Real-time forecasting of influenza virus evolution
Trevor Bedford (@trvrb)
16 Jan 2018
CREST International Symposium on Big Data Applications
Tokyo, Japan
Want to forecast the make up of the future flu population from the population that exists today
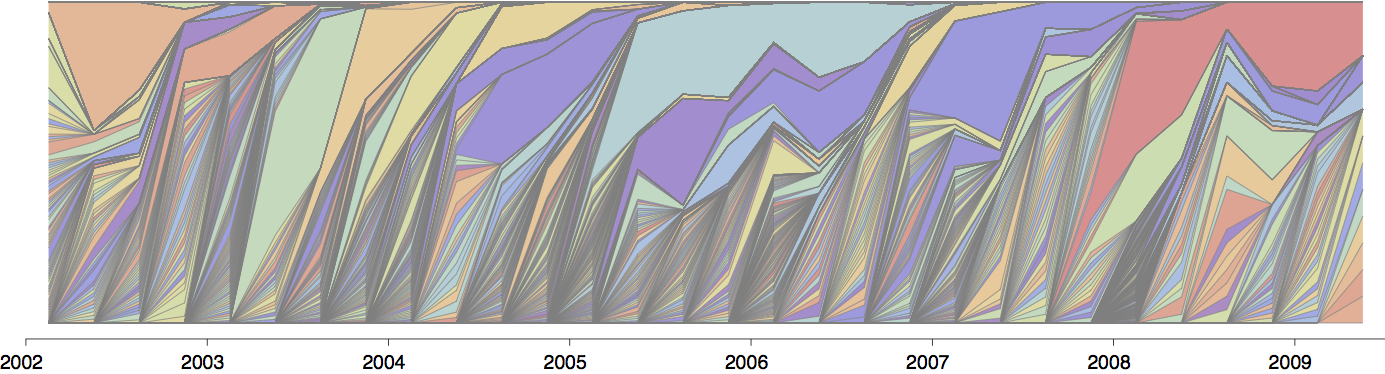
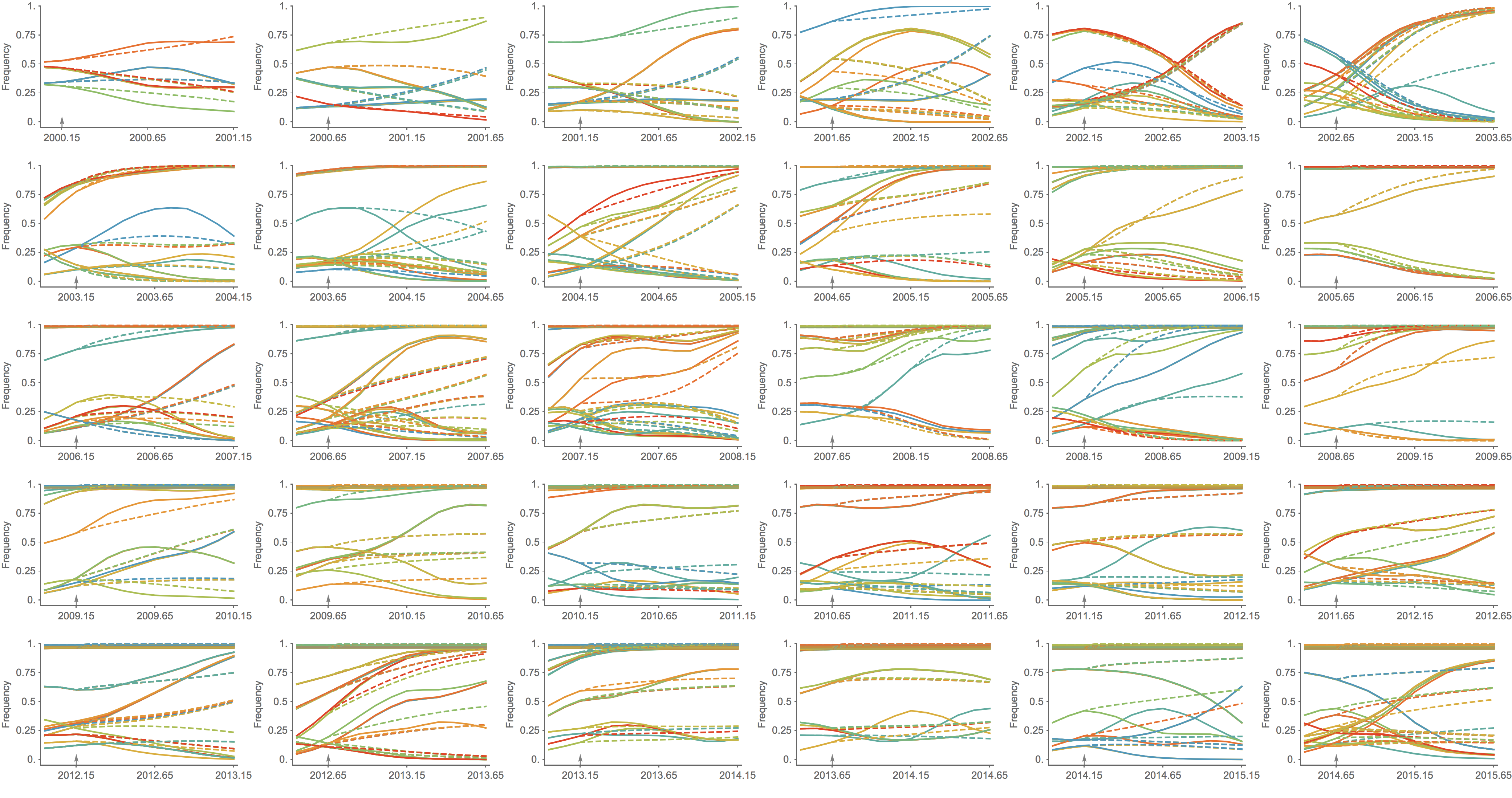
Population turnover (in H3N2) is extremely rapid
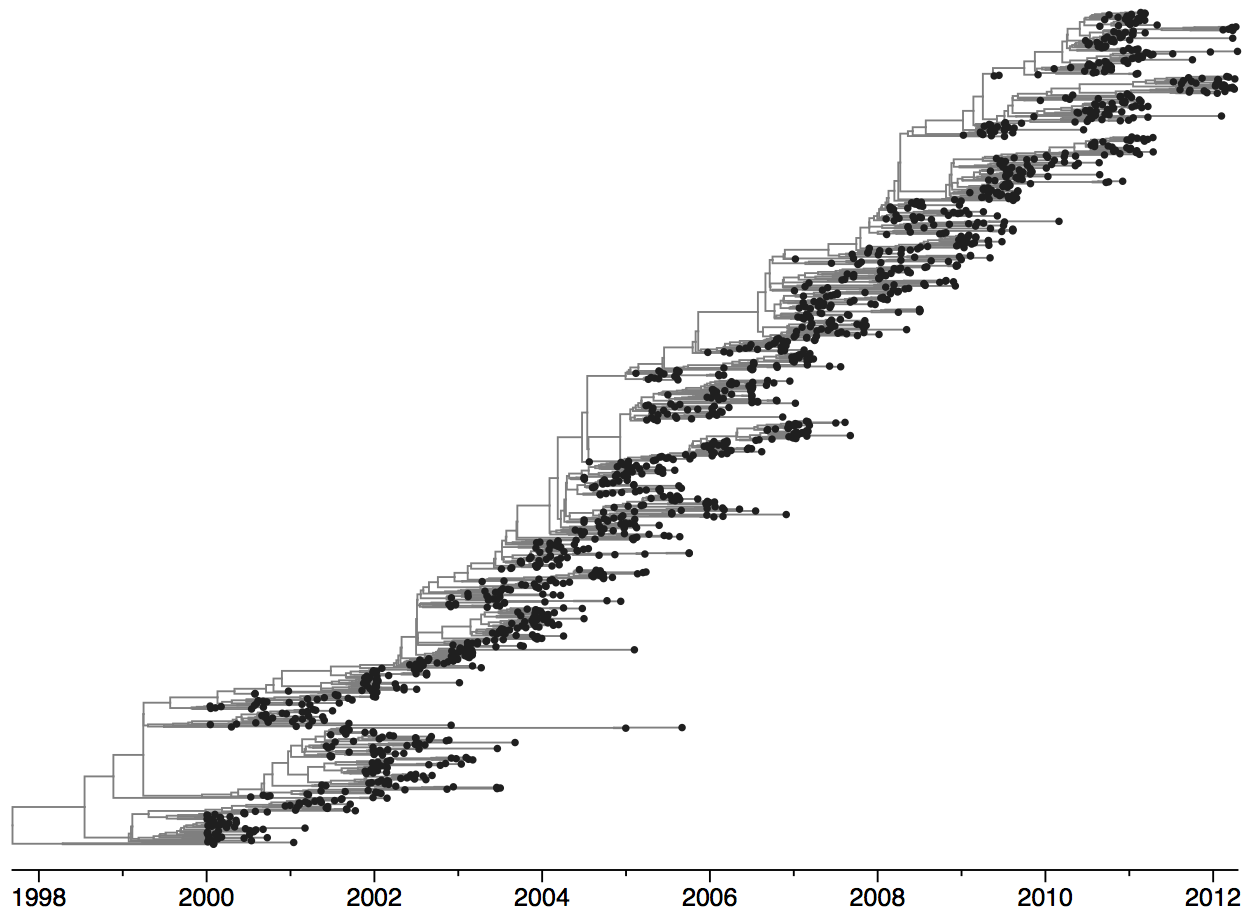
Clades emerge, die out and take over
