Real-time tracking of virus evolution
Trevor Bedford (@trvrb)
22 Oct 2015
Research Week Symposium
BCCDC
Slides at bedford.io/talks/
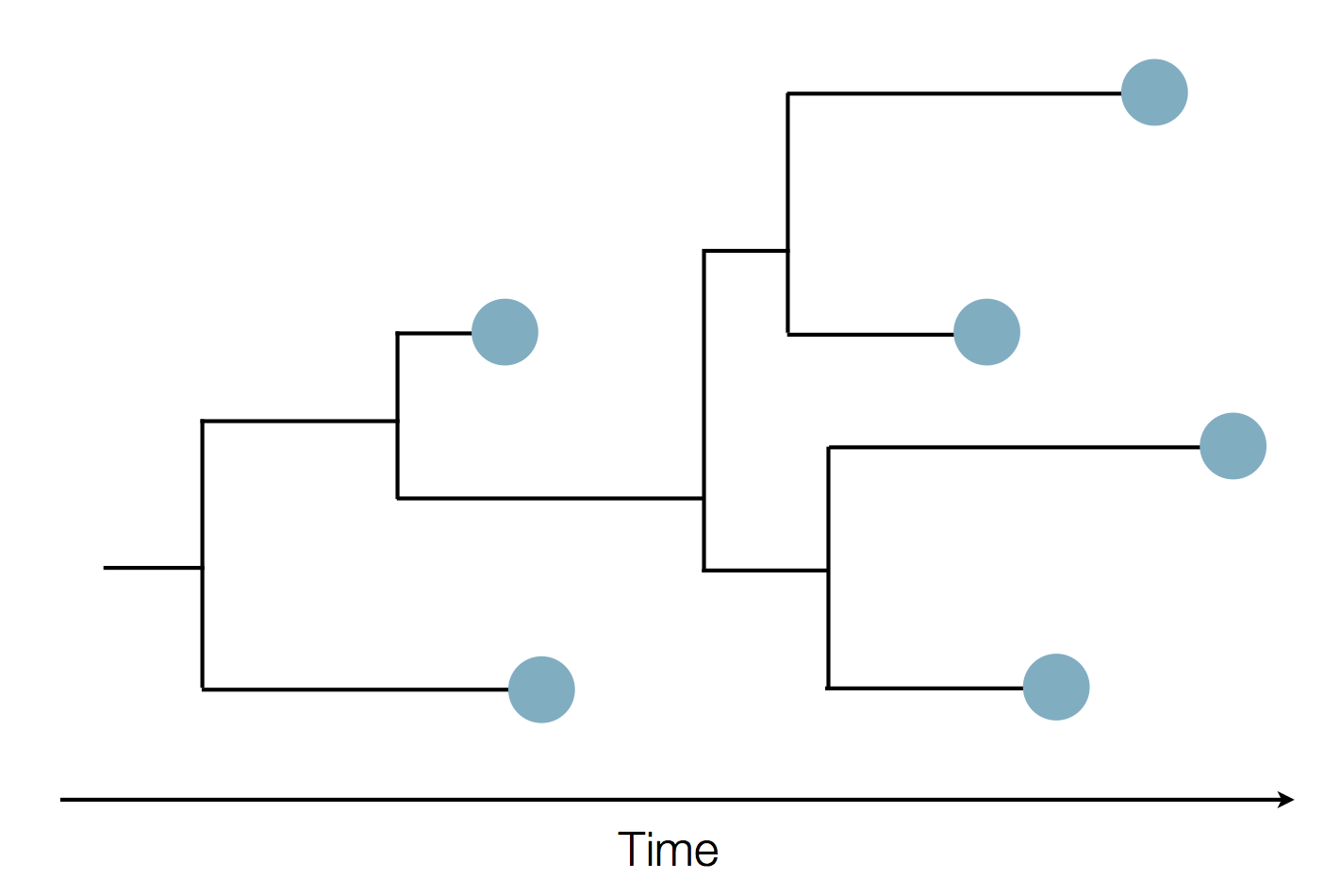
Phylogenies
Phylogenies describe history
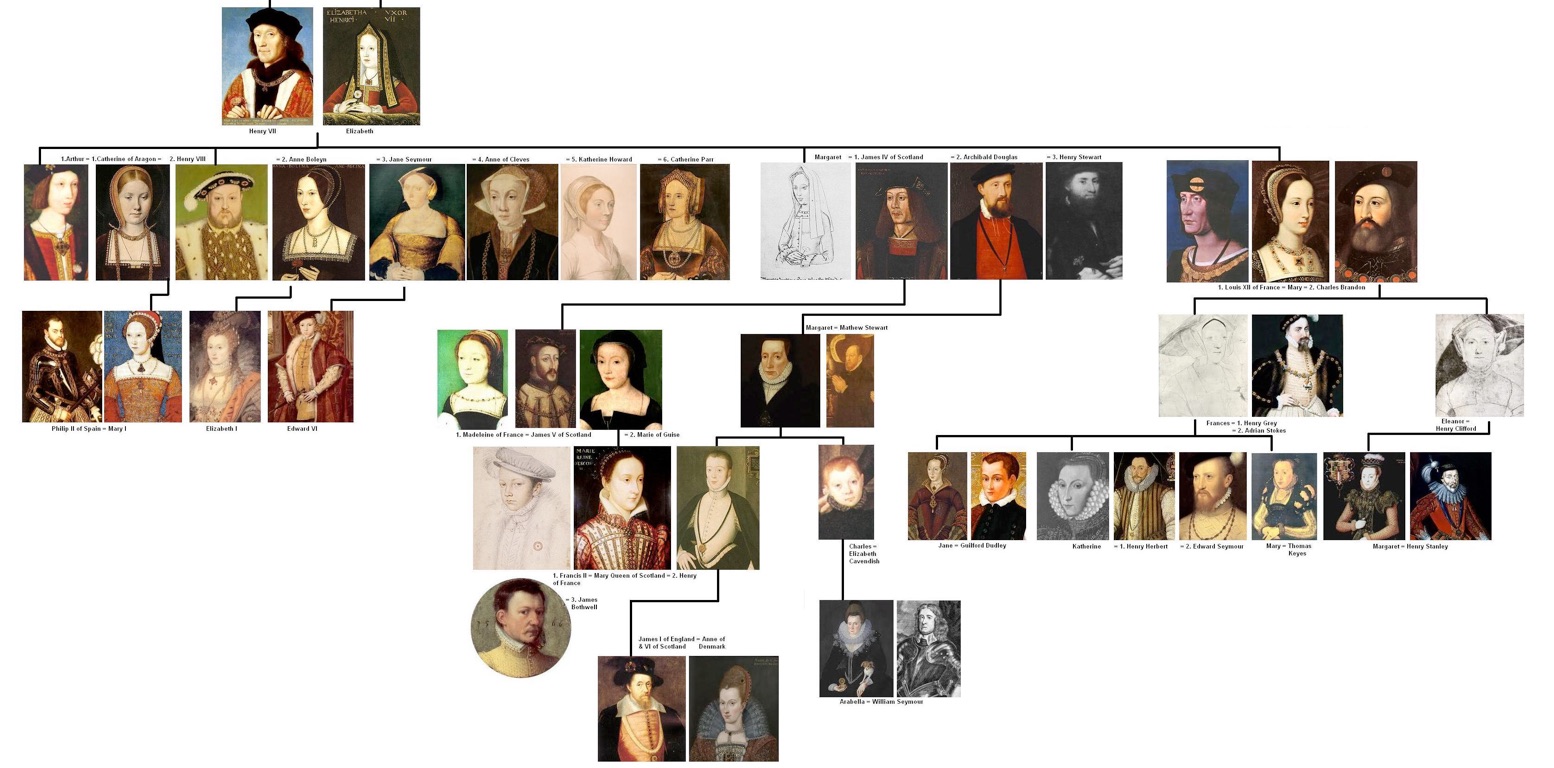
Phylogenies describe history
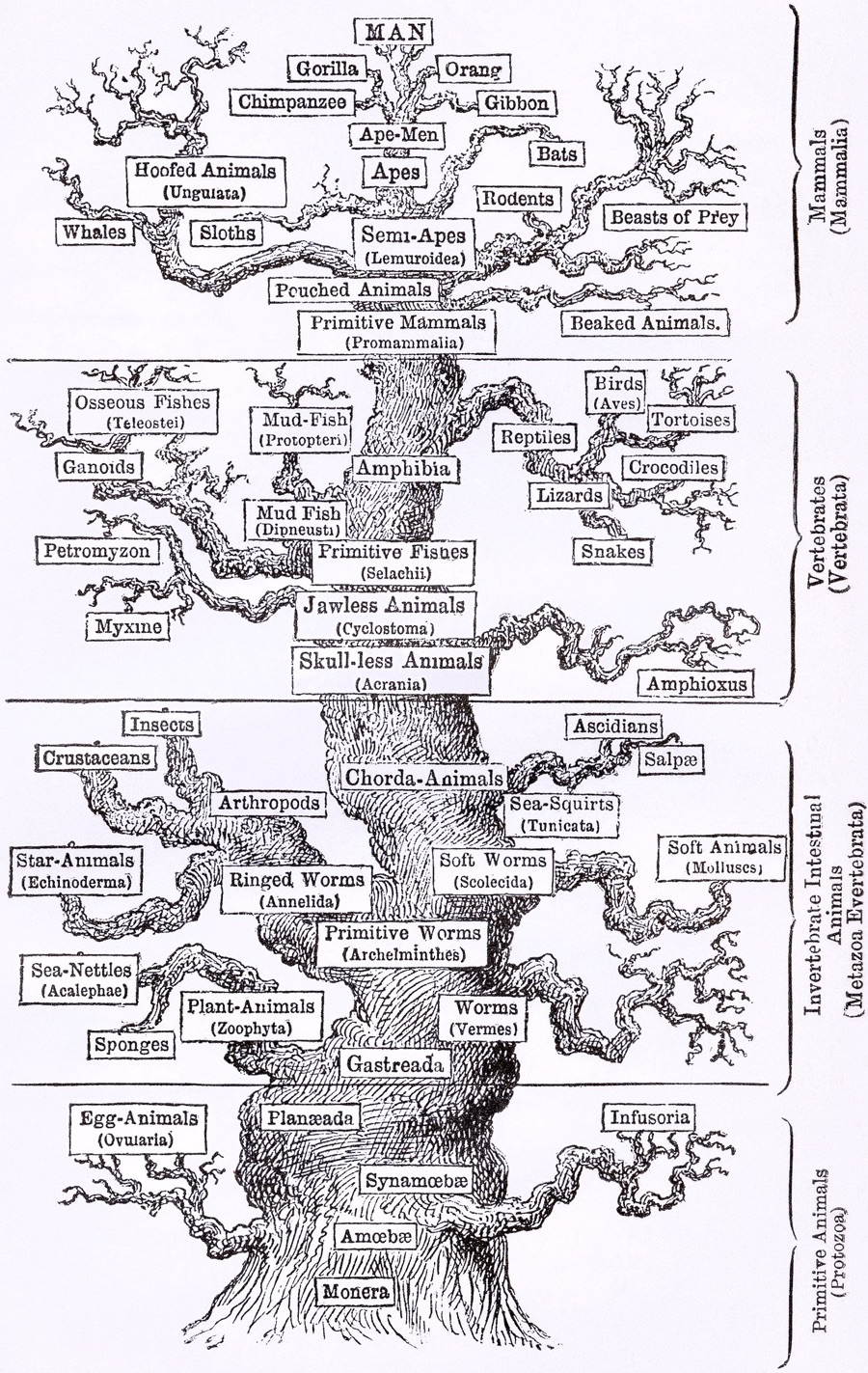
Phylogenies describe history
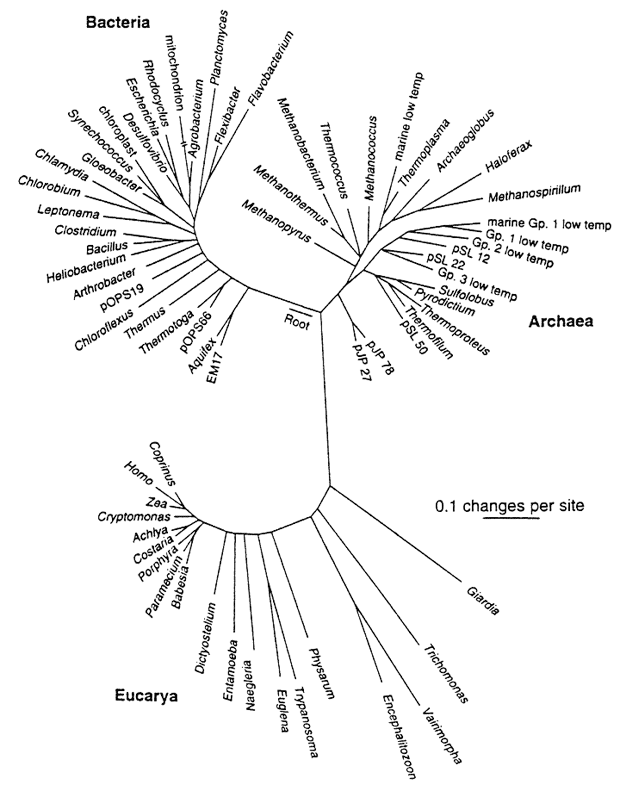
Phylogenies reveal process
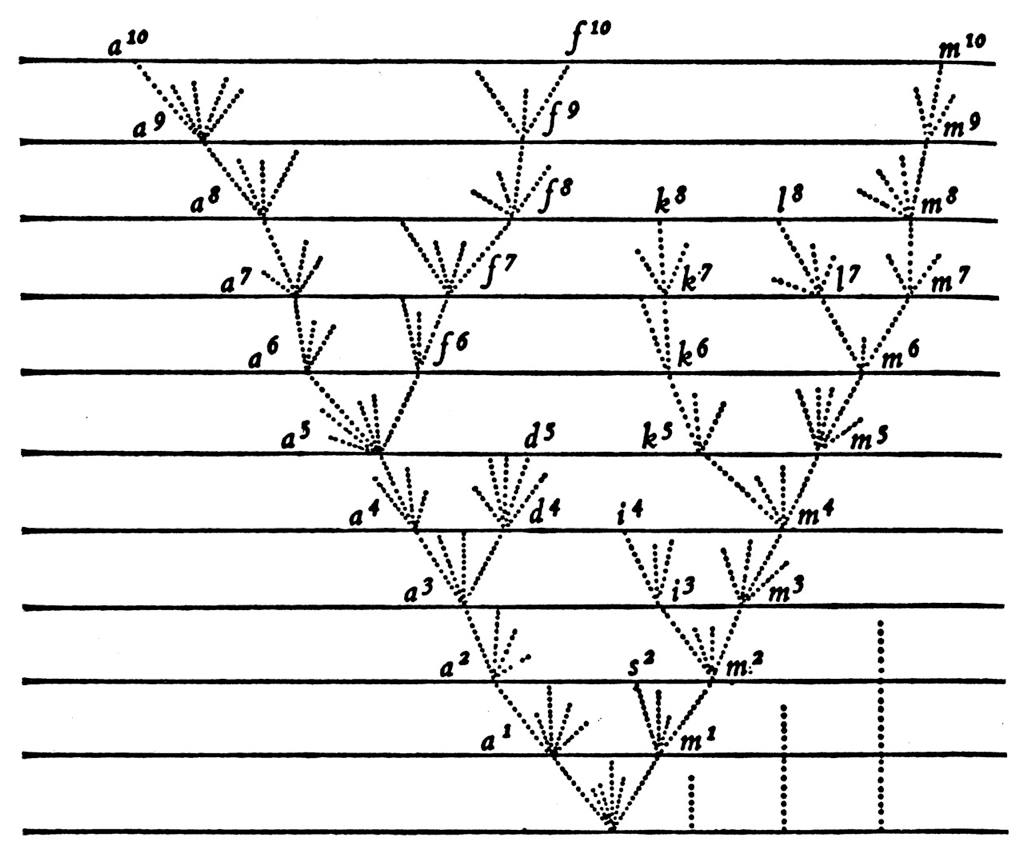
Epidemic process
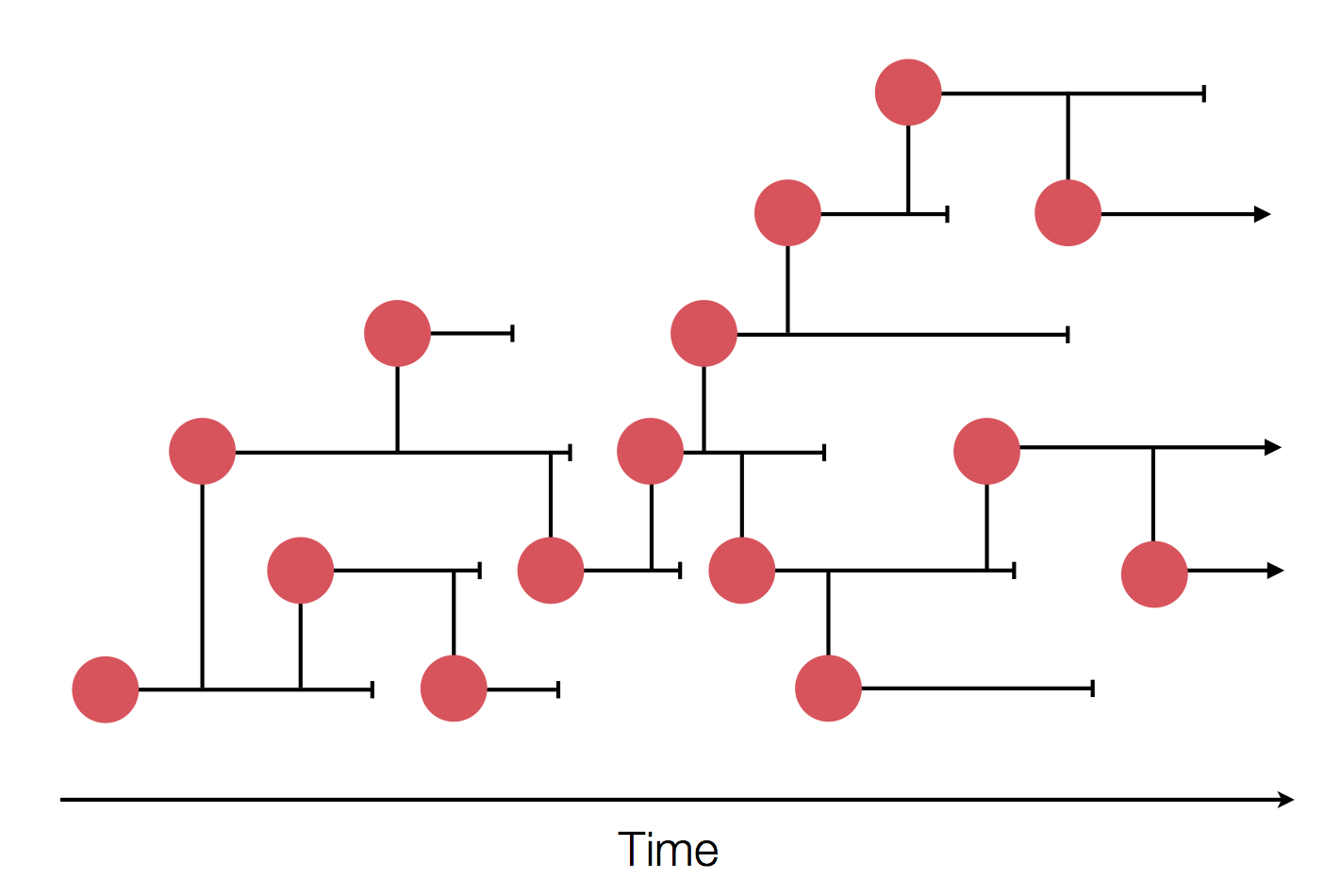
