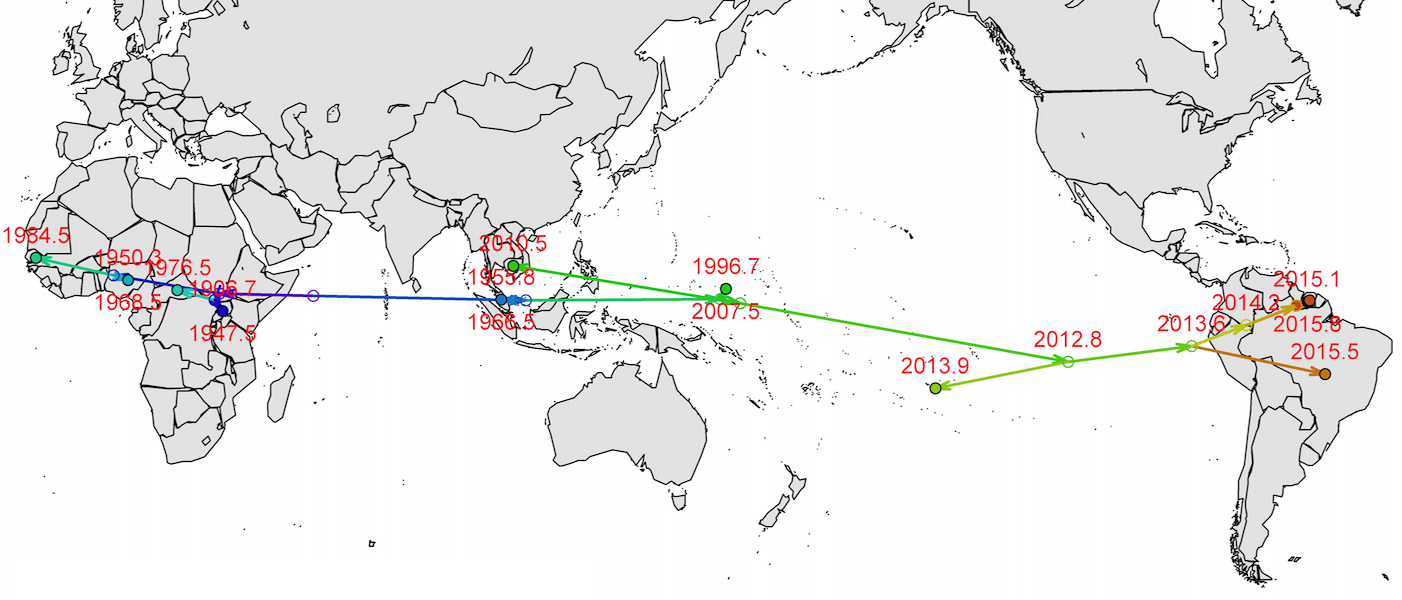
Isolated epidemics in the South Pacific

Single arrival into the Americas in early 2014
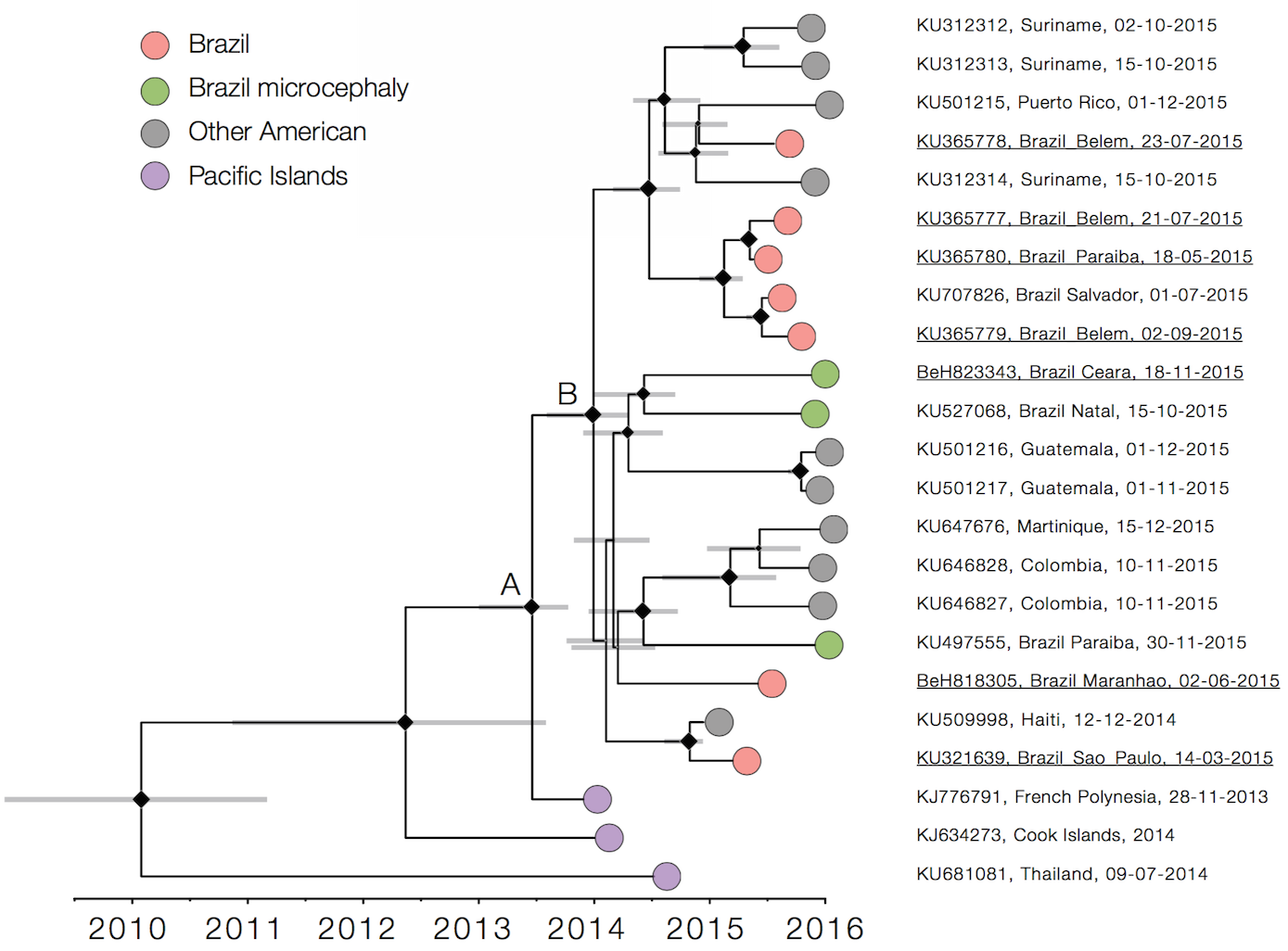
Working on analysis of ongoing evolution:
nextstrain.org/zika/
ZiBRA: Project to do real-time sequencing of Zika in Brazil
Road trip through northeast Brazil to collect samples and sequence
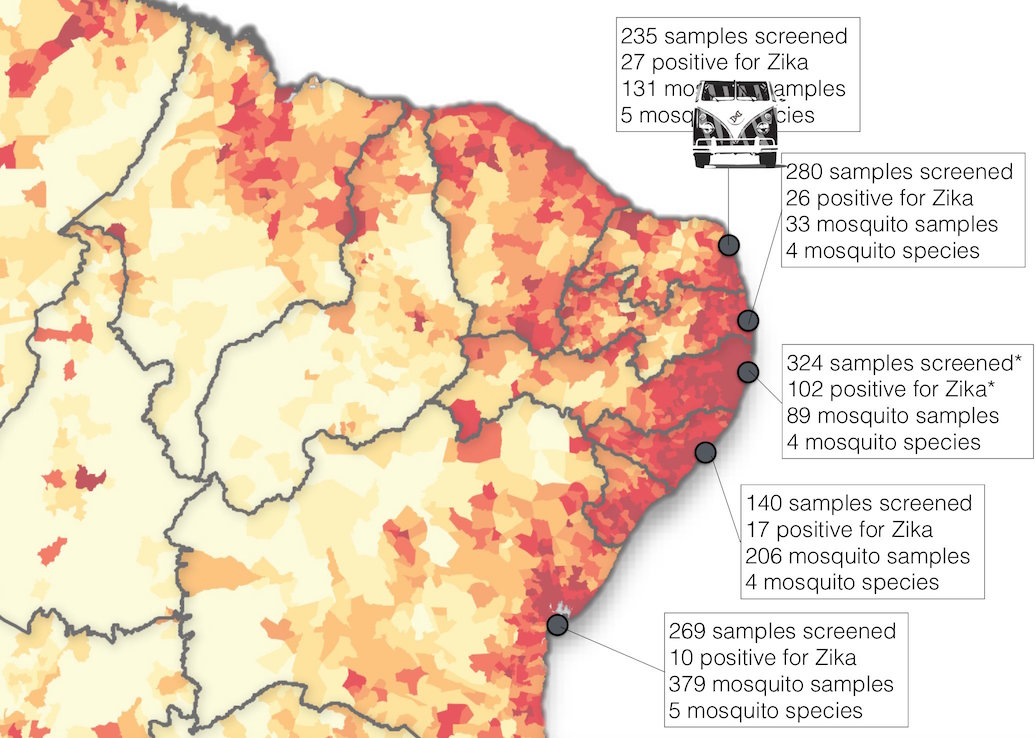
Sequencing on the MinION nanopore sequencer
Moving forward, genetically-informed outbreak response requires:
- Rapid sharing of sequence data, genetic context critical
- Technologies for rapid diagnostics and sequencing
- Technologies to rapidly conduct phylogenetic inference
- Technologies to explore genetic relationships and inform epidemiological investigation
Future work

Acknowledgements
Influenza: WHO Global Influenza Surveillance Network, GISAID, Worldwide Influenza Centre at the Francis Crick Institute, Richard Neher, Colin Russell, Andrew Rambaut
Ebola: data producers, Gytis Dudas, Andrew Rambaut, Philipe Lemey, Richard Neher, Nick Loman, Ian Goodfellow, Paul Kellam, Danny Park, Kristian Andersen, Pardis Sabeti
Zika: data producers, Nick Loman, Nuno Faria, Andrew Rambaut, Oliver Pybus, Richard Neher, Charlton Callender, Allison Black, Luiz Alcantara and the rest of the ZiBRA team
Contact
- Website: bedford.io
- Twitter: @trvrb
- Slides: bedford.io/talks/real-time-tracking-fmkbms/