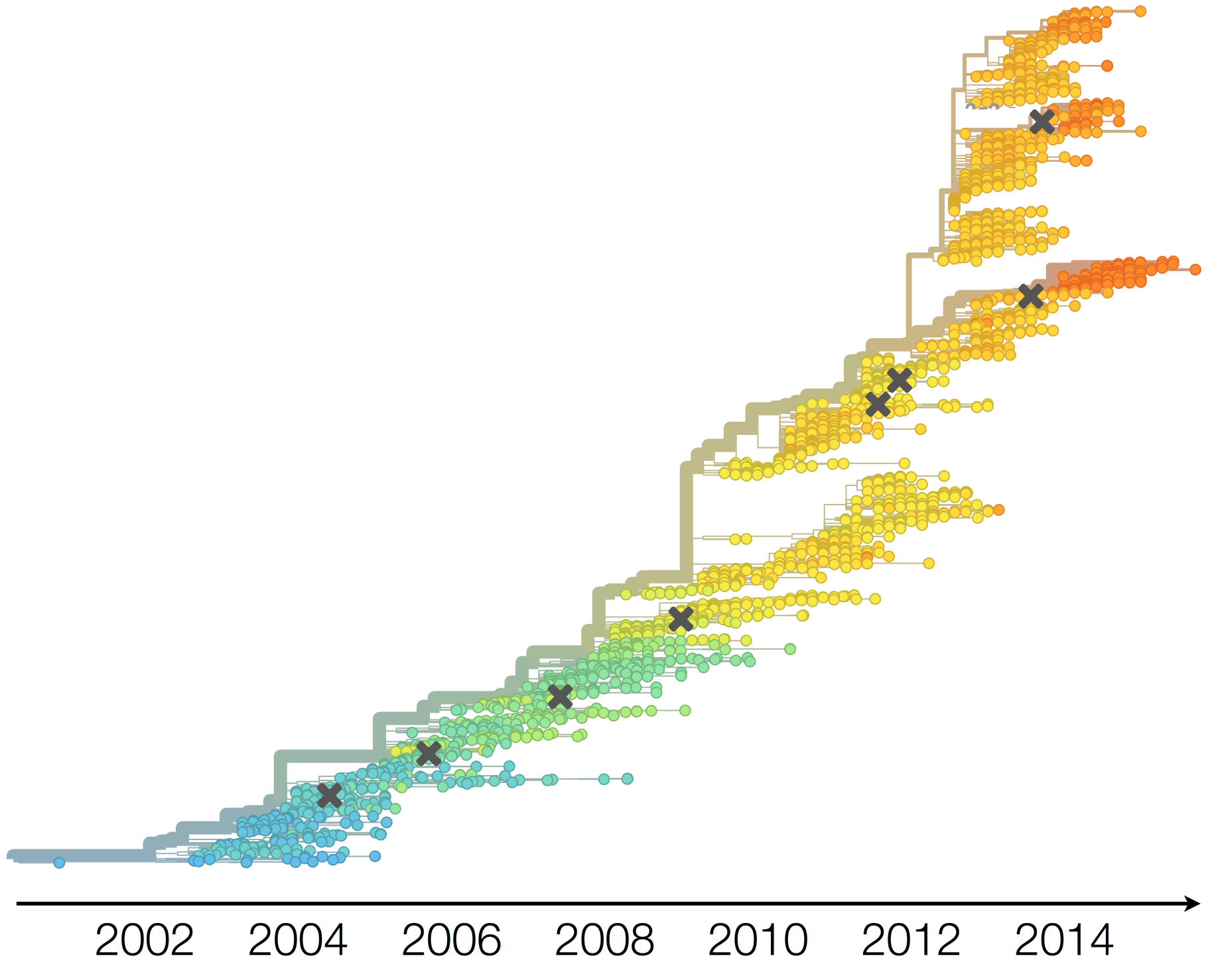
Clades show rapid turnover

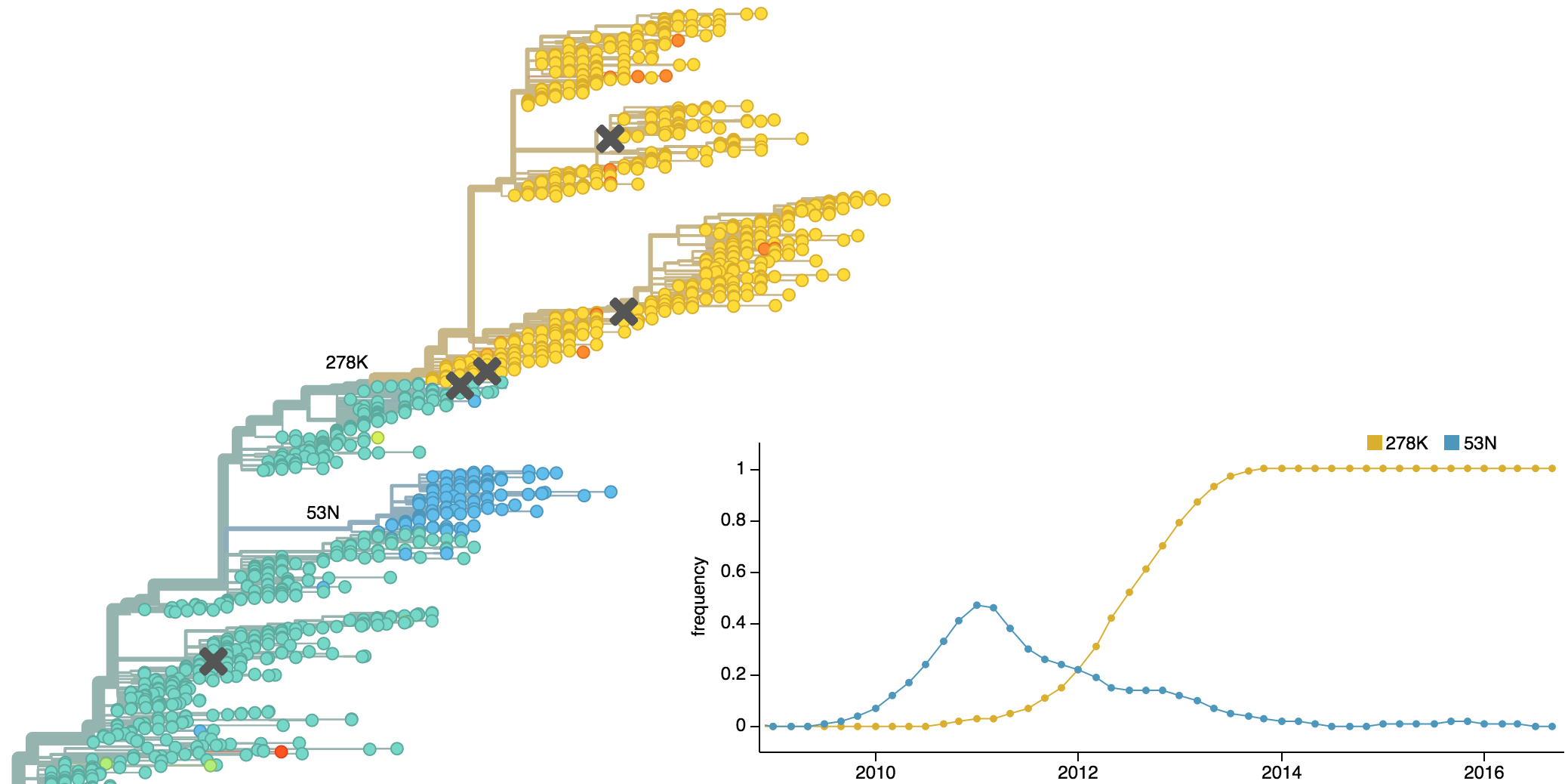
Dynamics driven by antigenic drift
Drift variants emerge and rapidly take over in the virus population
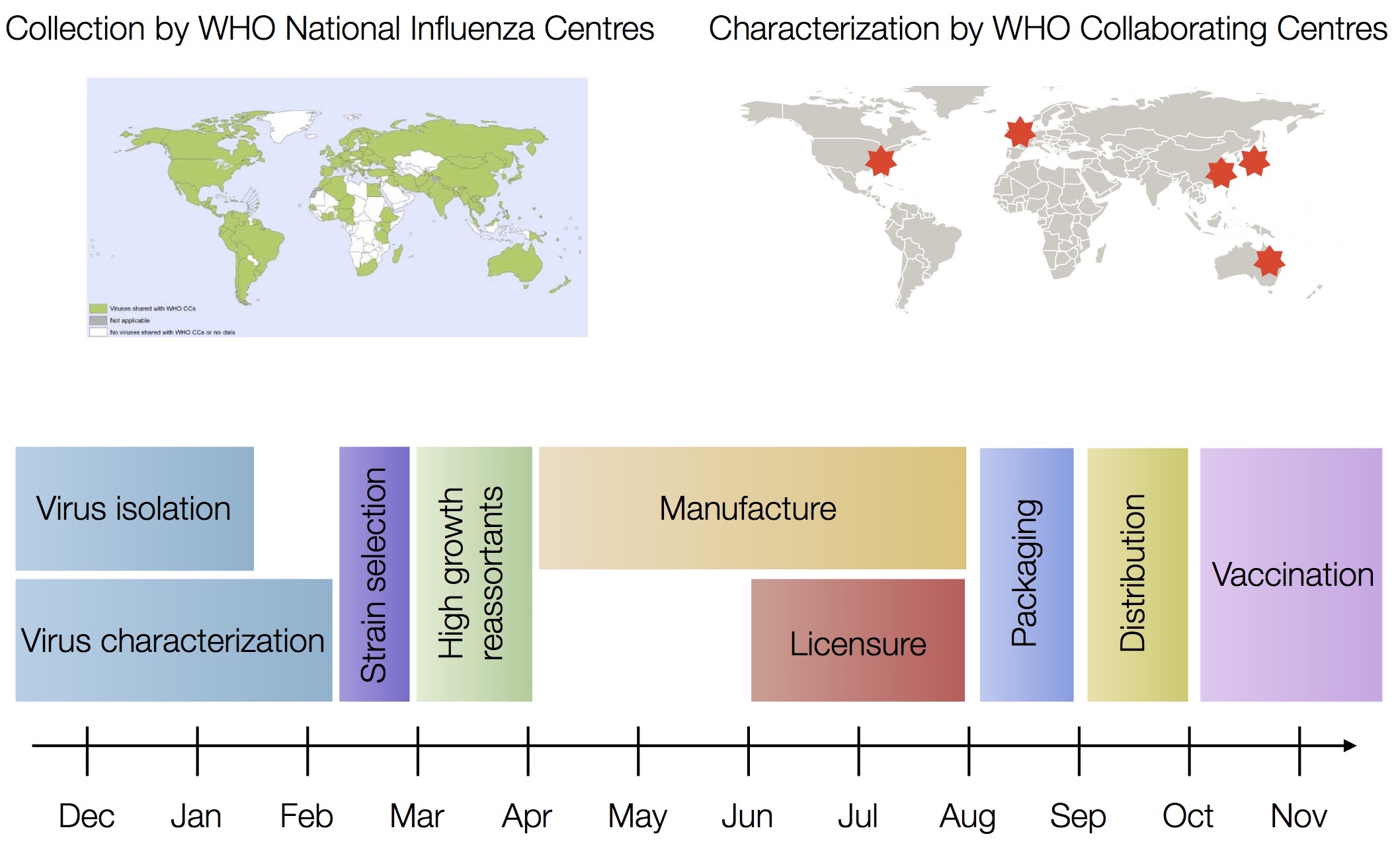
Drift necessitates vaccine updates
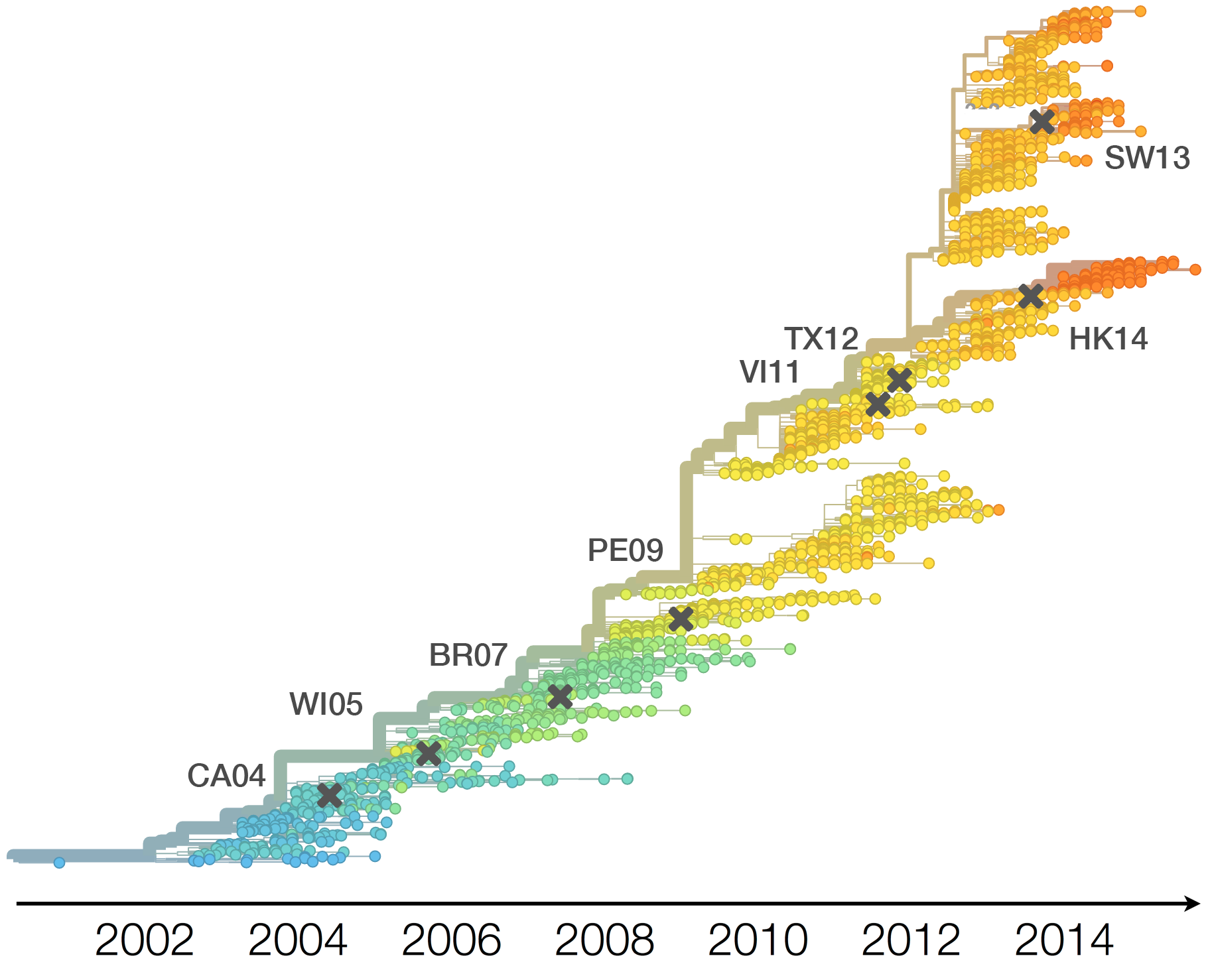
H3N2 vaccine updates occur every ~2 years
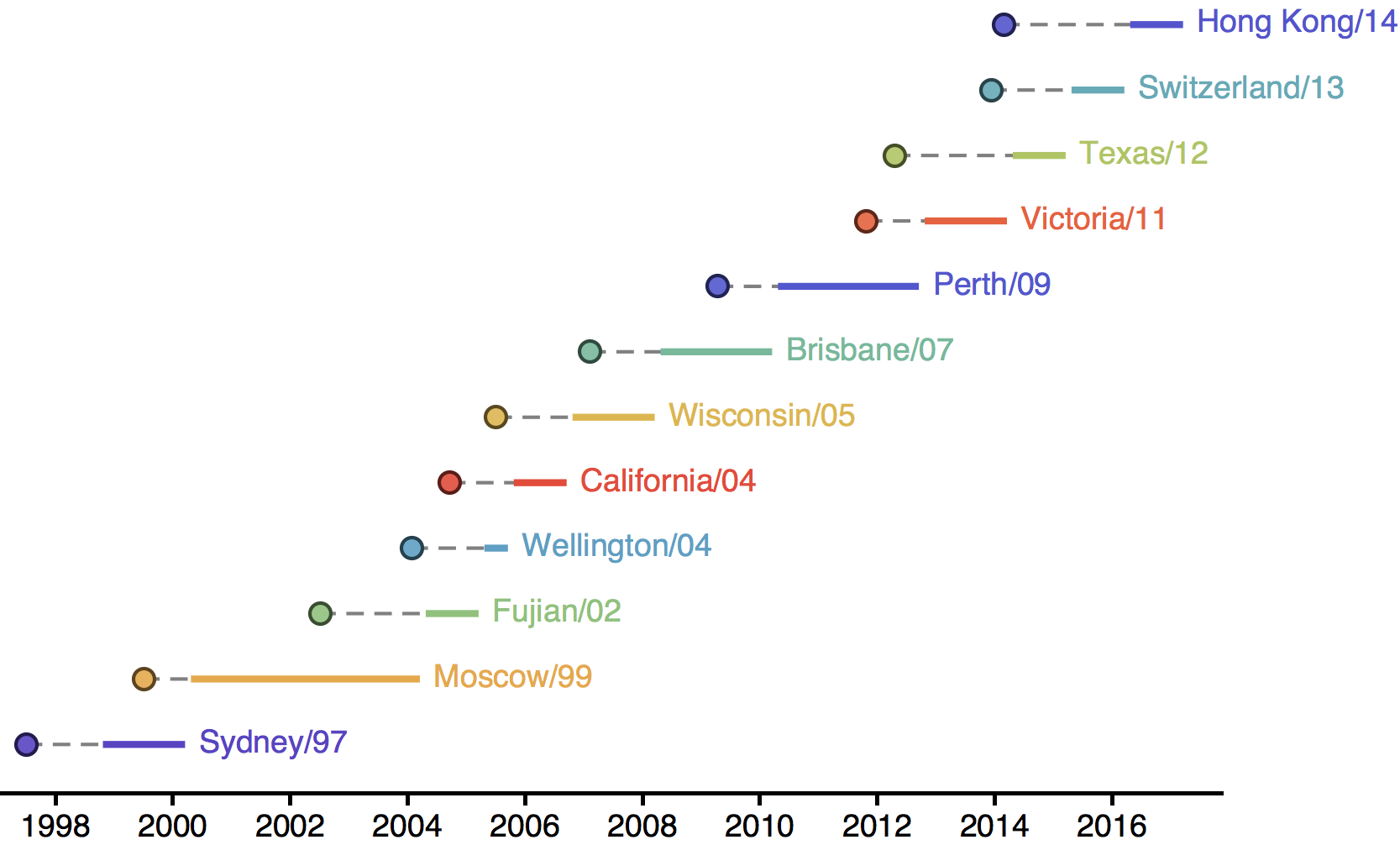
Timely surveillance and rapid analysis essential to vaccine strain selection