Sample some individuals
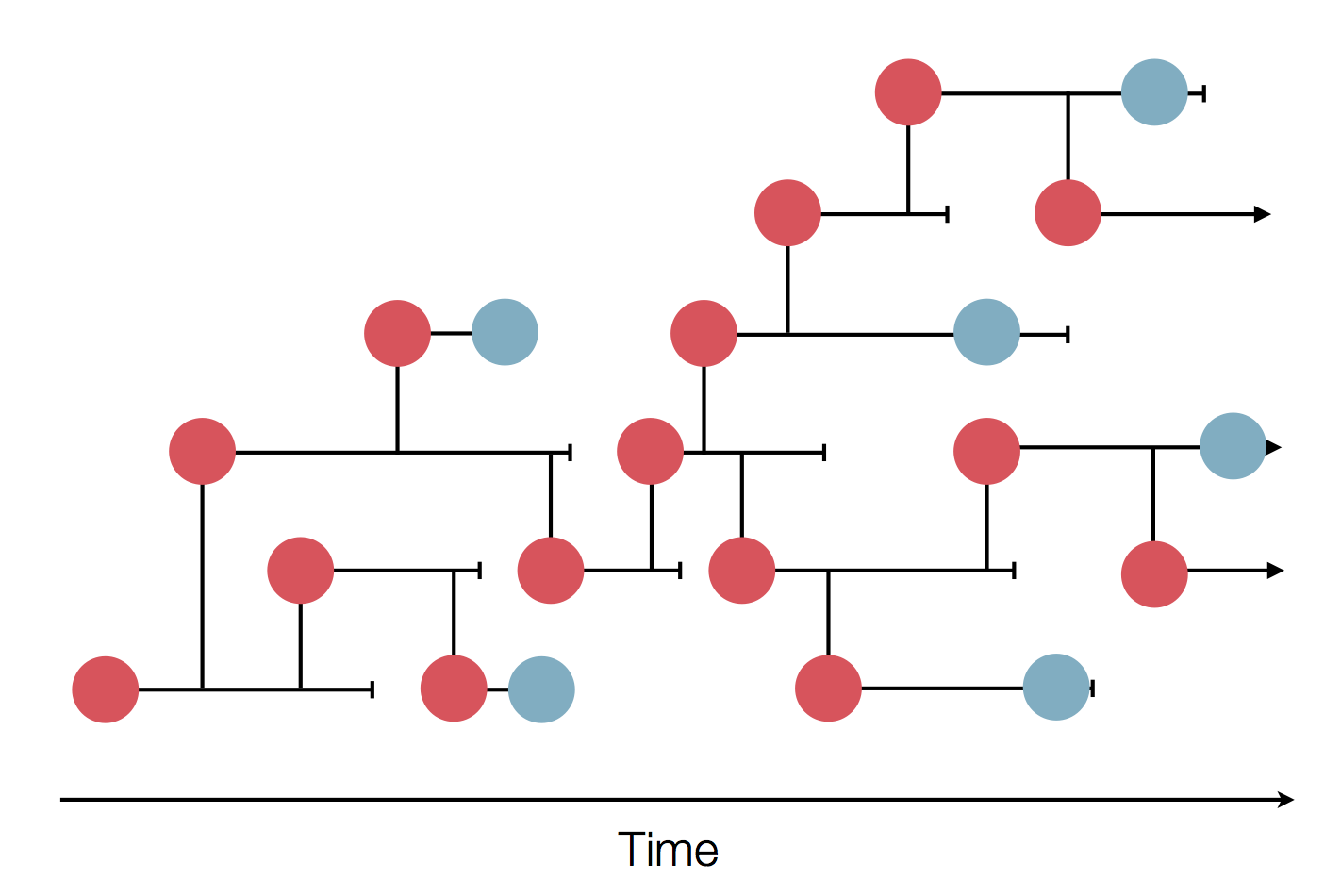
Sequence and determine phylogeny
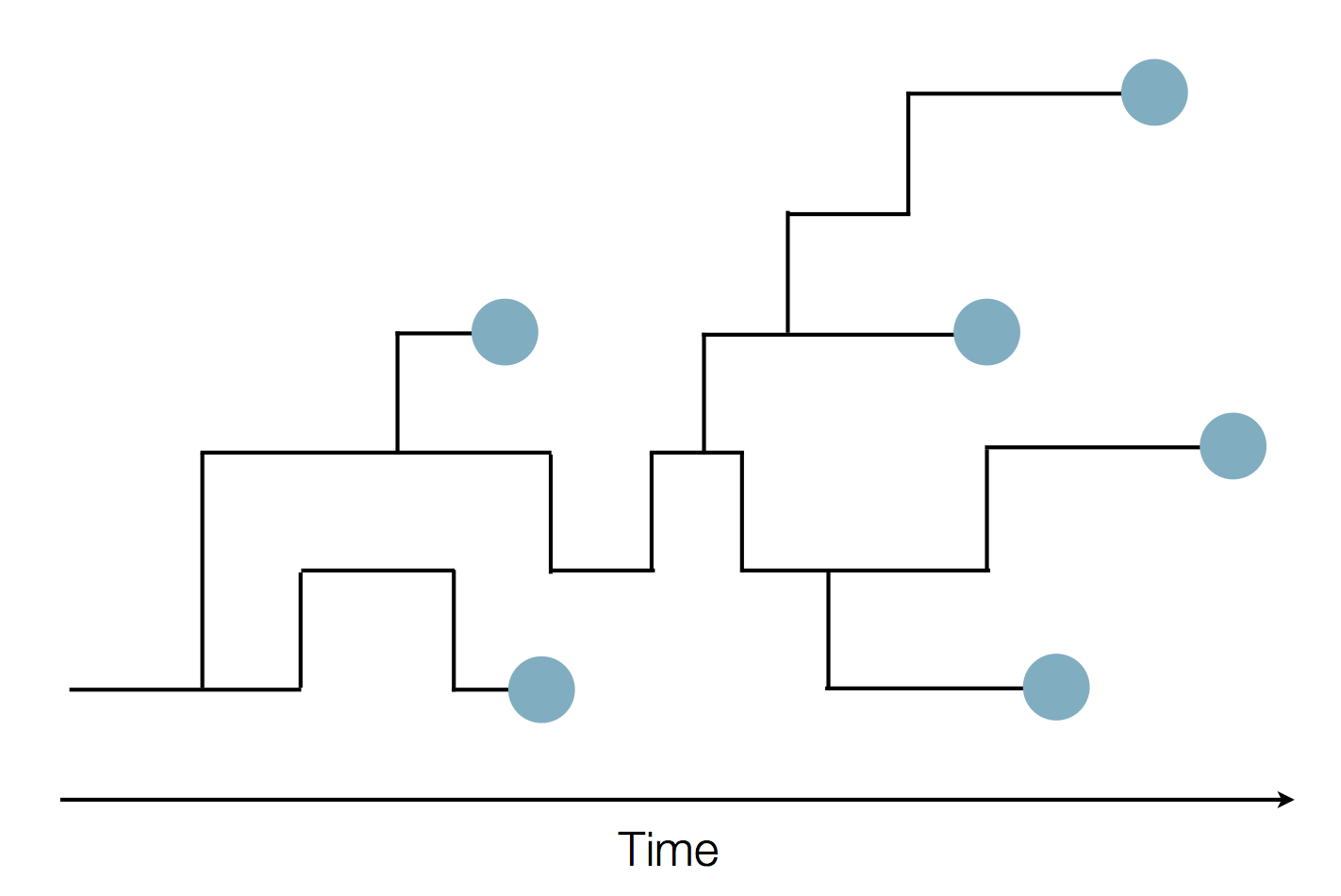
Sequence and determine phylogeny
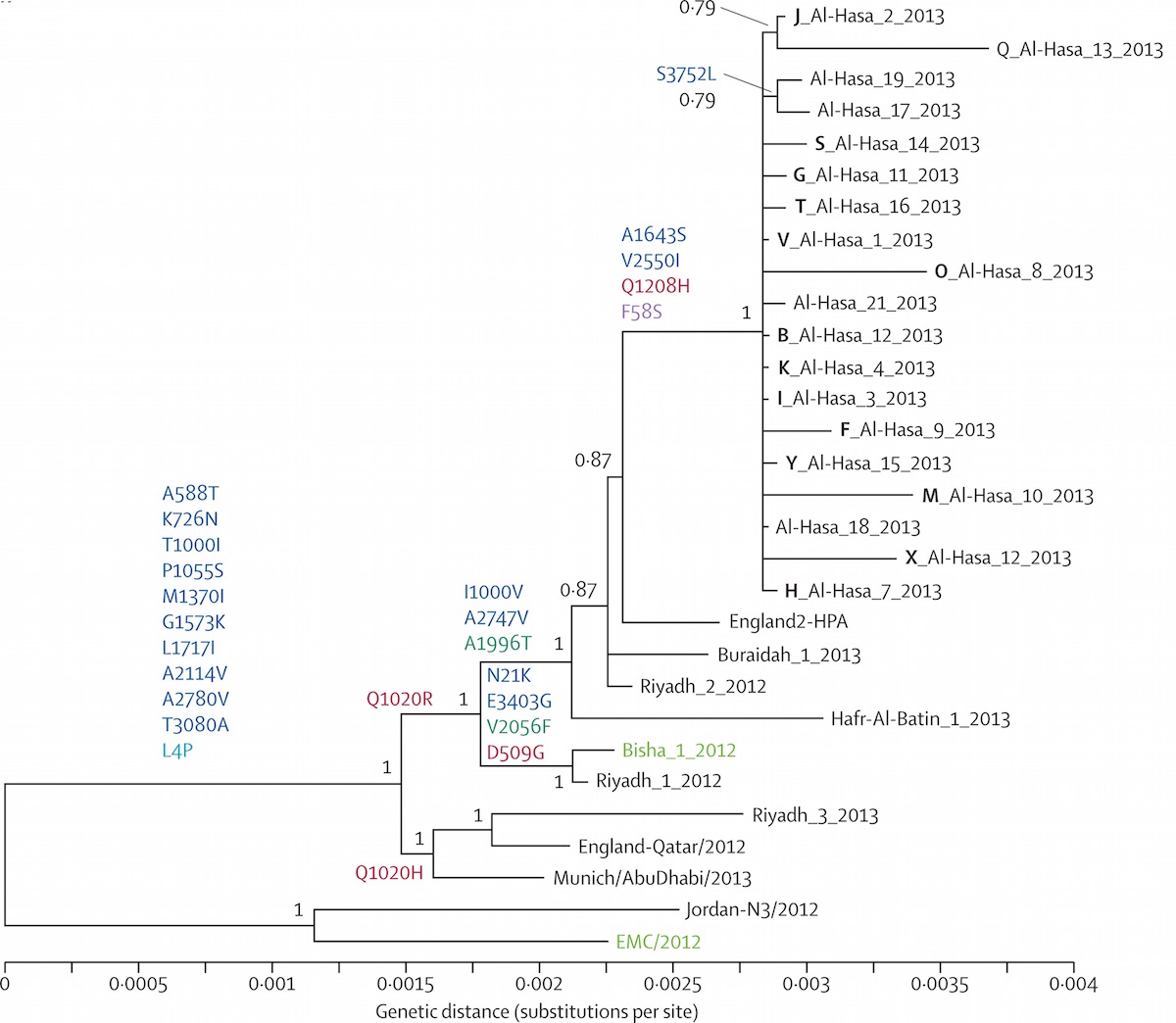
Middle Eastern MERS-CoV phylogeny
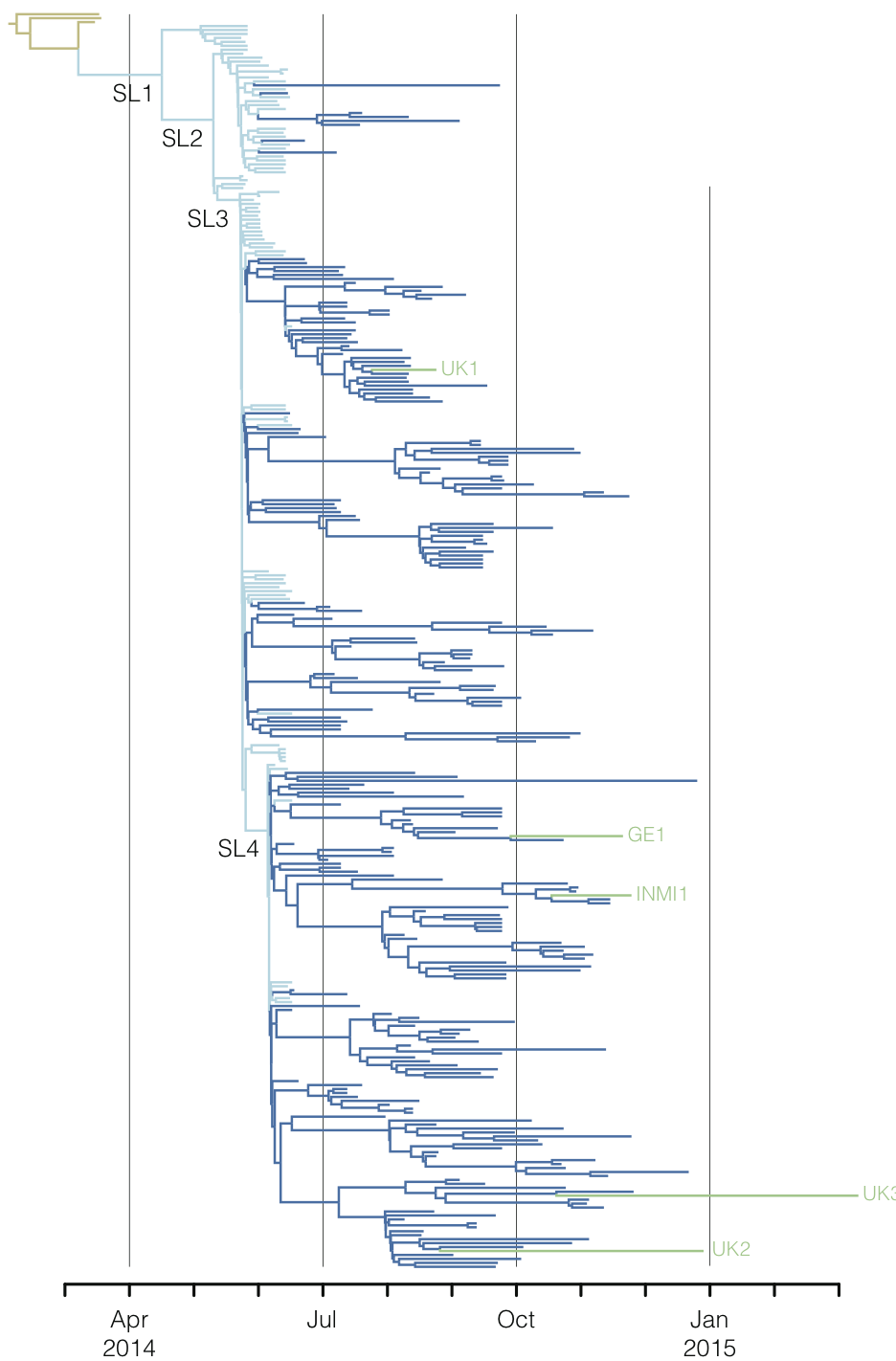
West African Ebola phylogeny
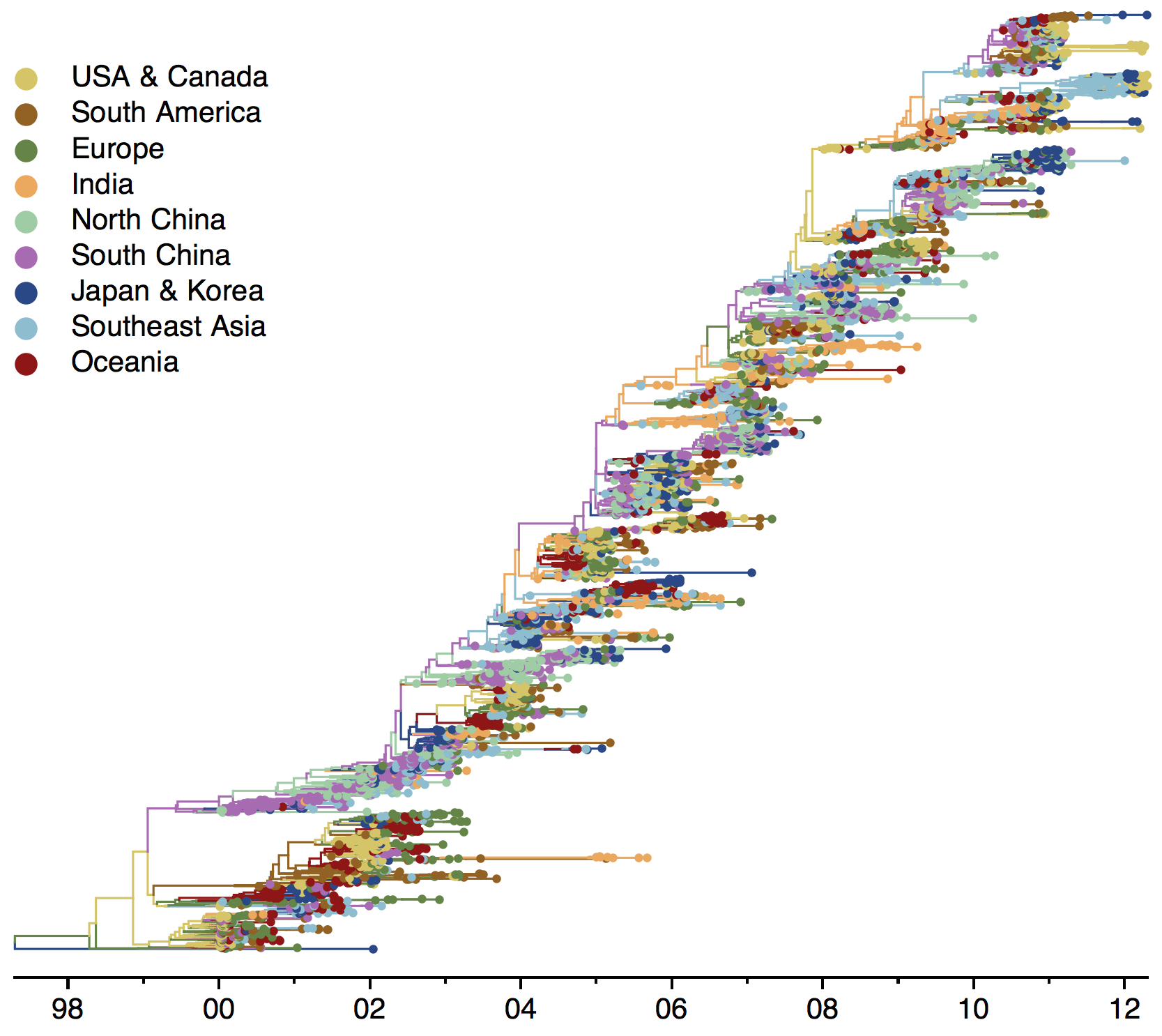
Global influenza phylogeny
Applications of evolutionary analysis for vaccine strain selection in influenza and charting epidemic spread in Ebola
Influenza
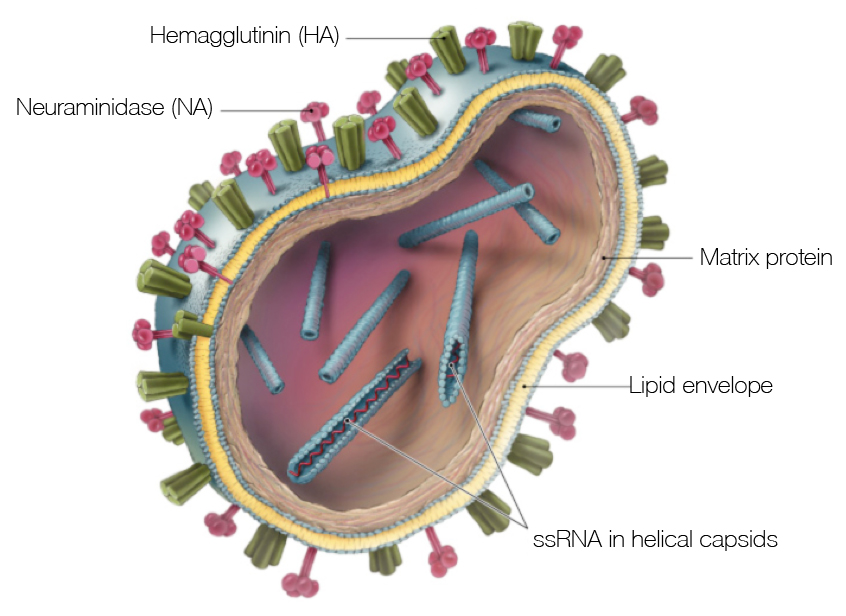
Influenza virion
Flu pandemics caused by host switch events
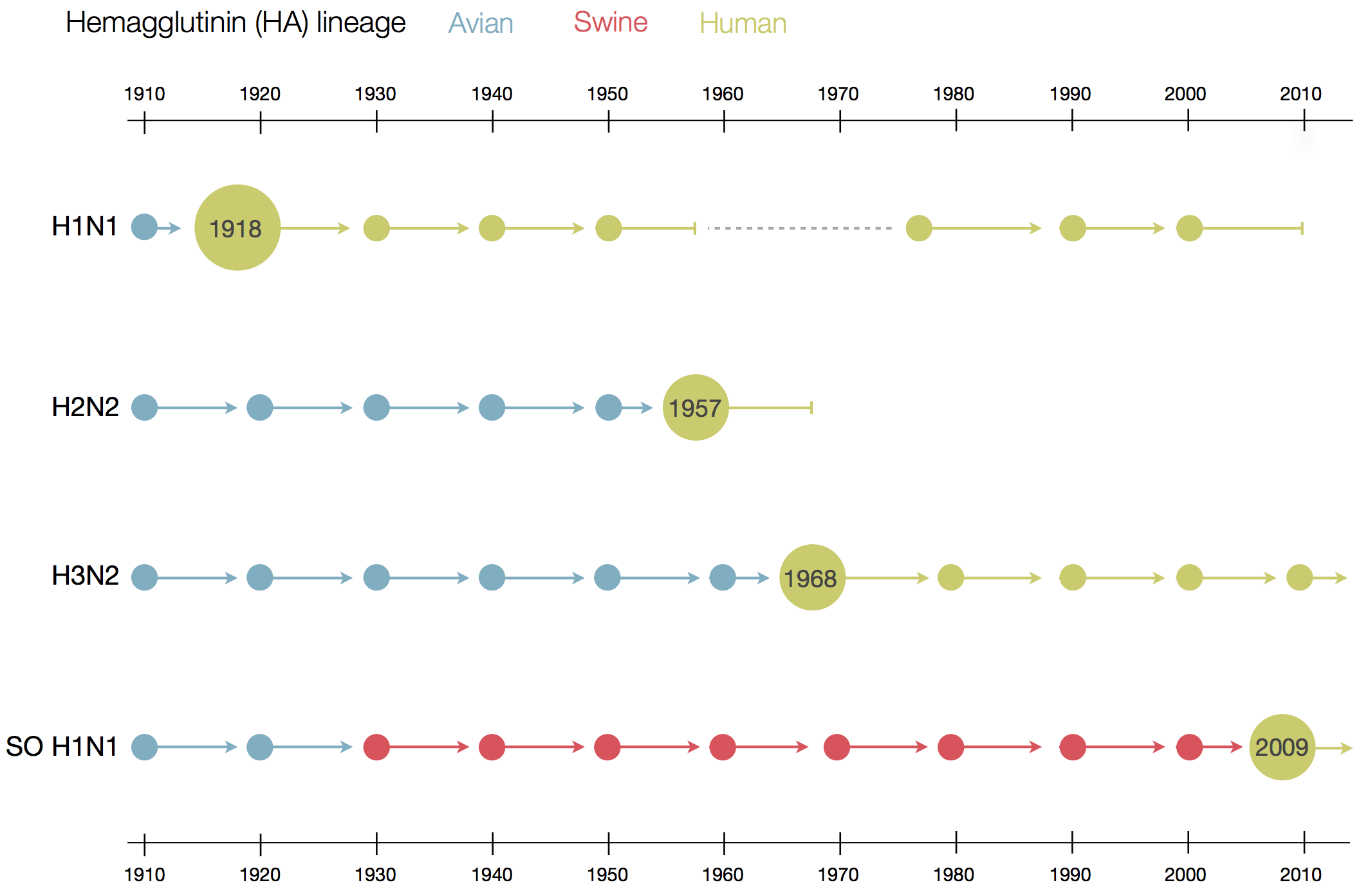
Influenza B does not have pandemic potential
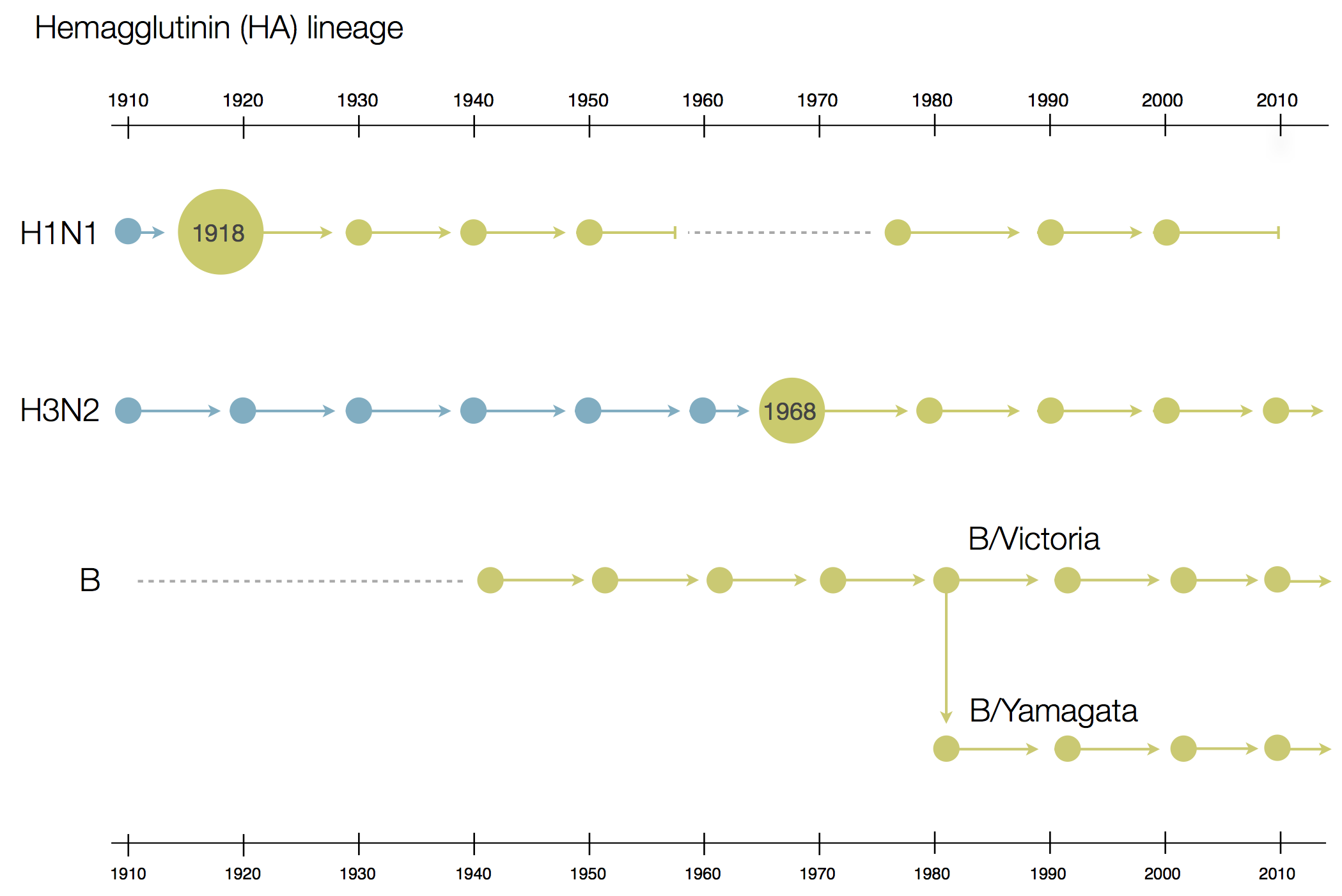
Phylogenetic trees of different influenza lineages
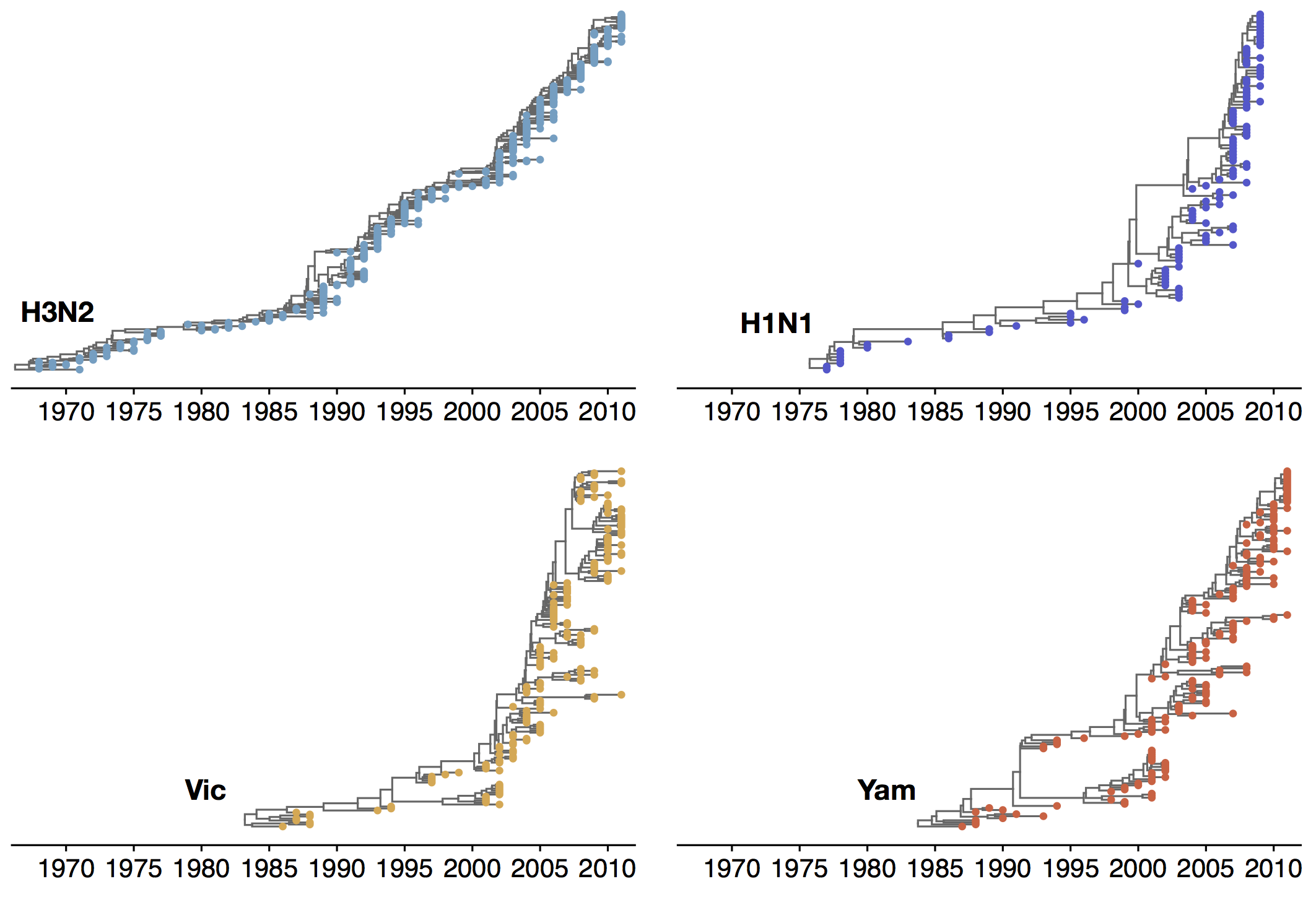
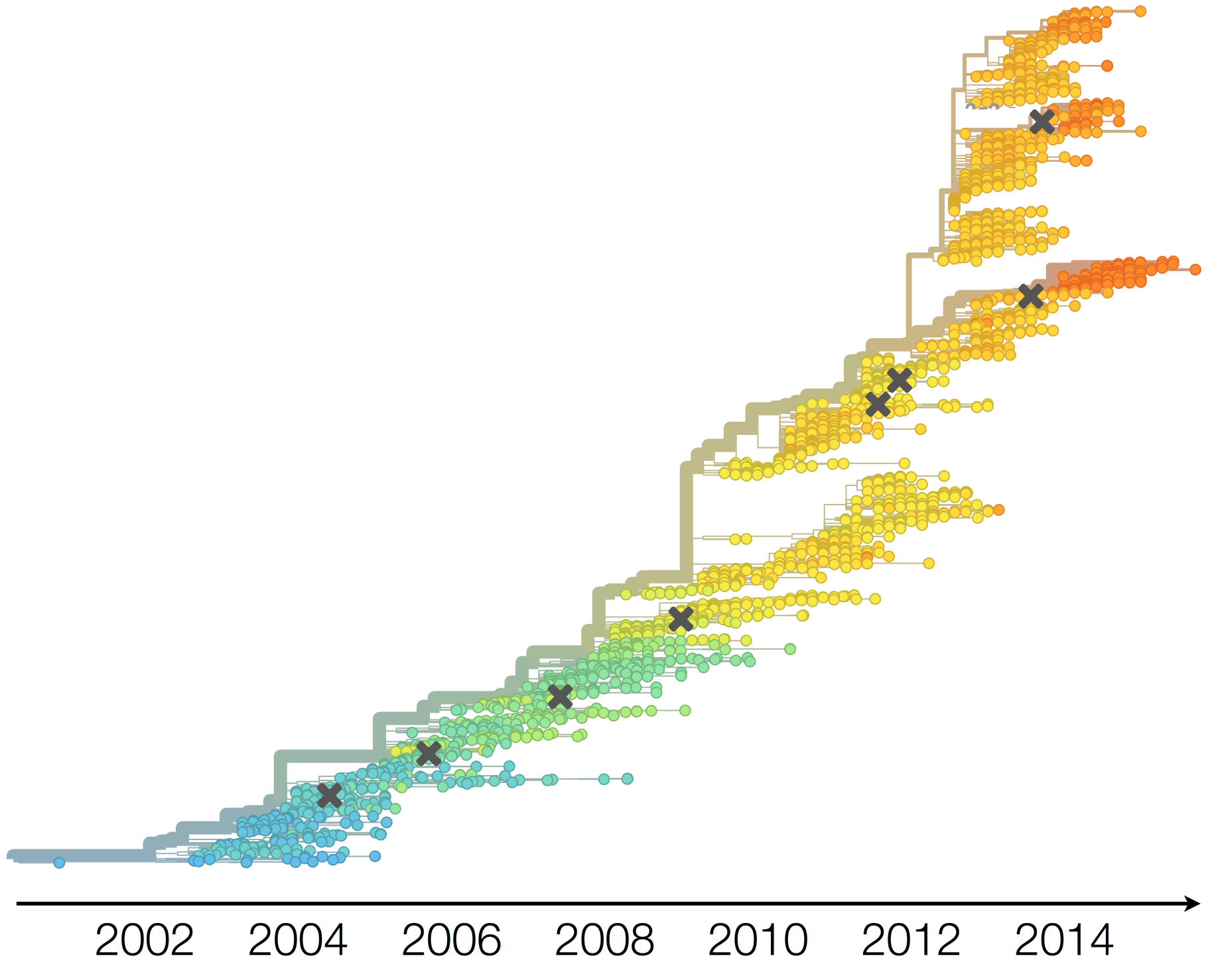
Antigenic evolution drives viral dynamics
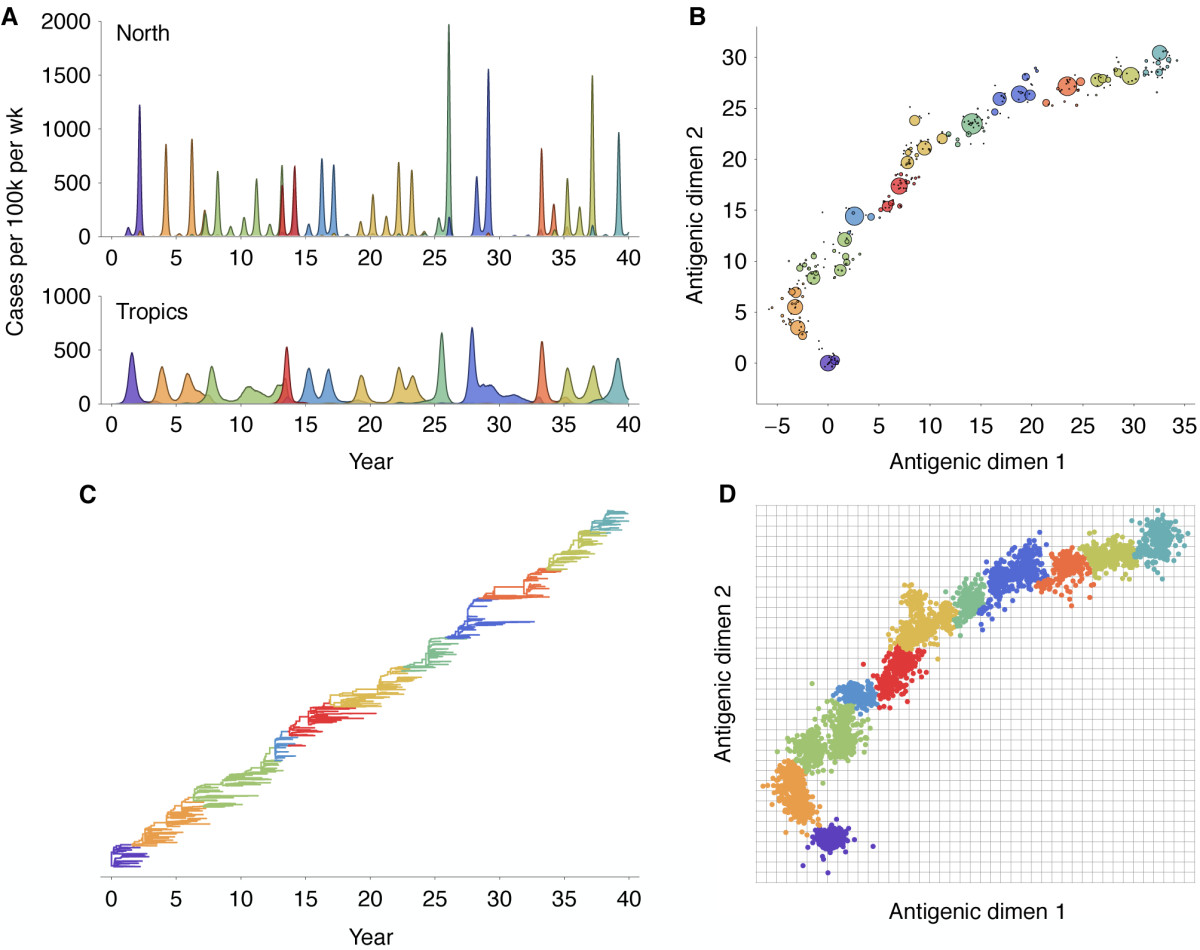
Antigenic evolution in H3N2