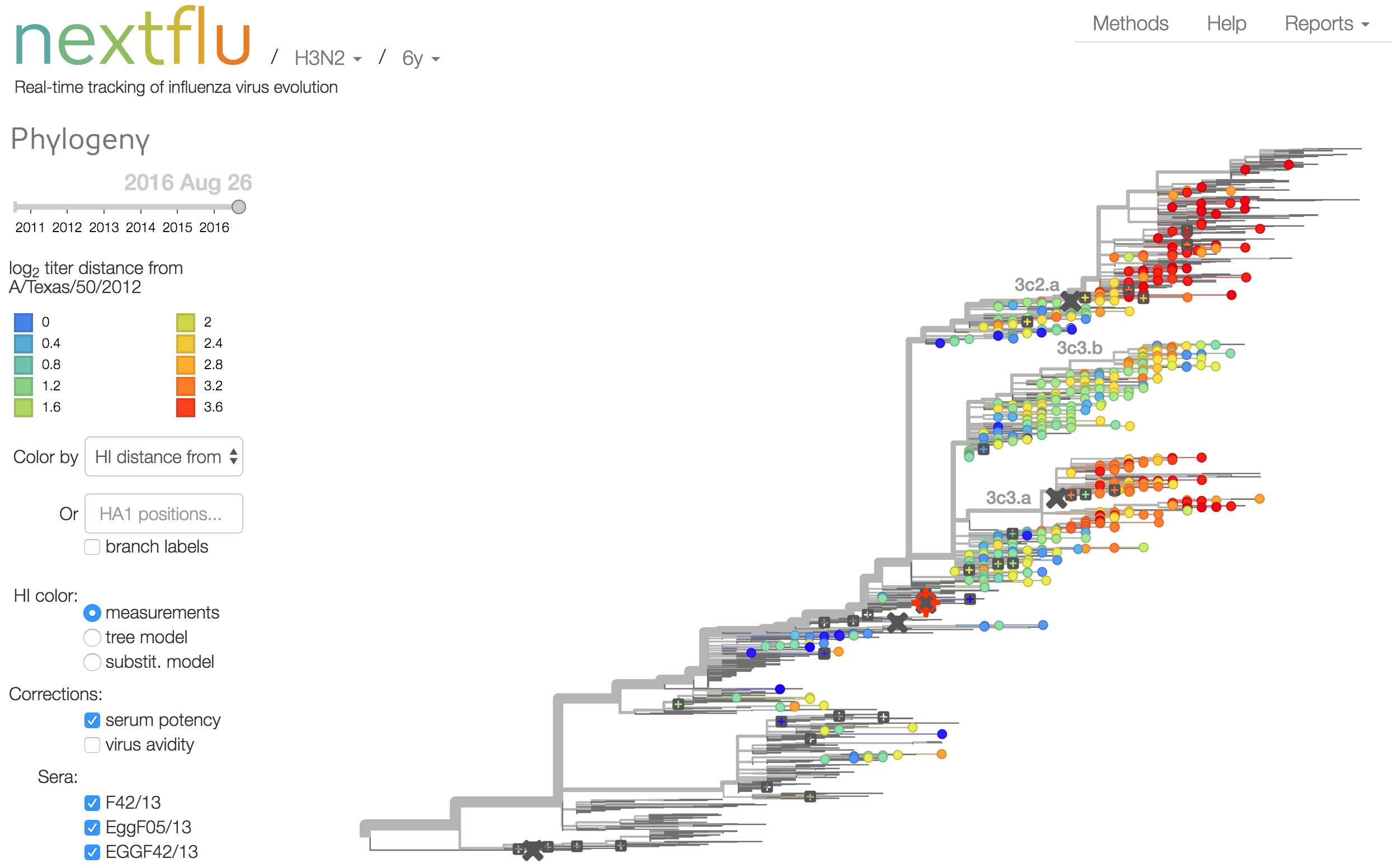
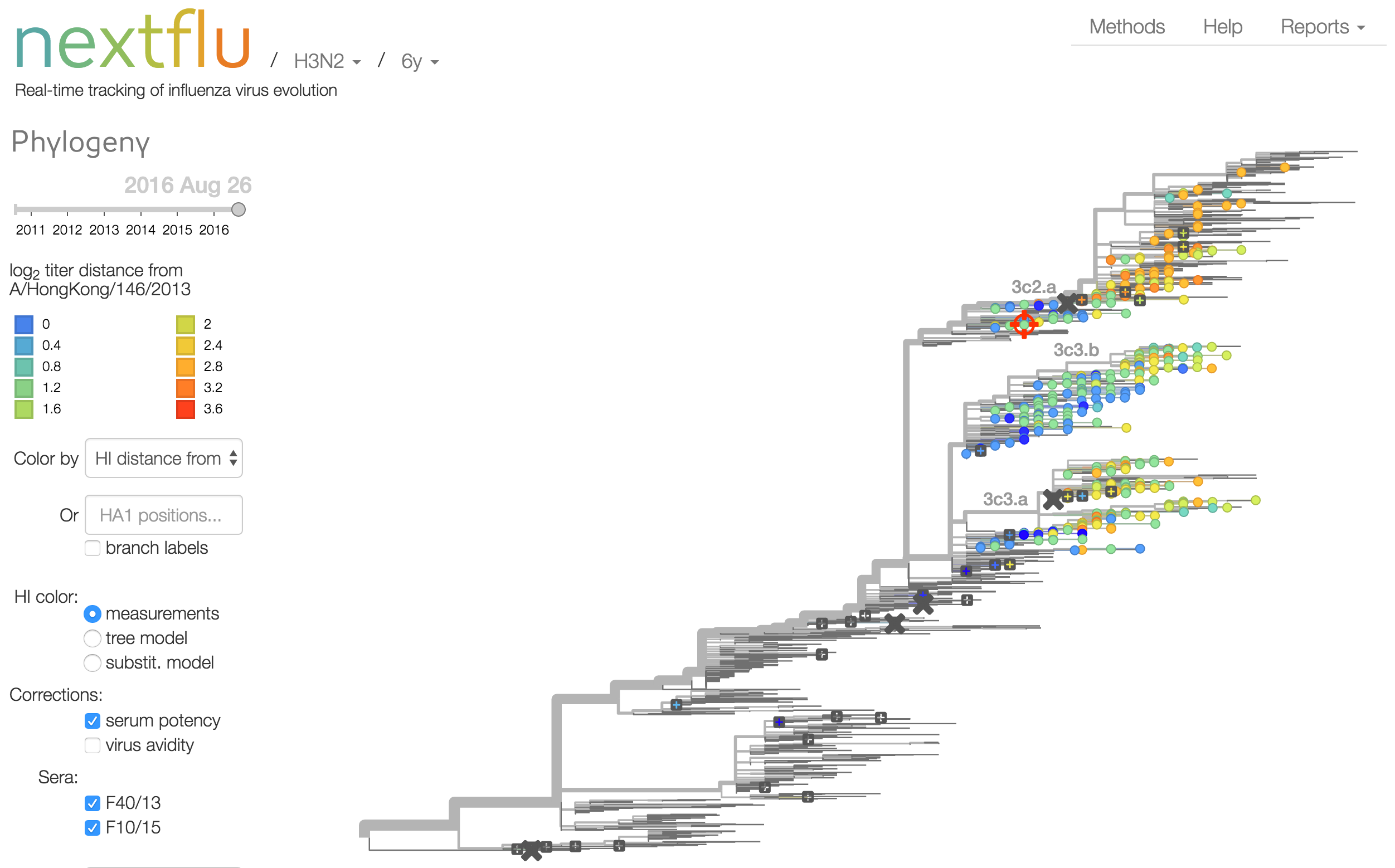
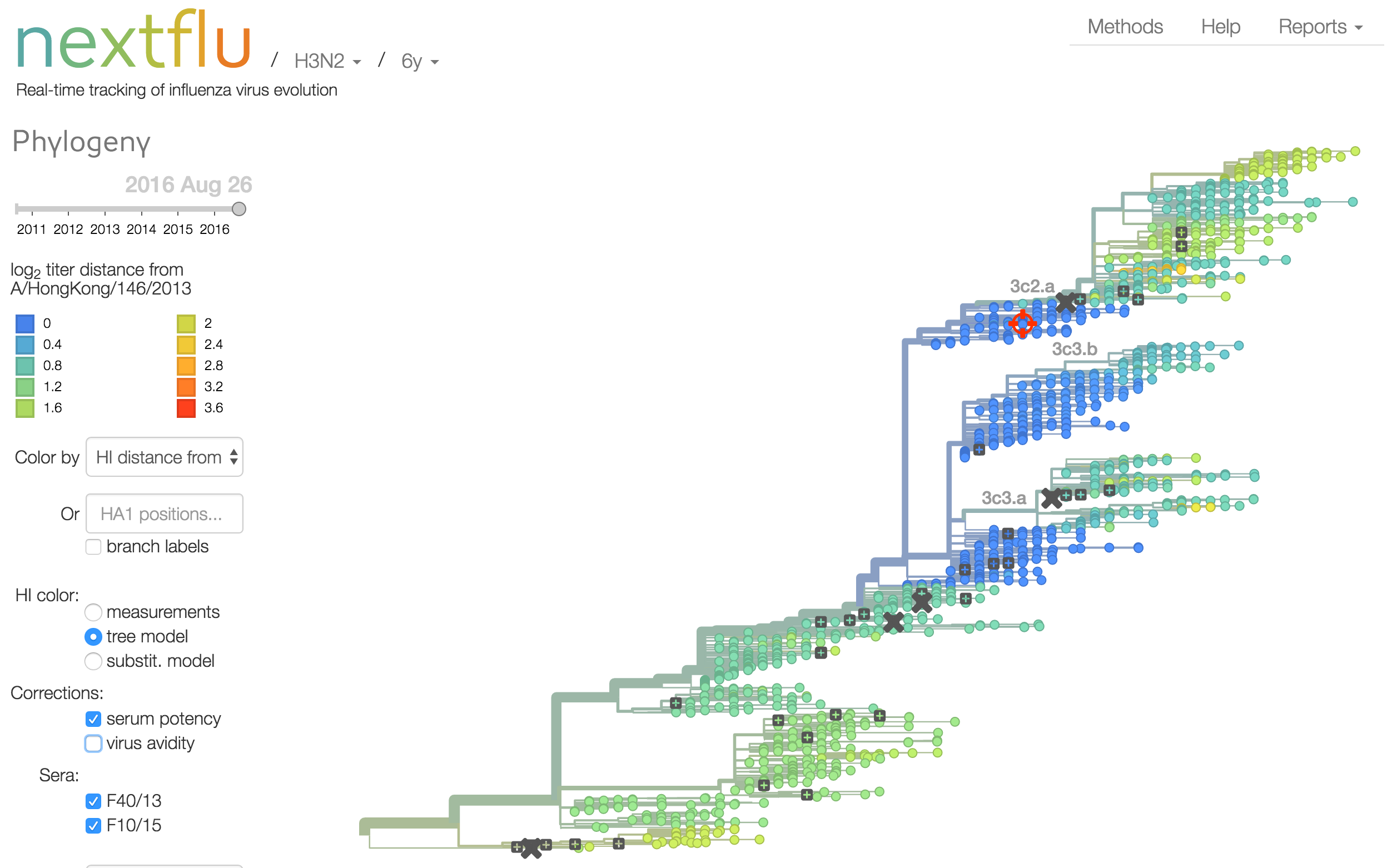
nextflu
Project to provide a real-time view of the evolving influenza population
nextflu
Project to provide a real-time view of the evolving influenza population
All in collaboration with Richard Neher

nextflu pipeline
- Download all recent HA sequences from GISAID
- Filter to remove outliers