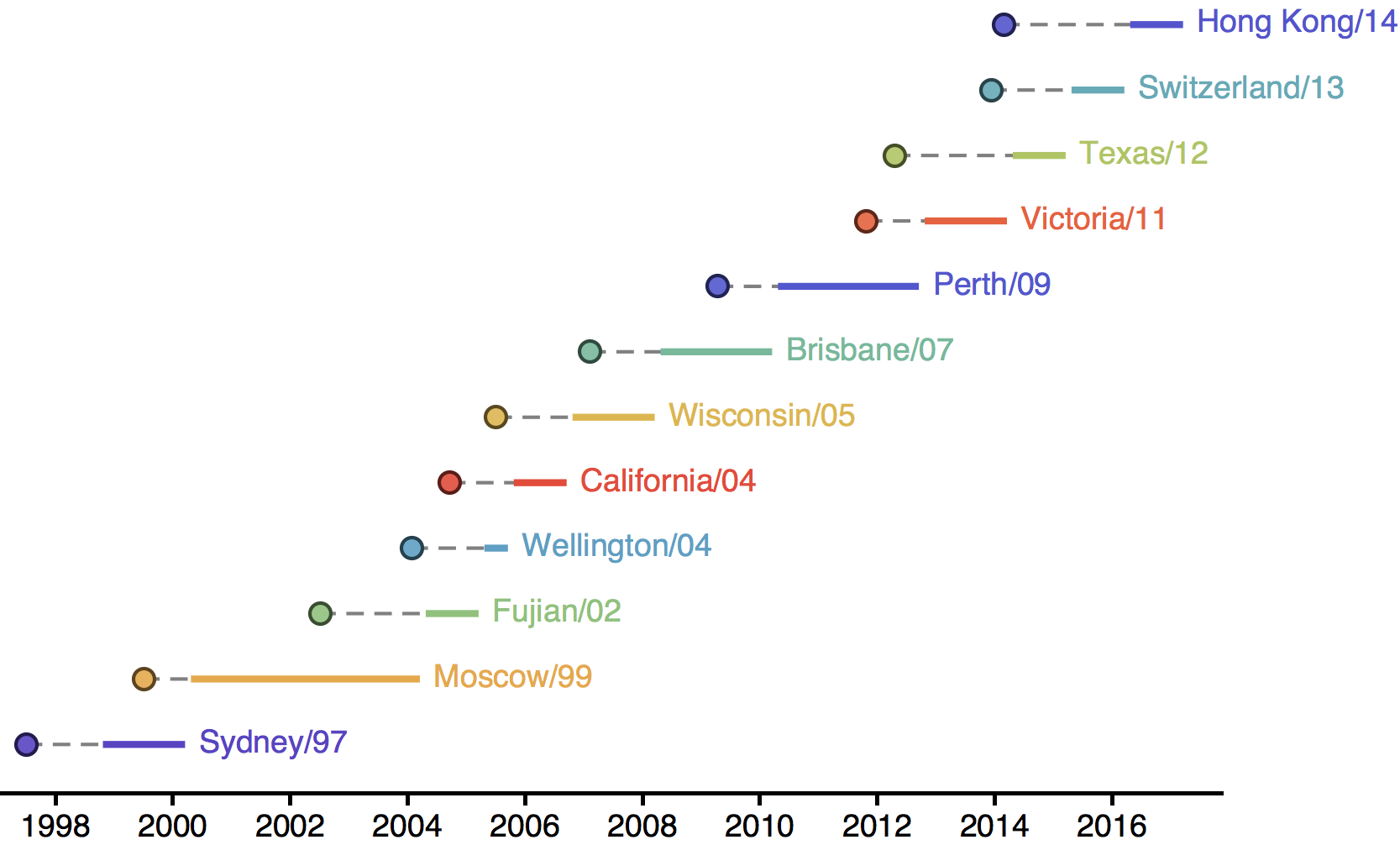
Influenza H3N2 vaccine updates
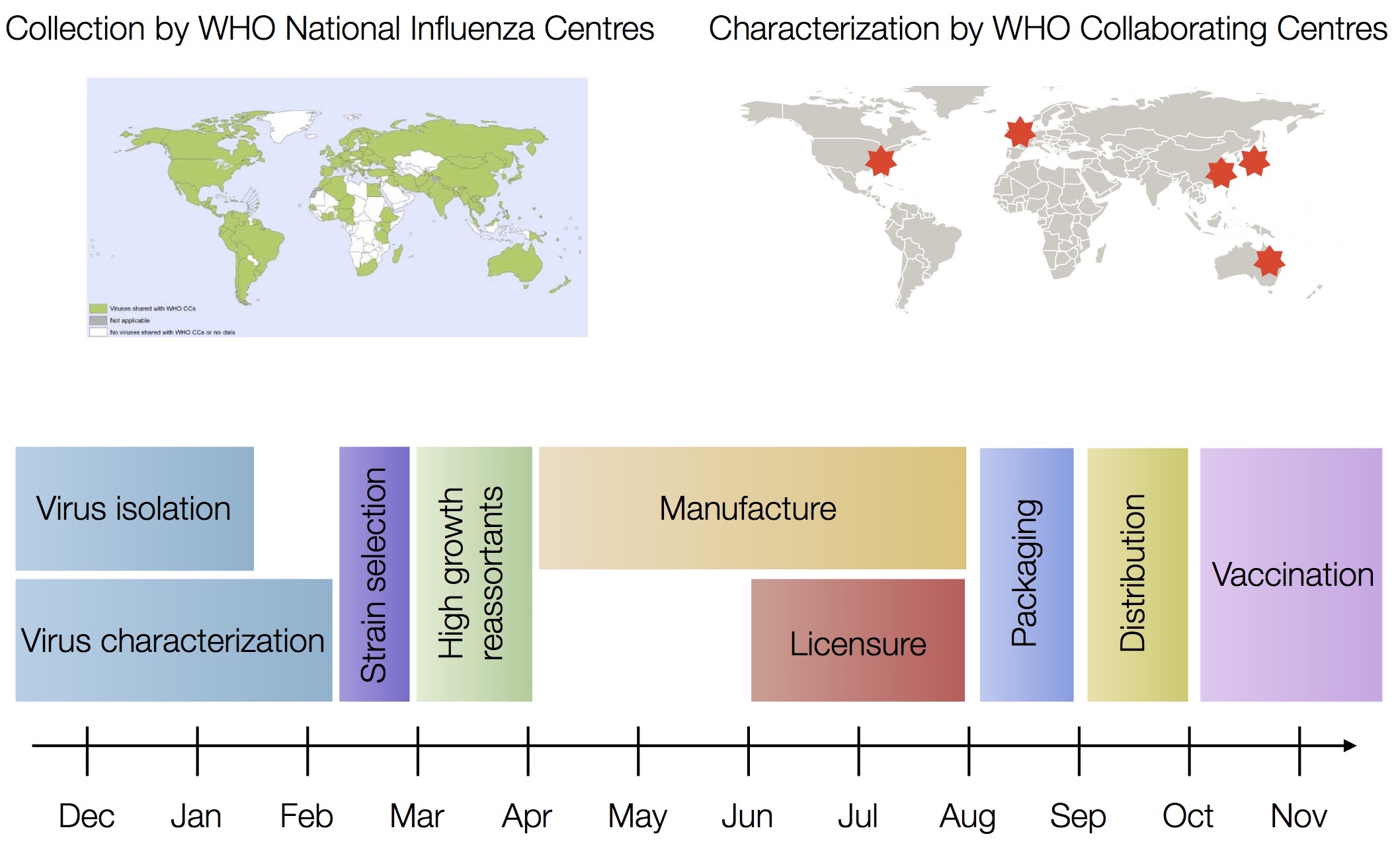
Vaccine strain selection timeline
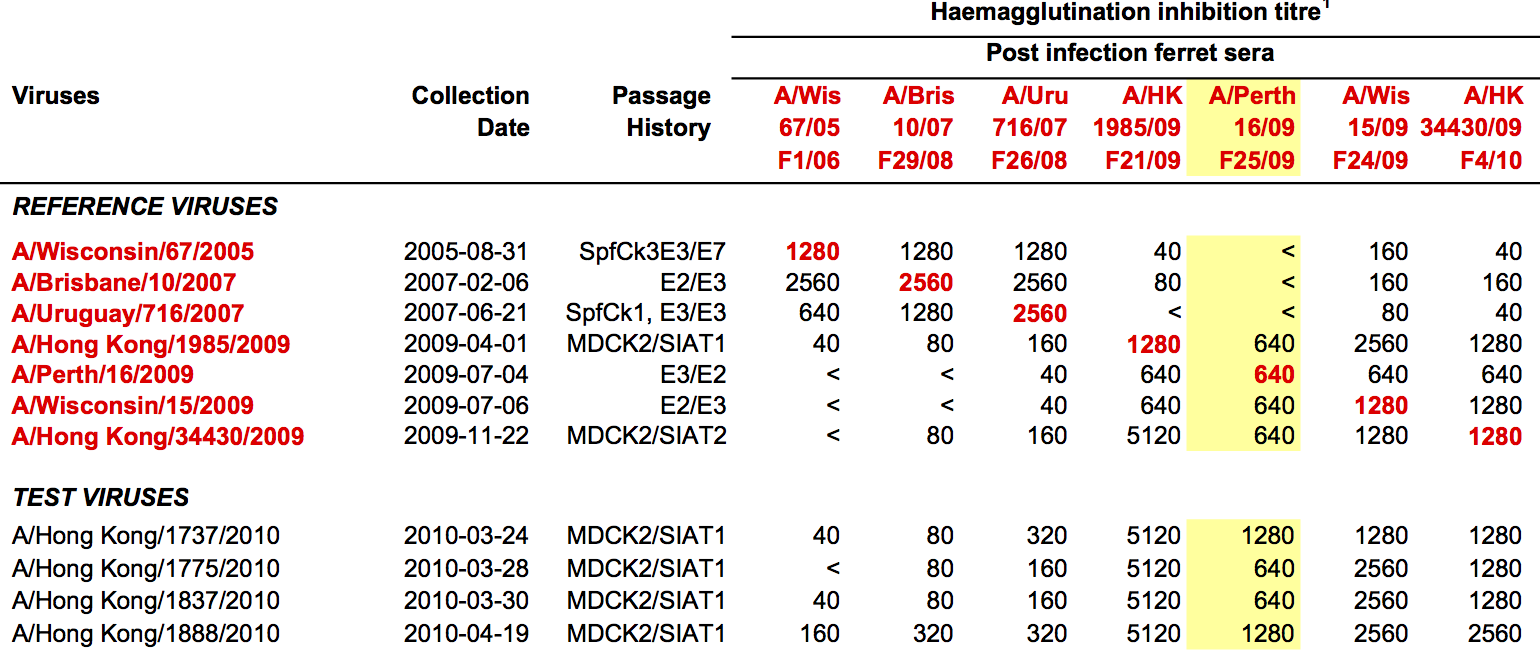
Antigenic "match" of key importance
Antibodies by vaccination should effectively bind to circulating viruses. This requires:
- Identification of antigenically distinct clades of virus
- Prediction of clade growth/decline
Hemagglutination inhibition (HI) assays measure binding
H3N2 population in Feb 2014