- Subsample across time and space
- Align sequences
- Build tree
- Estimate clade frequencies
- Infer antigenic phenotypes
- Export for visualization
Up-to-date analysis publicly available at:
nextflu.org
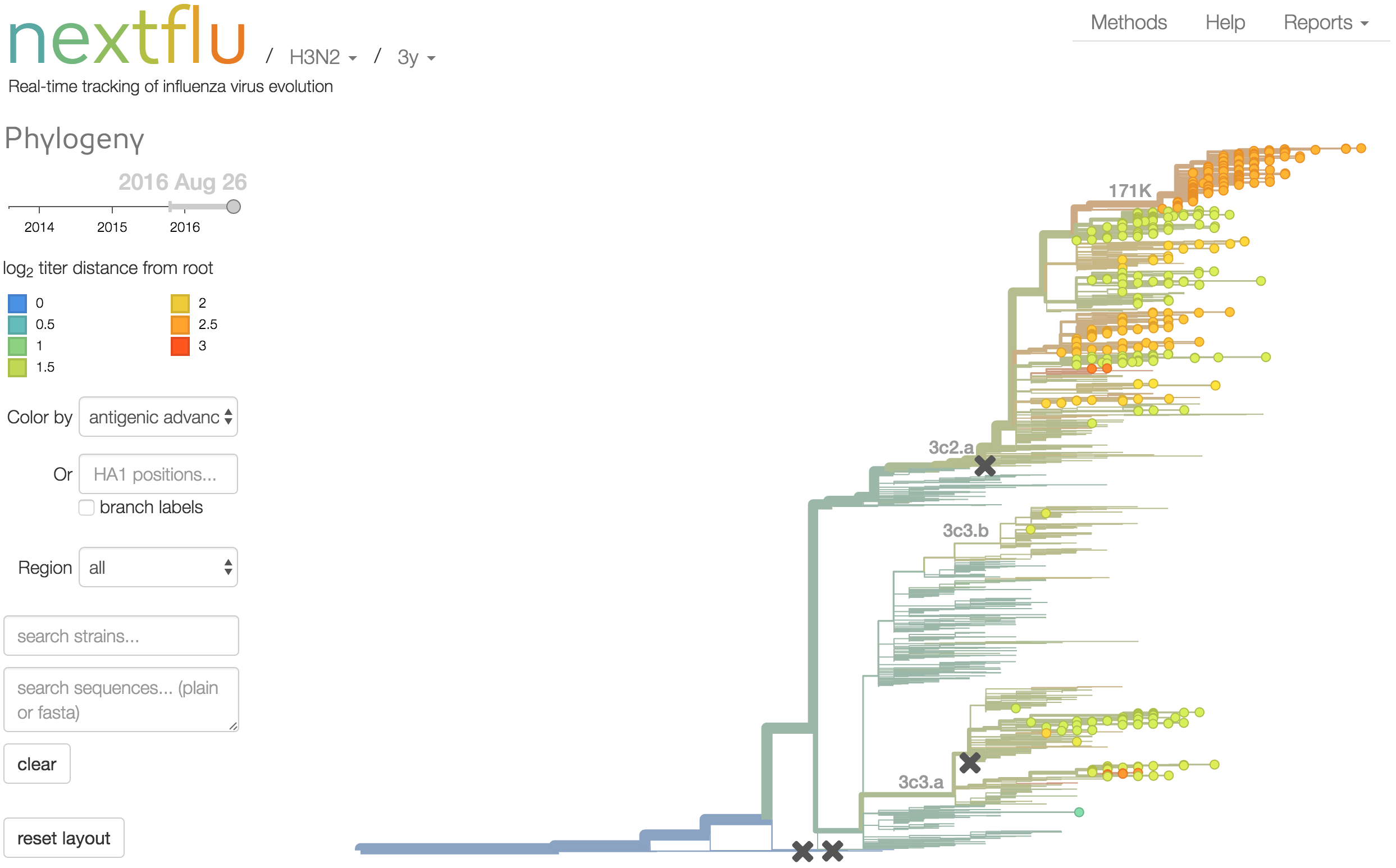
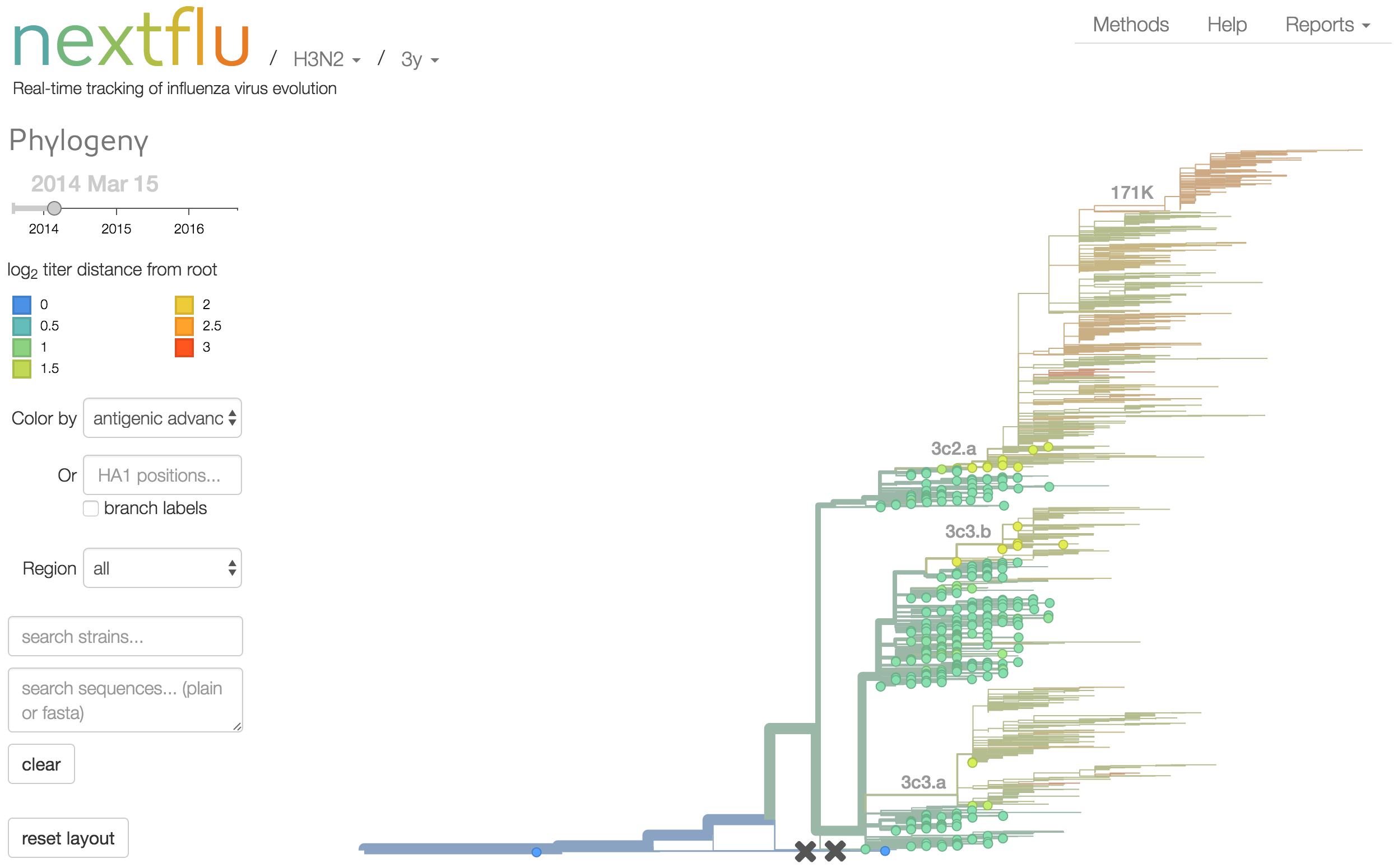
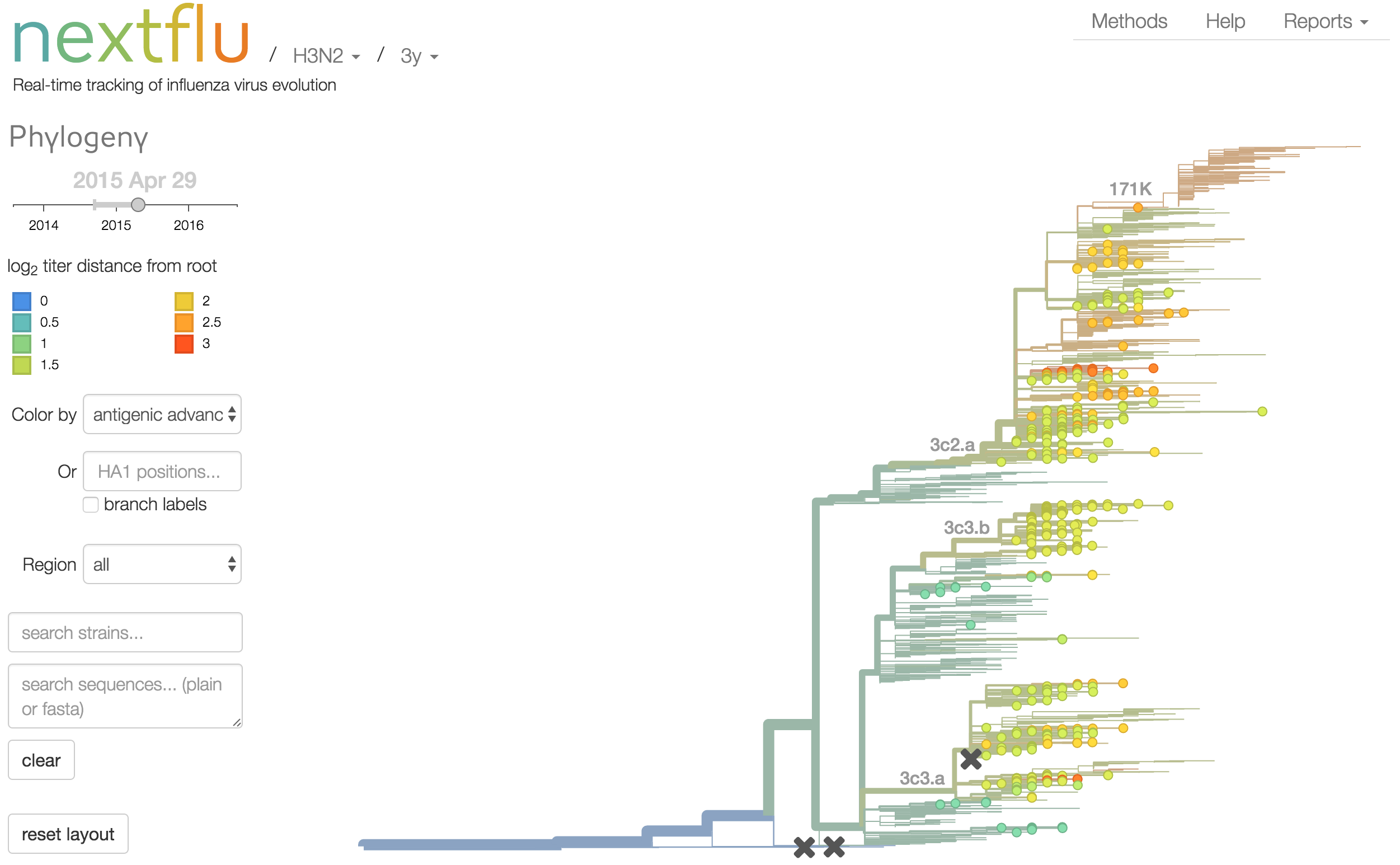
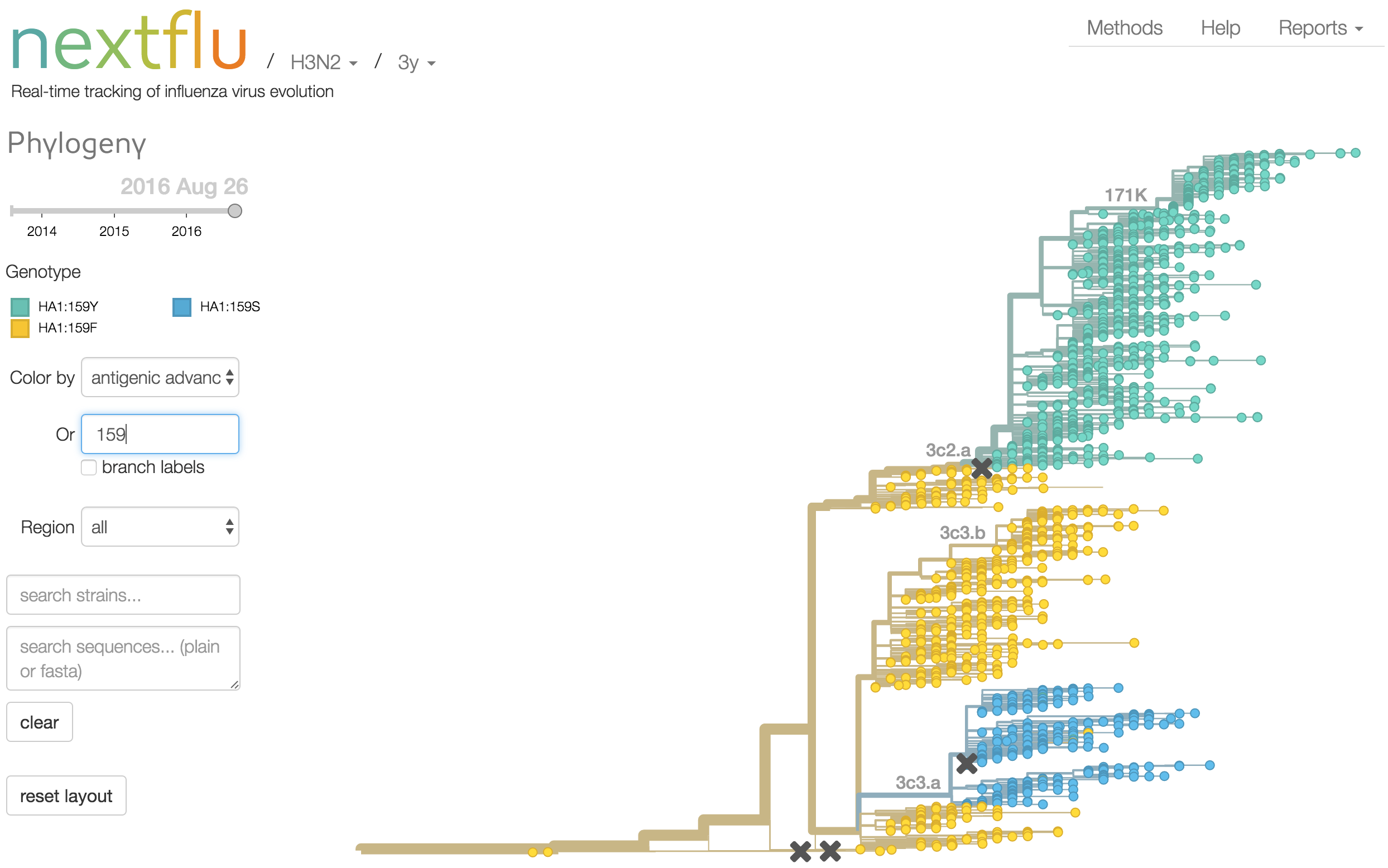
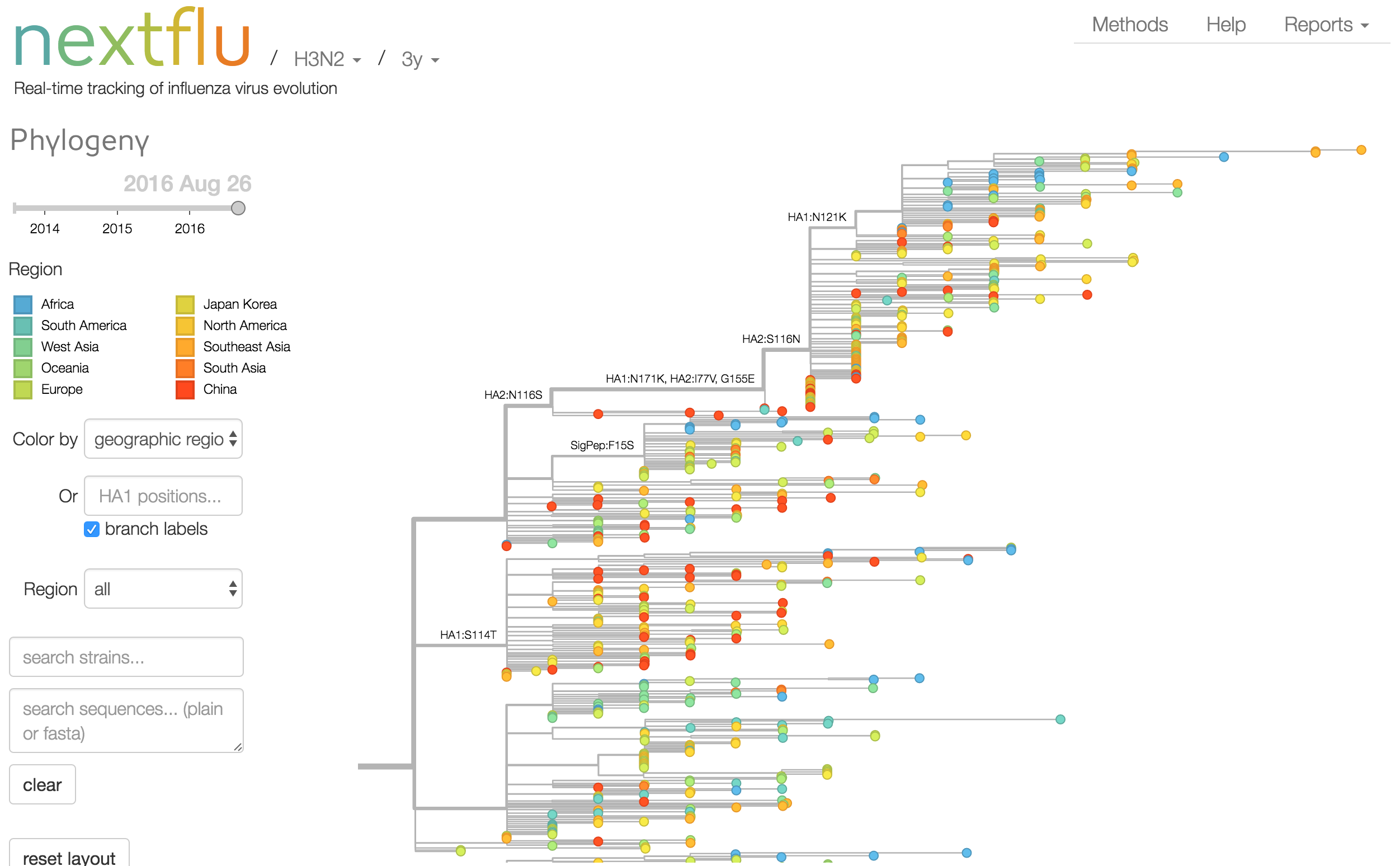
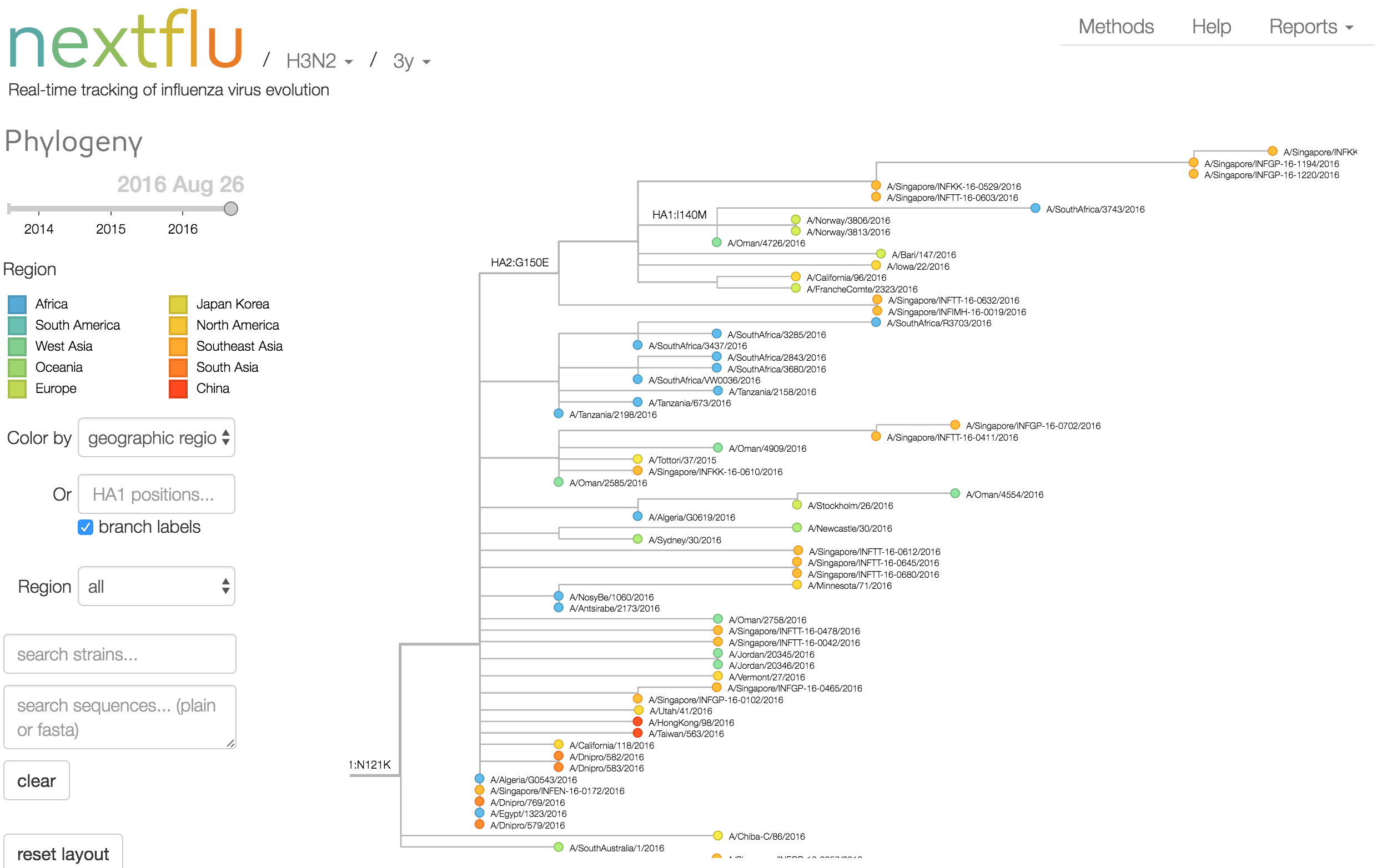
Antigenic analysis
Influenza hemagglutination inhibition (HI) assay
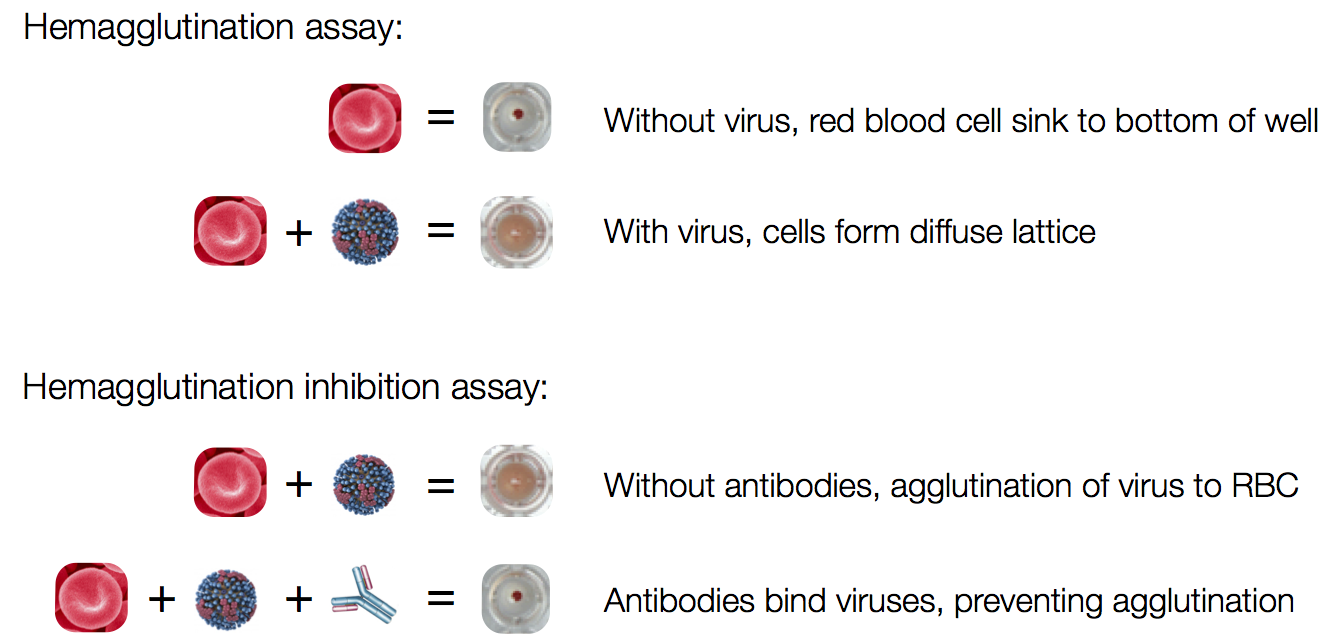
HI measures cross-reactivity across viruses
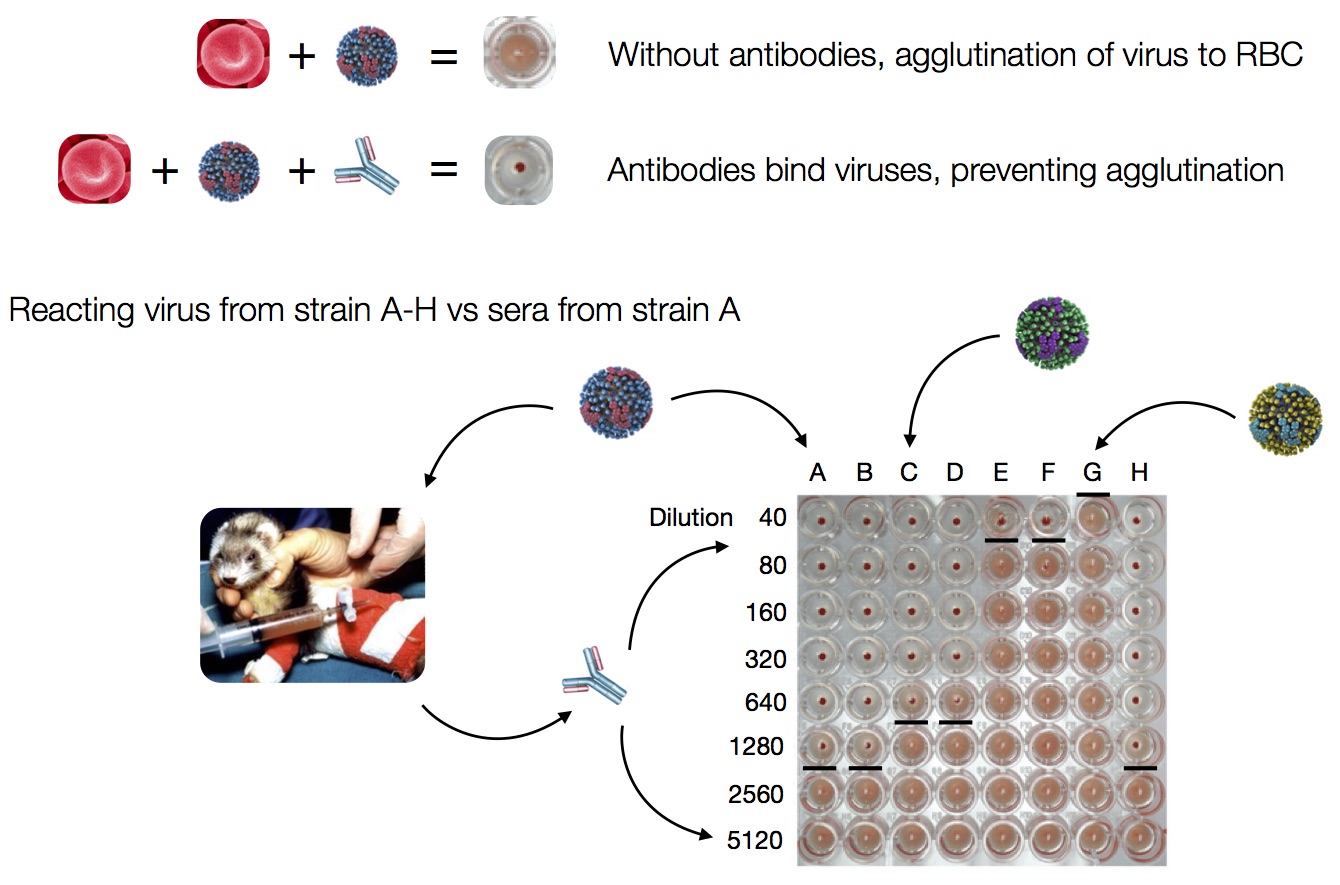
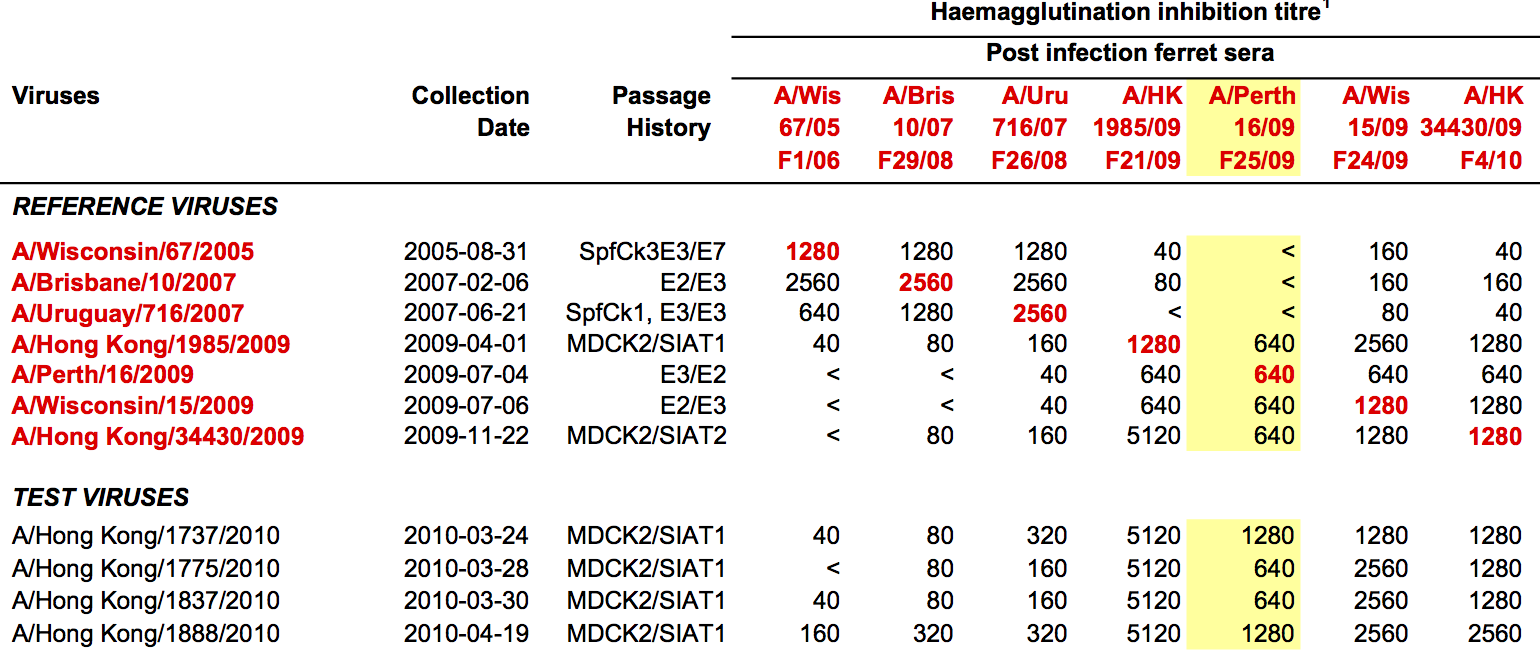
Data in the form of table of maximum inhibitory titers
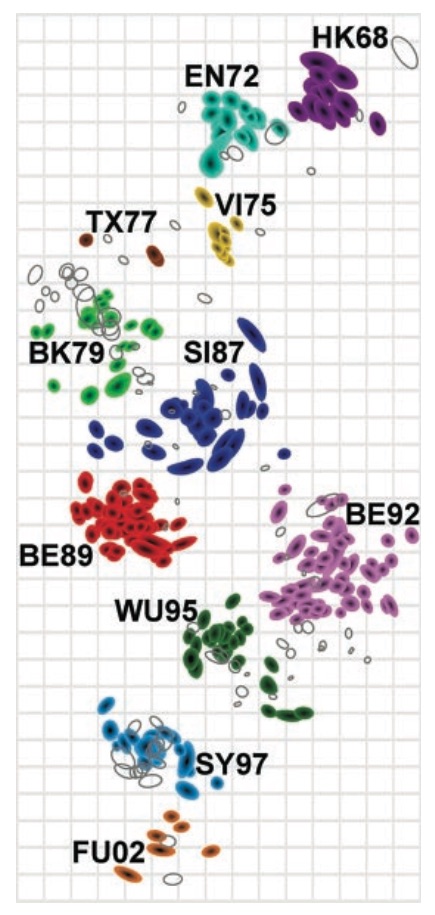
Antigenic cartography compresses HI measurements into an interpretable diagram
Instead of a geometric model, we sought a phylogenetic model of HI titer data