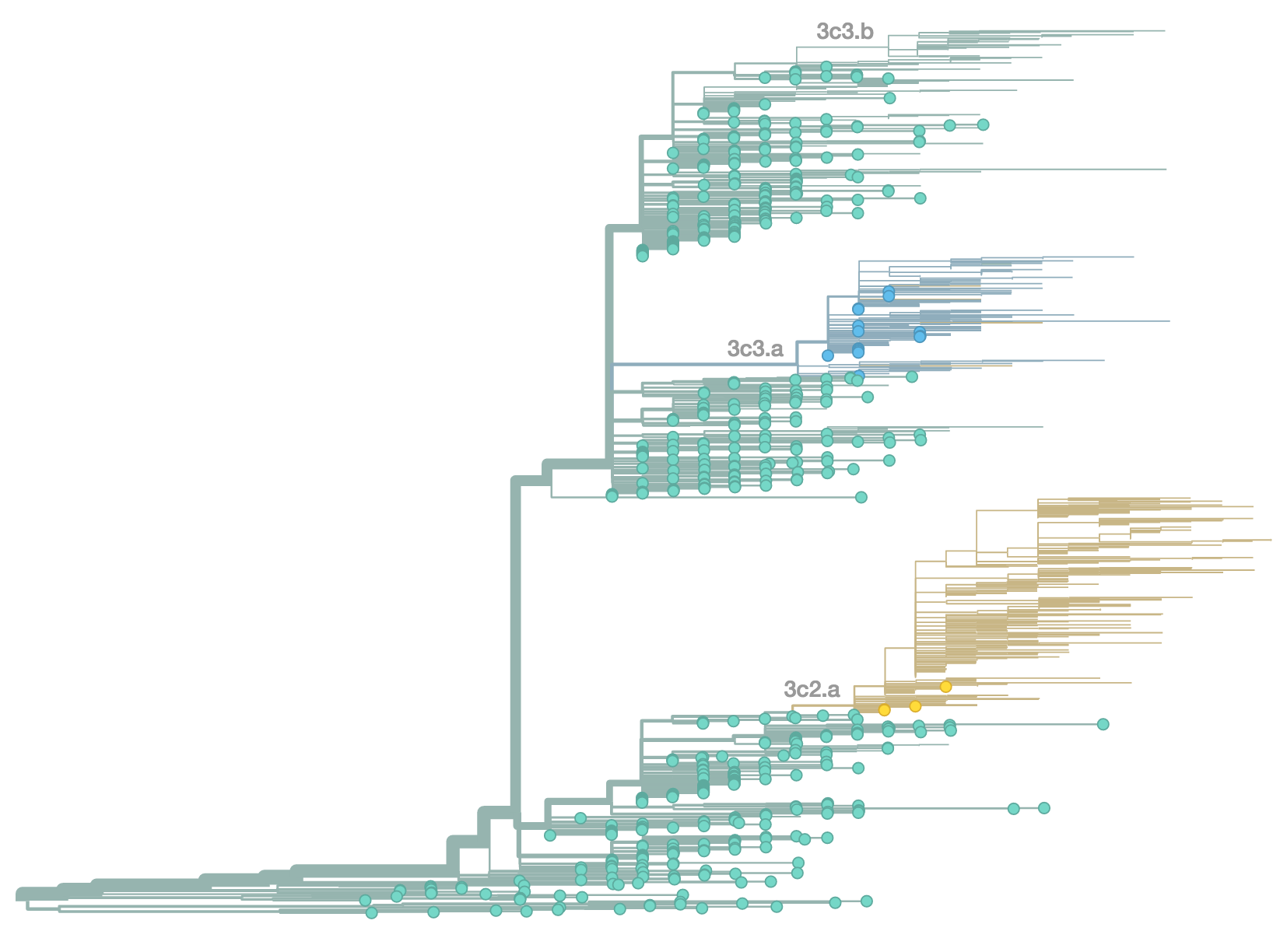
H3N2 population in Jun 2014
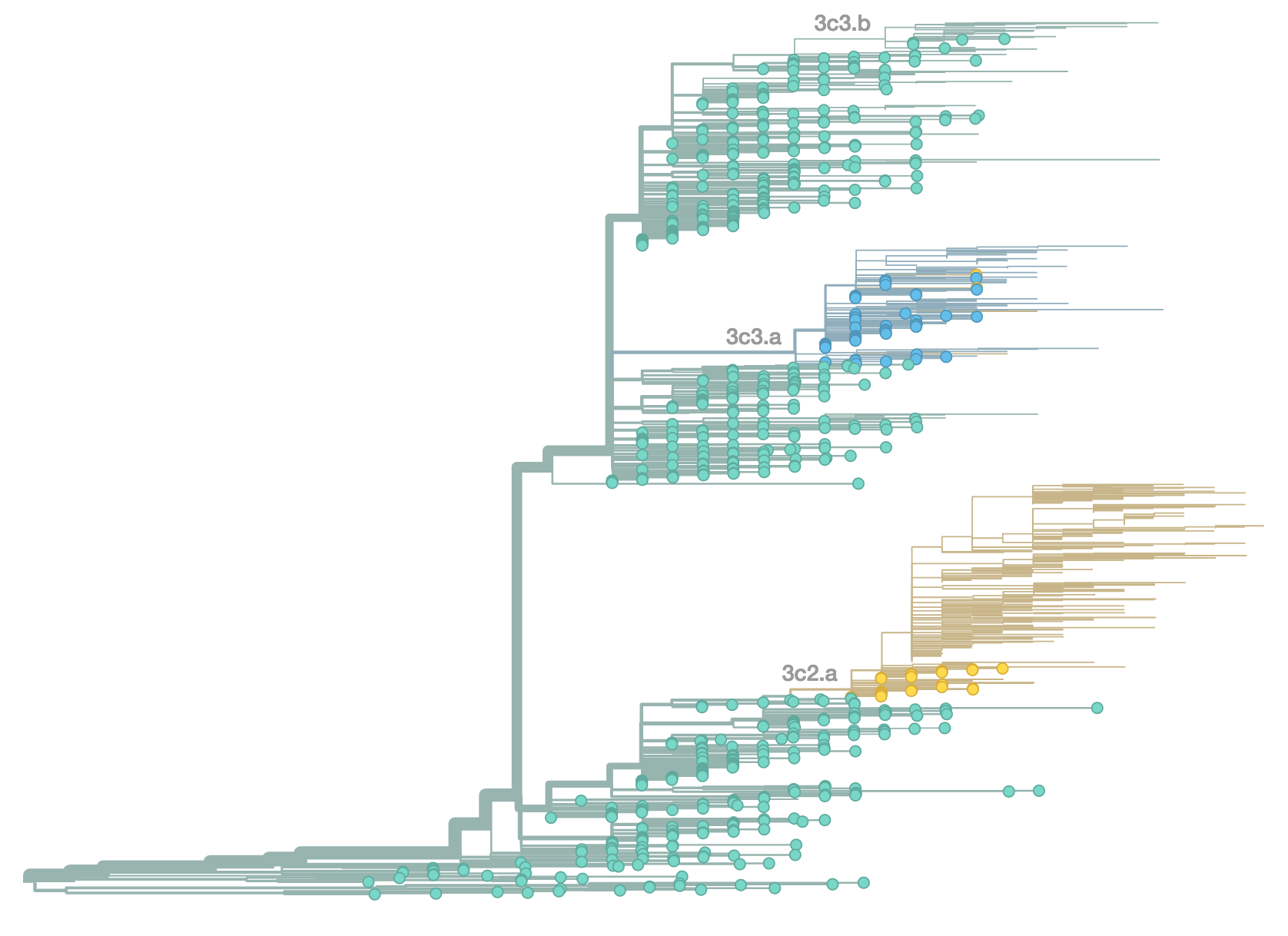
H3N2 population in Oct 2014
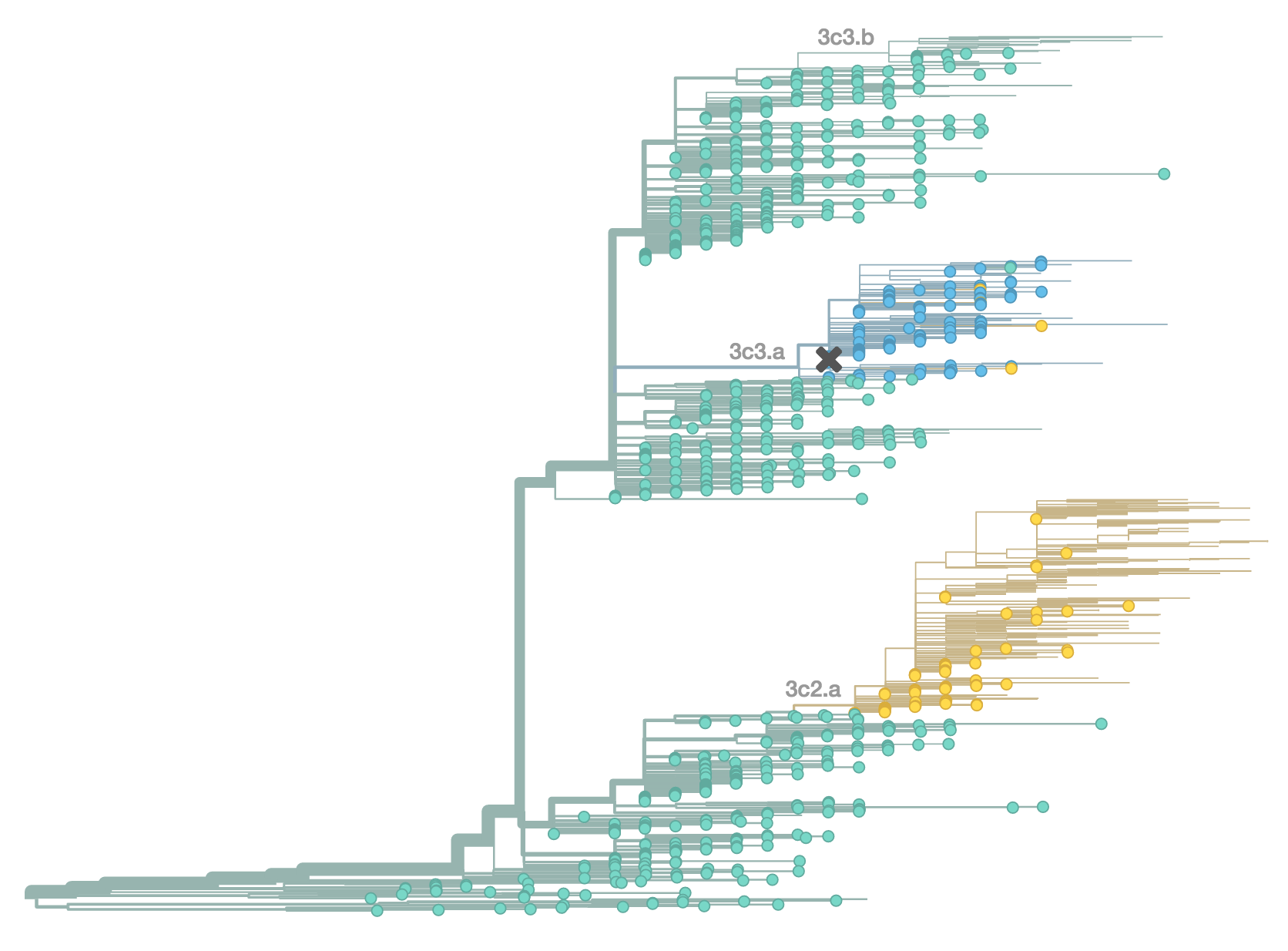
H3N2 population in Feb 2015
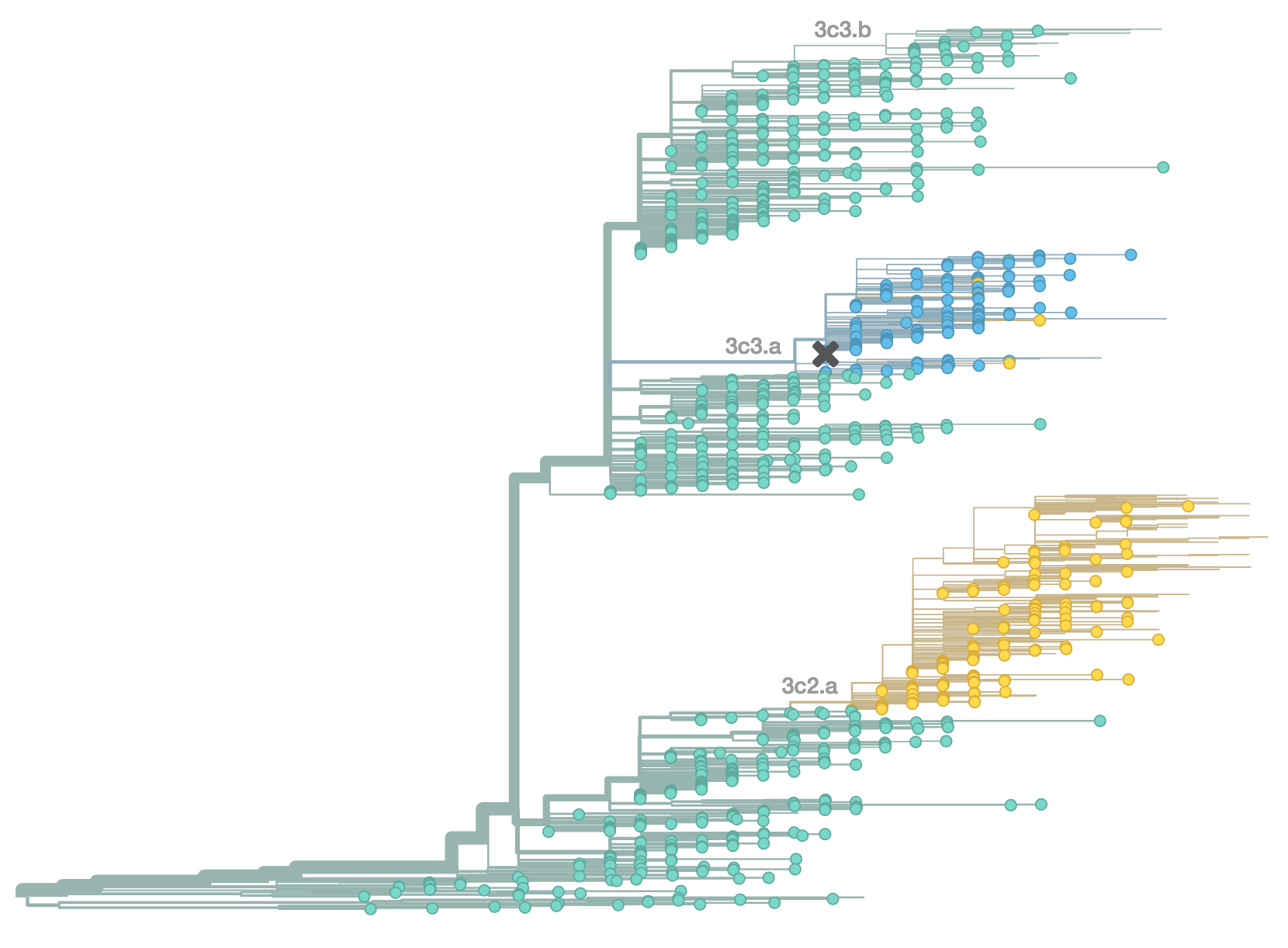
Resulted in a mismatched 2014-2015 vaccine
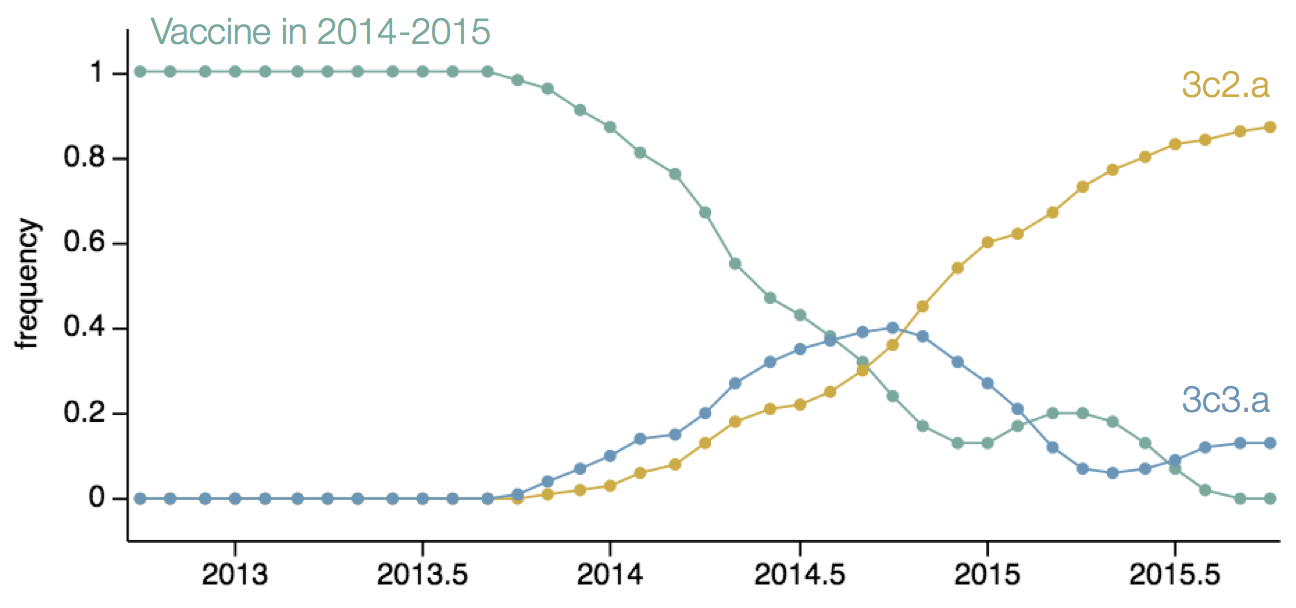
nextflu
Project to provide a real-time view of the evolving influenza population
All in collaboration with Richard Neher

nextflu pipeline
- Download all recent HA sequences from GISAID
- Filter to remove outliers
- Align sequences
- More filtering
- Build tree
- Estimate frequencies
- Export JSON for visualization
nextflu.org
Predictive models
A simple predictive model estimates the fitness $f$ of virus $i$ as
$$\hat{f}_i = \beta^\mathrm{ep} \, f_i^\mathrm{ep} + \beta^\mathrm{ne} \, f_i^\mathrm{ne}$$
where $f_i^\mathrm{ep}$ measures cross-immunity via substitutions at epitope sites and $f_i^\mathrm{ep}$ measures mutational load via substitutions at non-epitope sites.