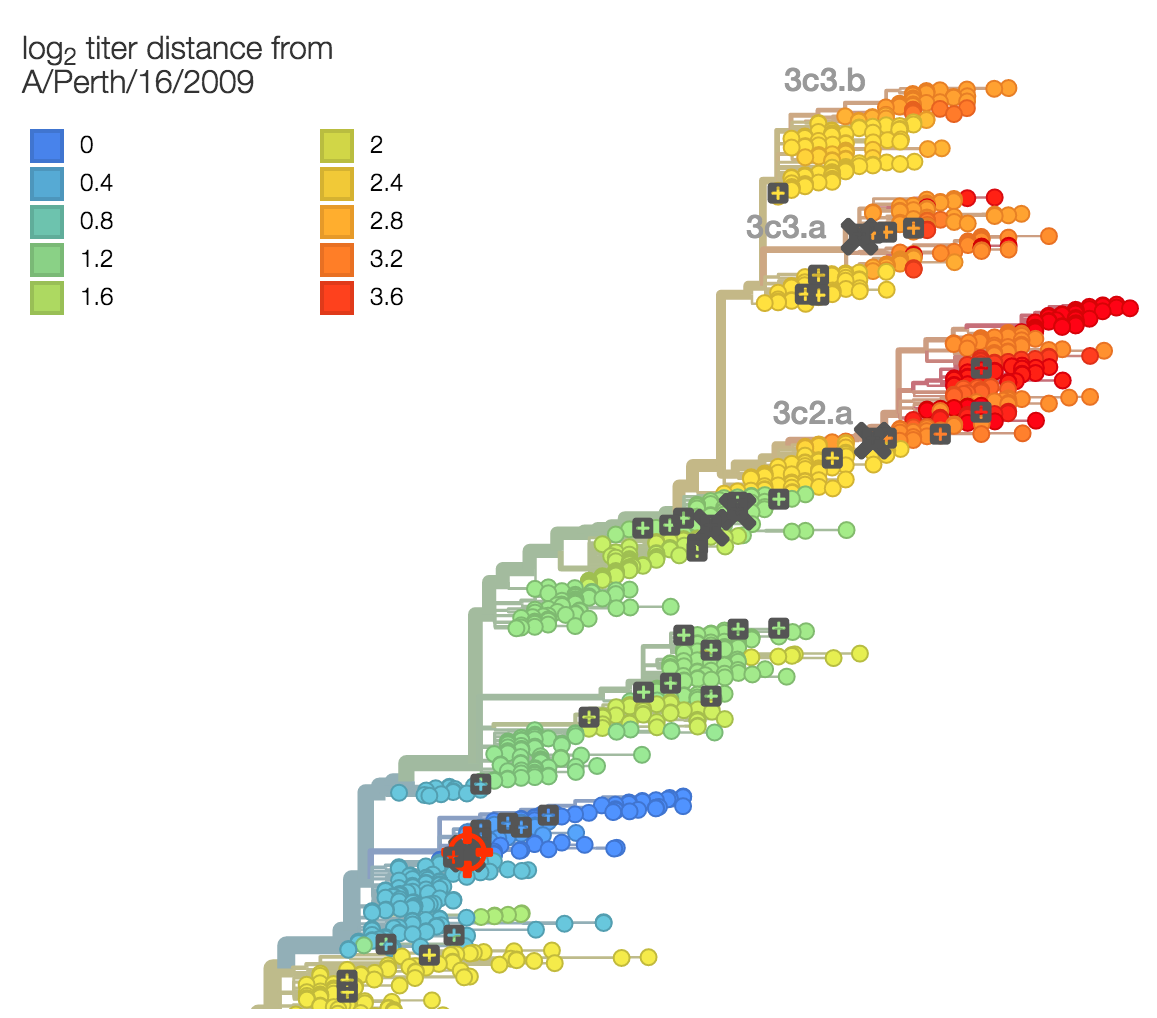
Identify phylogeny branches associated with drops in HI titer
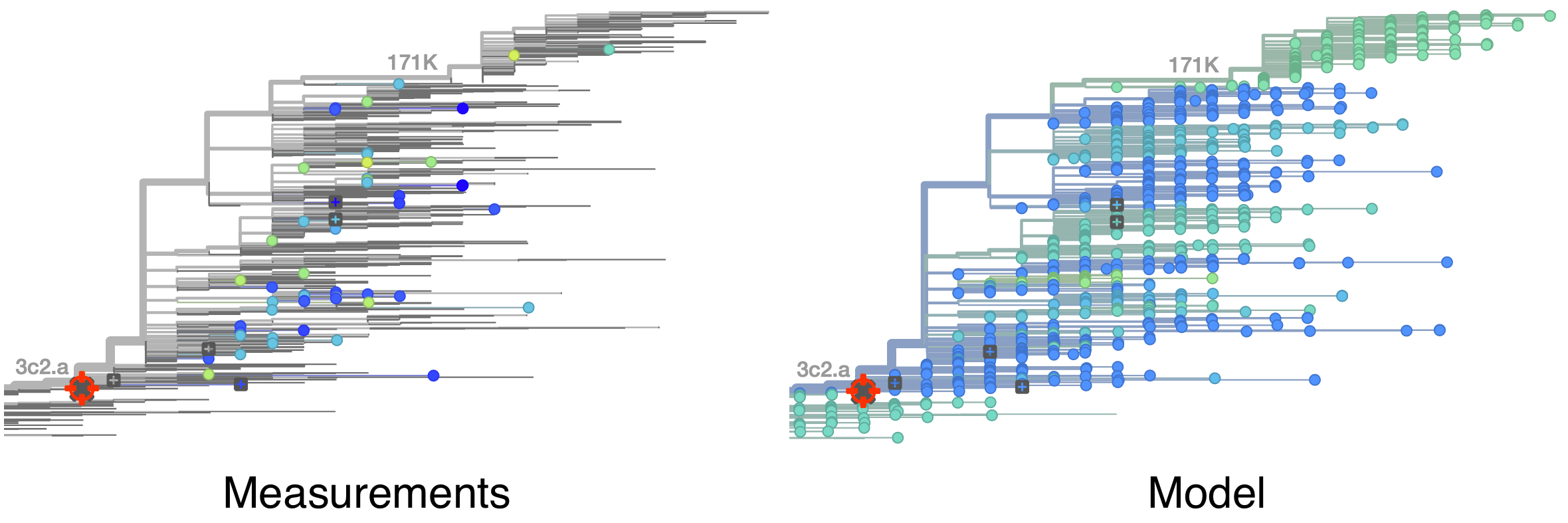
Model can be used to interpolate across tree and predict phenotype of untested viruses
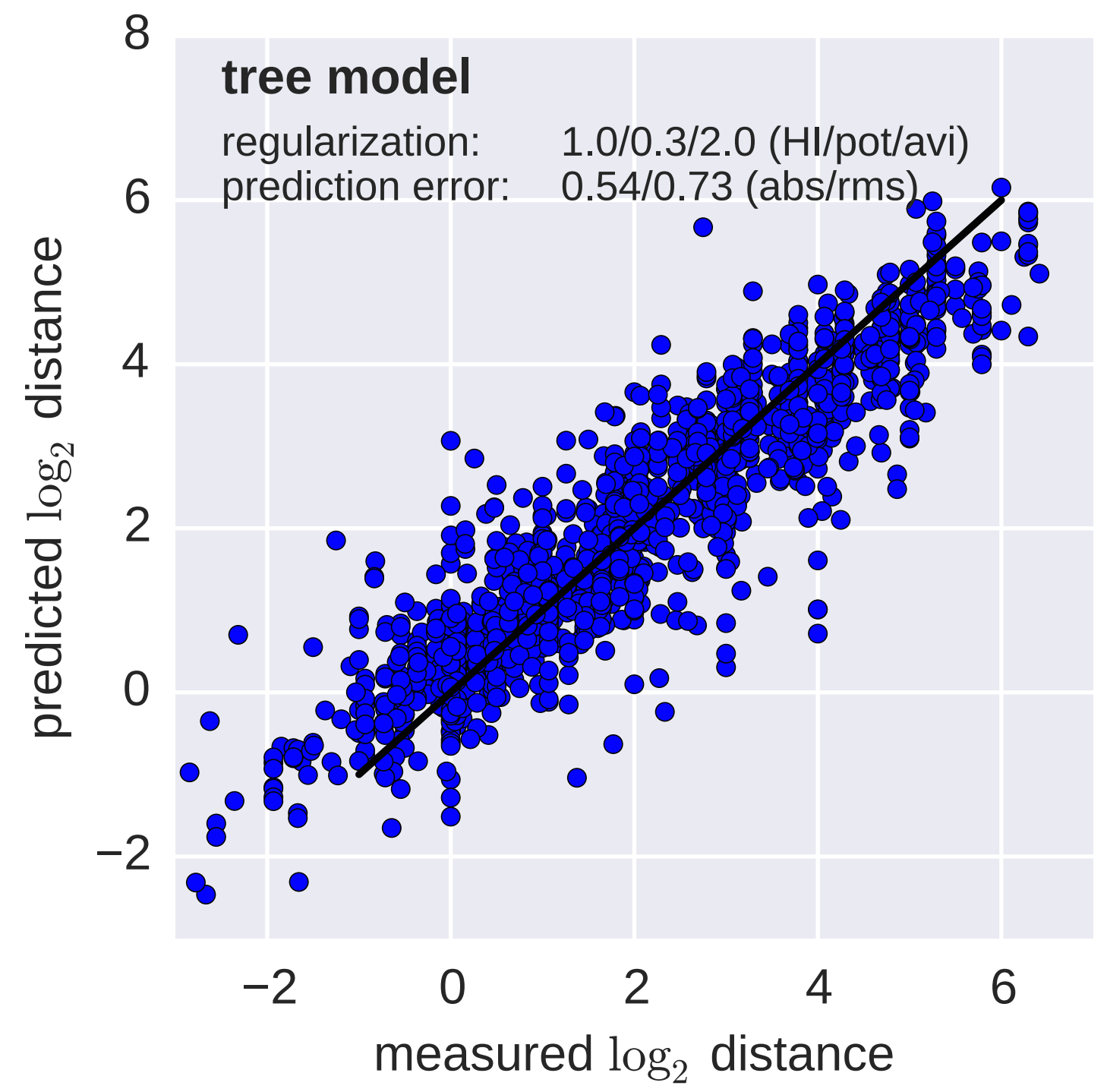
Model is highly predictive of missing titer values
Incorporate HI data from US Centers for Disease Control and Prevention
Up-to-date analysis at:
nextflu.org
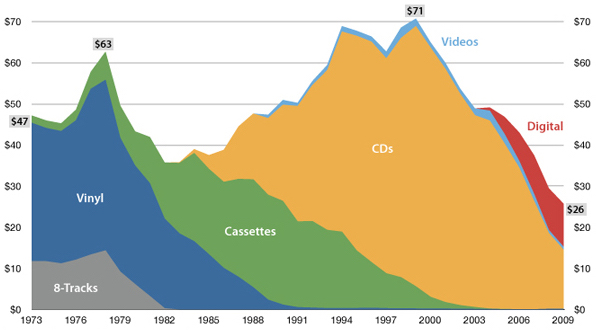