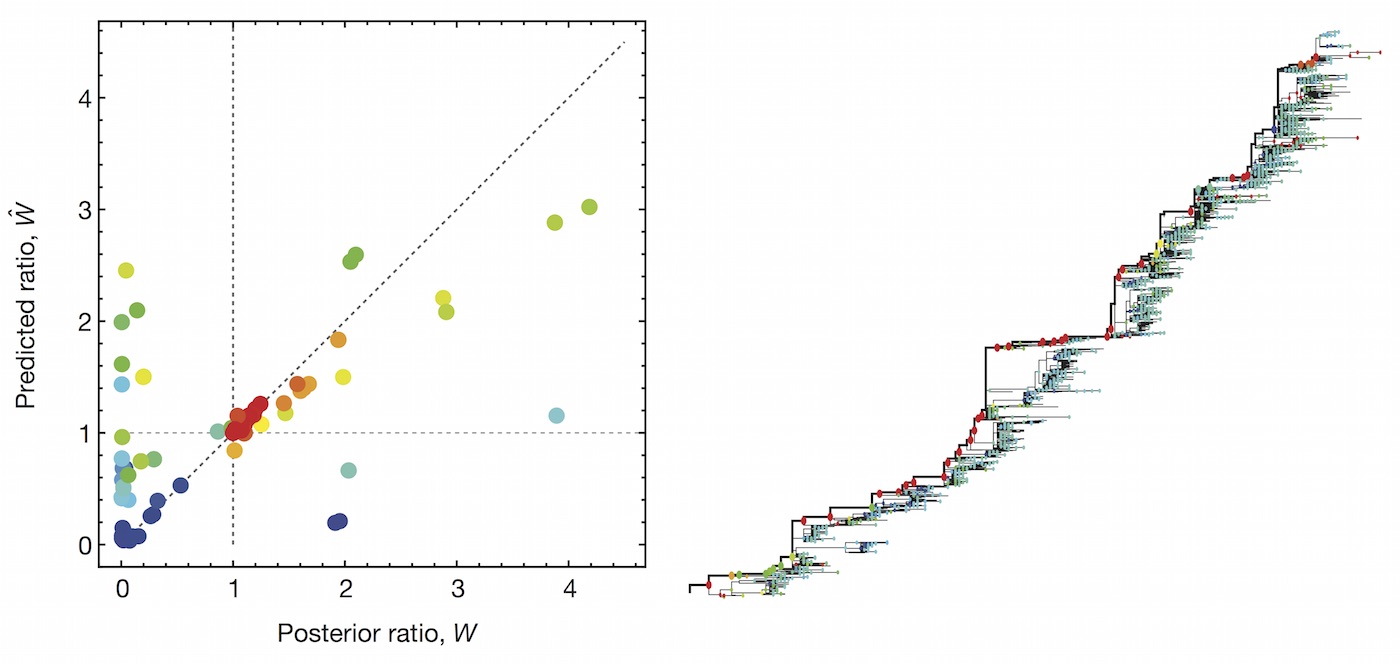
Predictive models
Another approach quantifies phylogenetic branching patterns
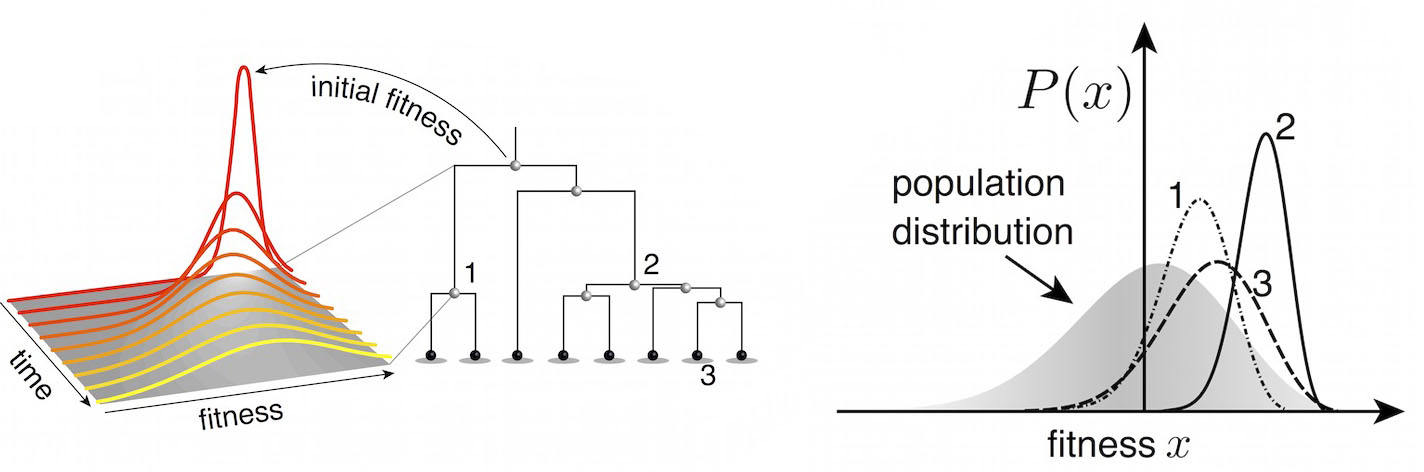
We're now working to include quantitative predictions of future clade behavior in nextflu
And also including other predictors, like geography:
Ebola
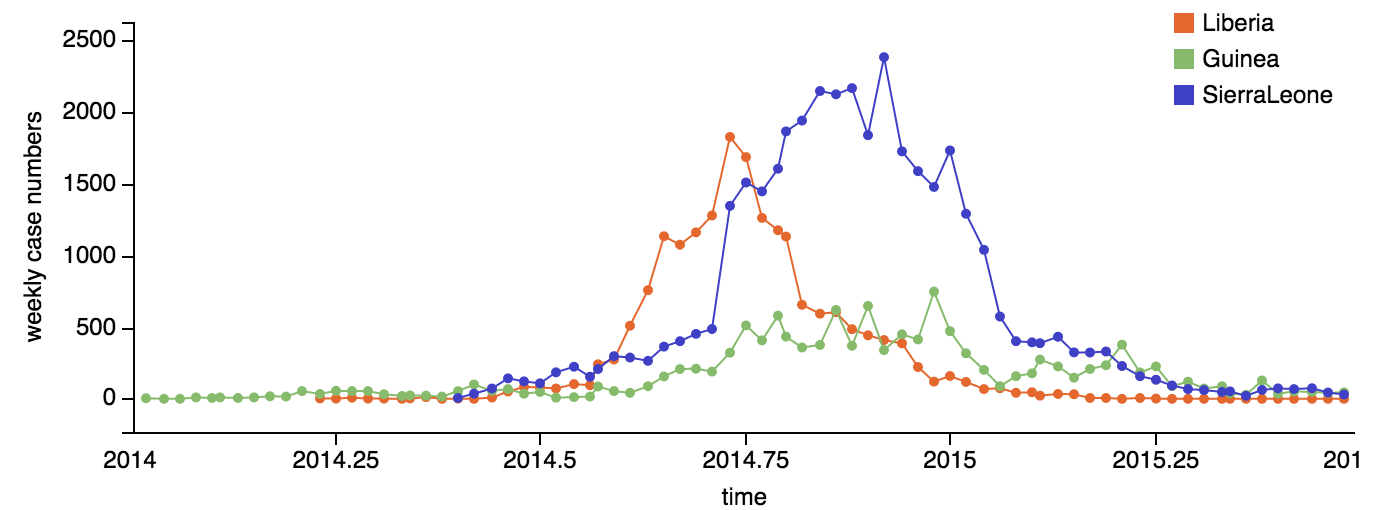
Epidemic nearly contained, but resulted in >28,000 confirmed cases and >11,000 deaths
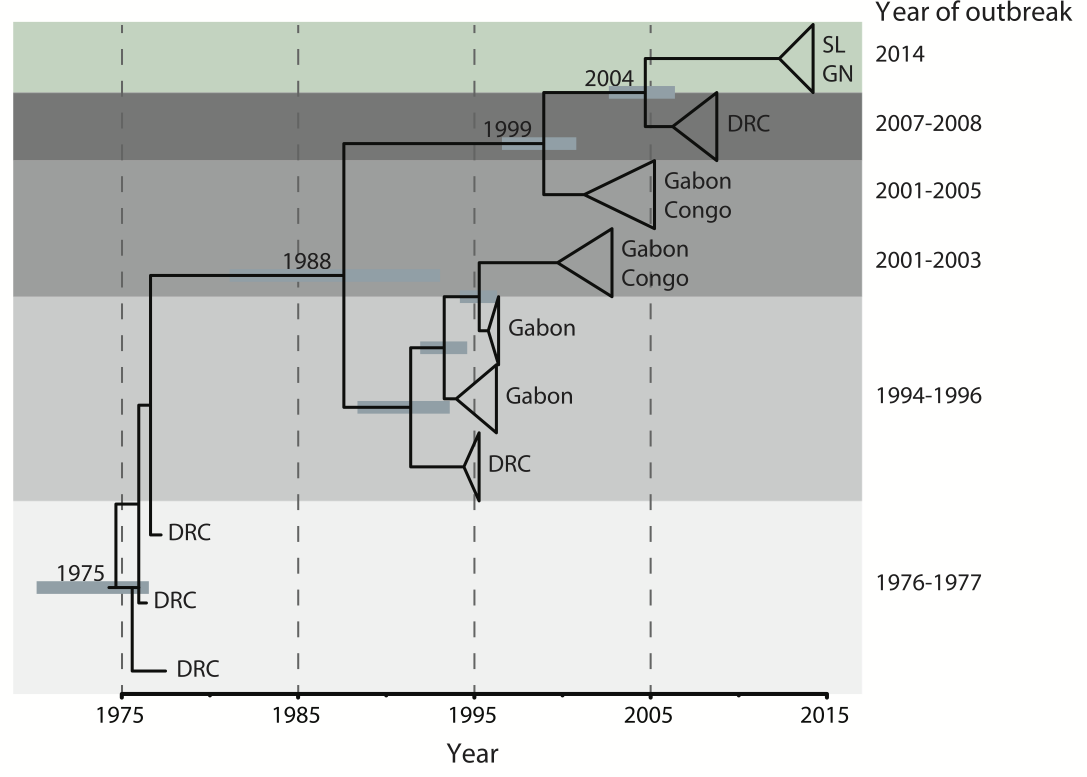
Outbreaks are independent spillovers from the animal reservoir
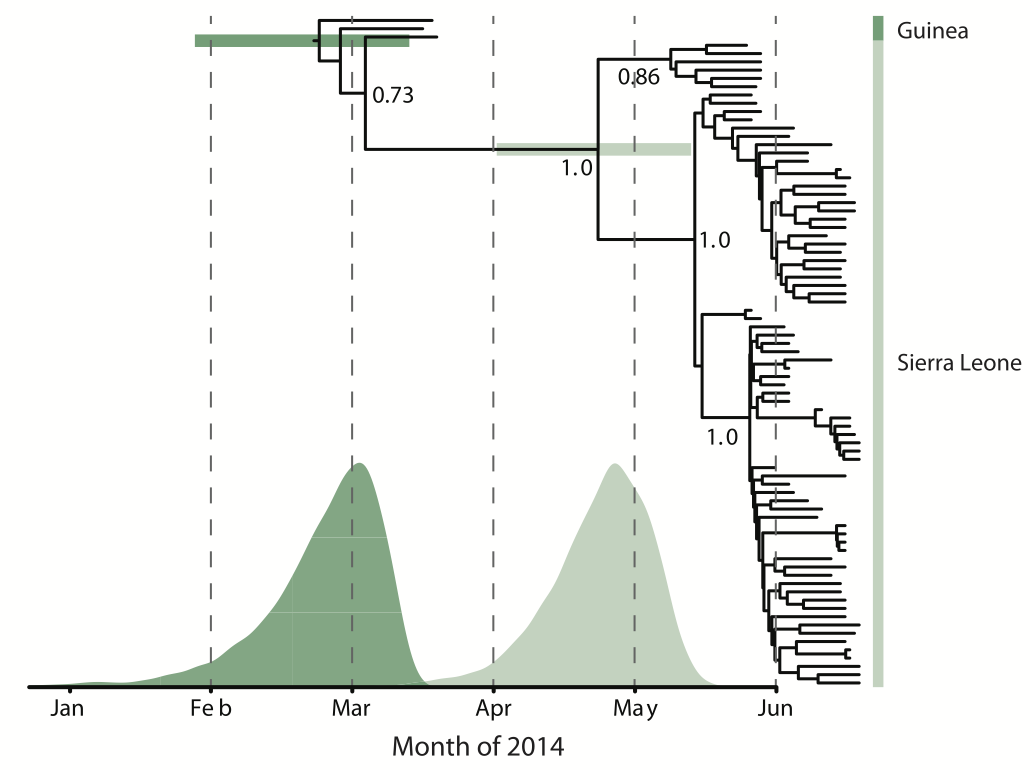
Person-to-person spread in the early West African outbreak
Continued spread through Dec 2014
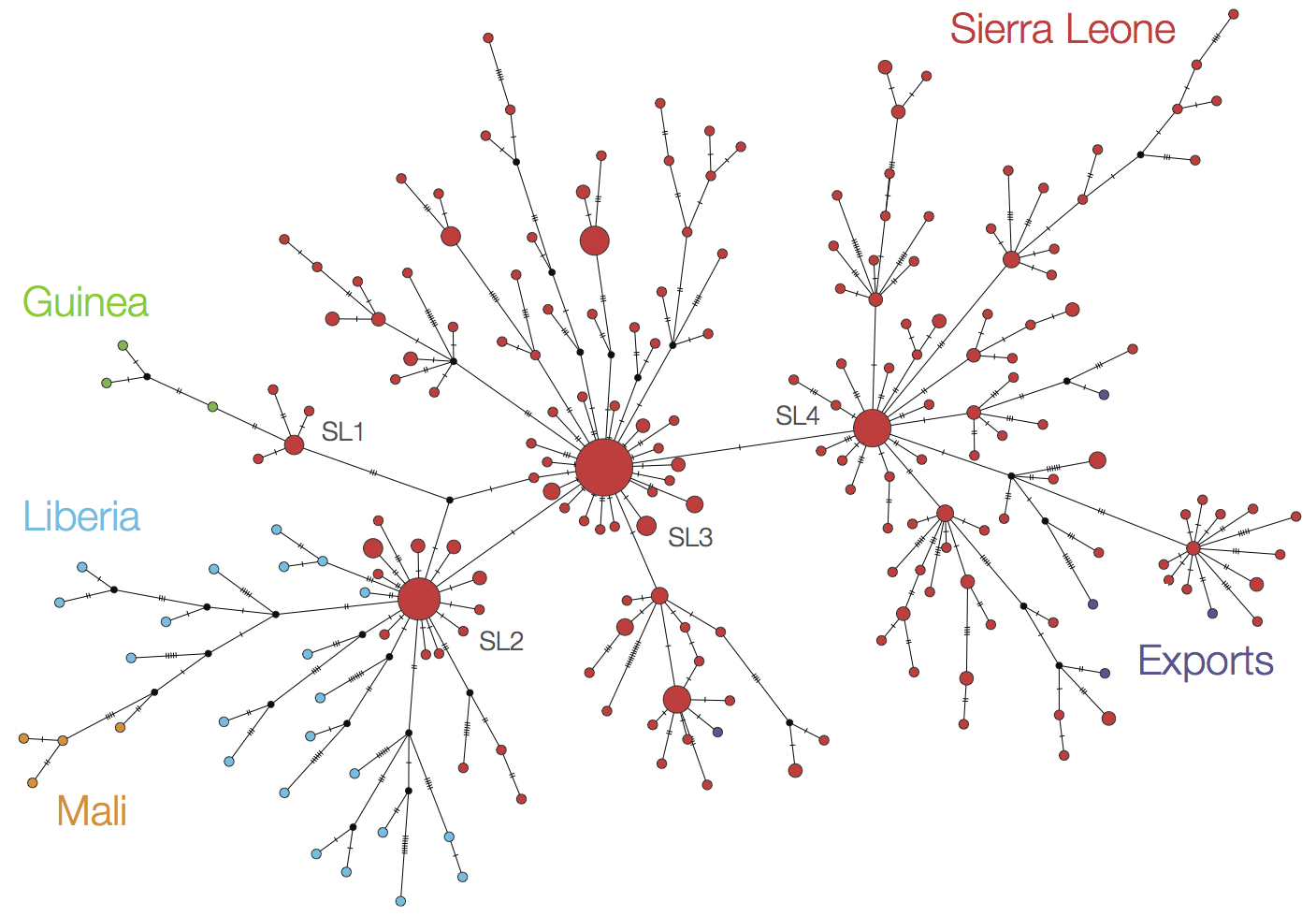
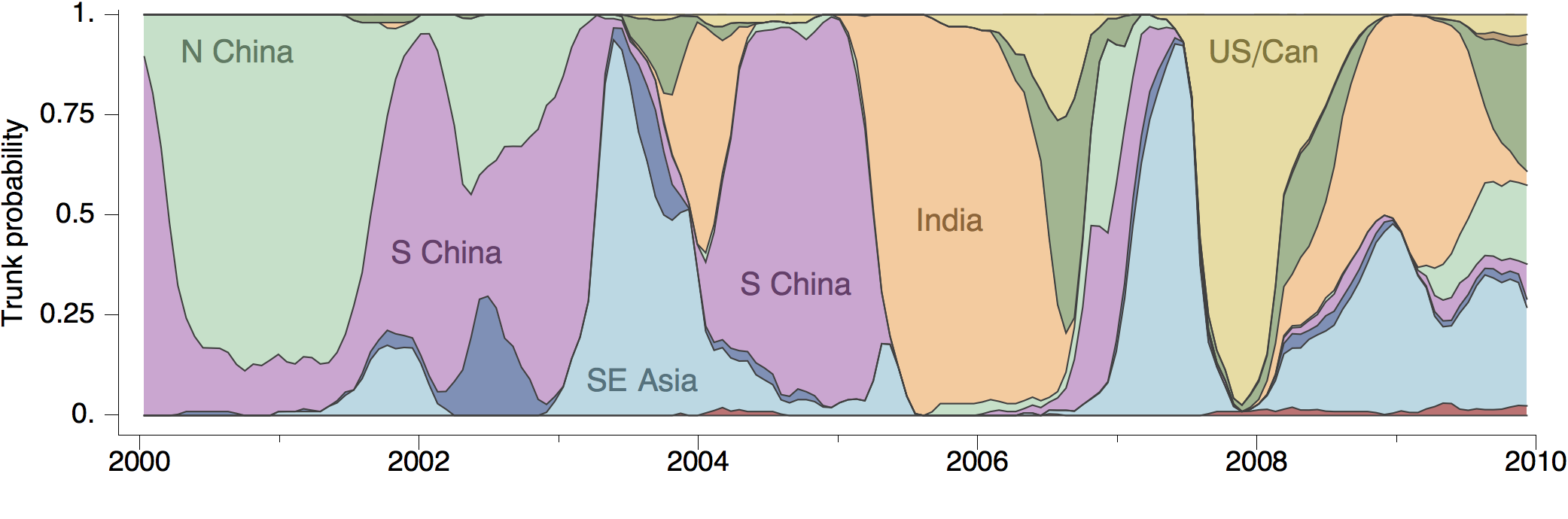
At epidemic height, geographic spread of particular interest
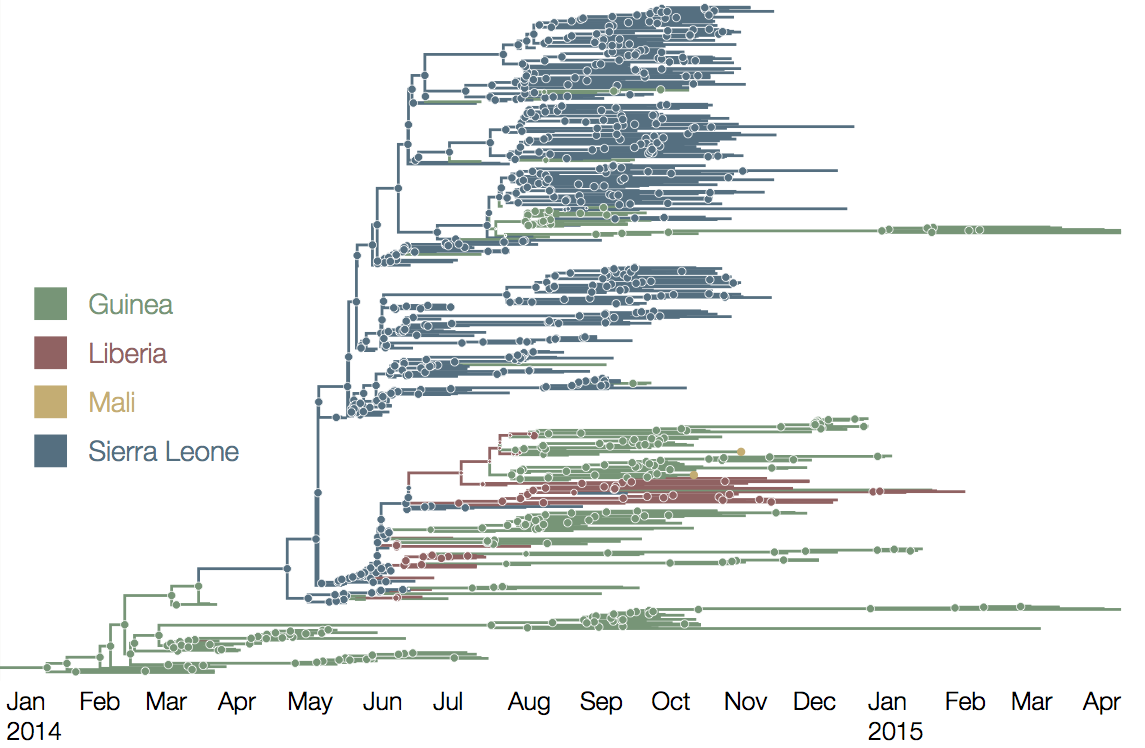
Later on, tracking transmission clusters of primary importance
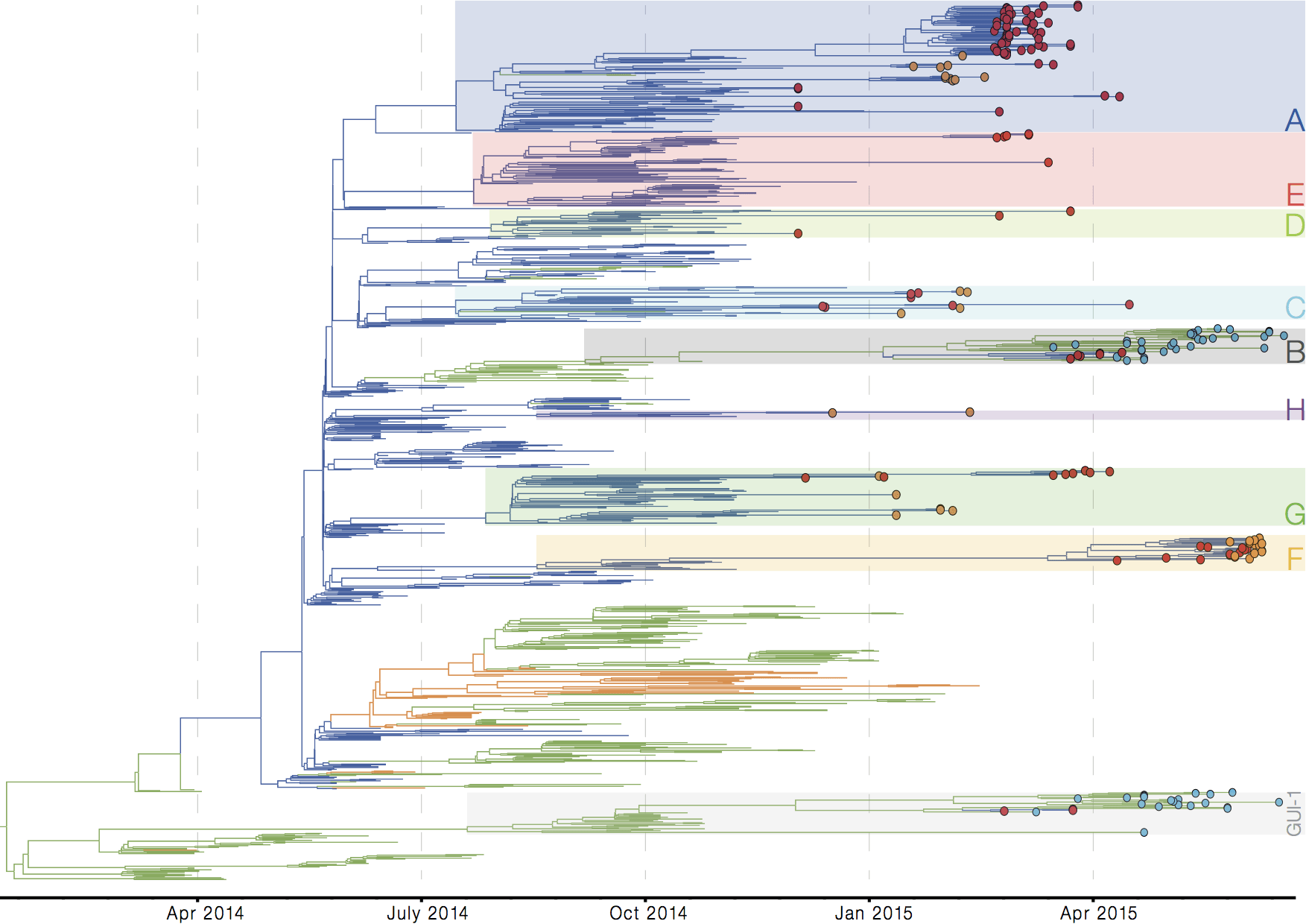
ebola.nextflu.org
Moving forward, genetically-informed outbreak response requires:
- Rapid sharing of sequence data, genetic context critical
- Technologies to rapidly conduct phylogenetic inference
- Technologies to explore genetic relationships and inform epidemiological investigation
Acknowledgements
Richard Neher (Max Planck Tübingen), Andrew Rambaut (University of Edinburgh), Colin Russell (Cambridge University), Michael Lässig (University of Cologne), Marta Łuksza (Institute for Advanced Study), Gytis Dudas (University of Edinburgh), Pardis Sabeti (Harvard University), Danny Park (Harvard University), Nick Loman (University of Birmingham) Matthew Cotten (Sanger Institute), Paul Kellam (Sanger Institute), WHO Global Influenza Surveillance Network, GISAID
Contact
- Website: bedford.io
- Twitter: @trvrb
- Slides: bedford.io/talks/real-time-tracking-bccdc/