Forecasting
"The future is here, it's just not evenly distributed yet"
— William Gibson
USA music industry, 2011 dollars per capita
Influenza population turnover
Vaccine strain selection timeline
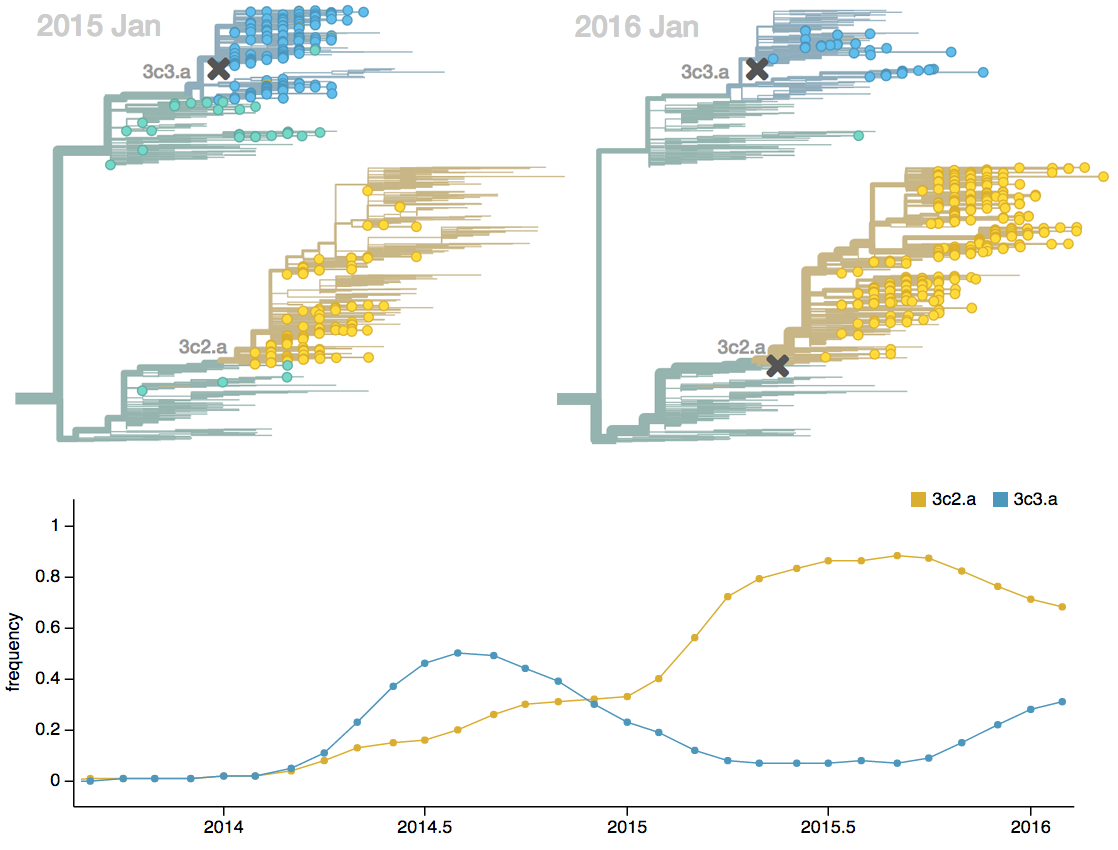
Seek to explain change in clade frequencies over 1 year
Fitness models can project clade frequencies
Clade frequencies $X$ derive from the fitnesses $f$ and frequencies $x$ of constituent viruses, such that
$$\hat{X}_v(t+\Delta t) = \sum_{i:v} x_i(t) \, \mathrm{exp}(f_i \, \Delta t)$$
This captures clonal interference between competing lineages
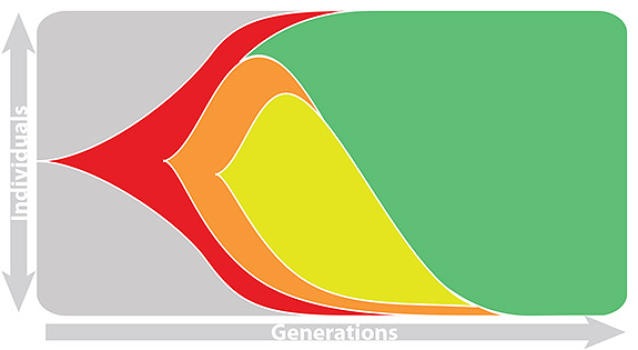
The question of forecasting becomes: how do we accurately estimate fitnesses of circulating viruses?
Fortunately, there's lots of training data and previously successful strains have had:
- Amino acid changes at epitope sites
- Antigenic novelty based on HI
- Rapid phylogenetic growth
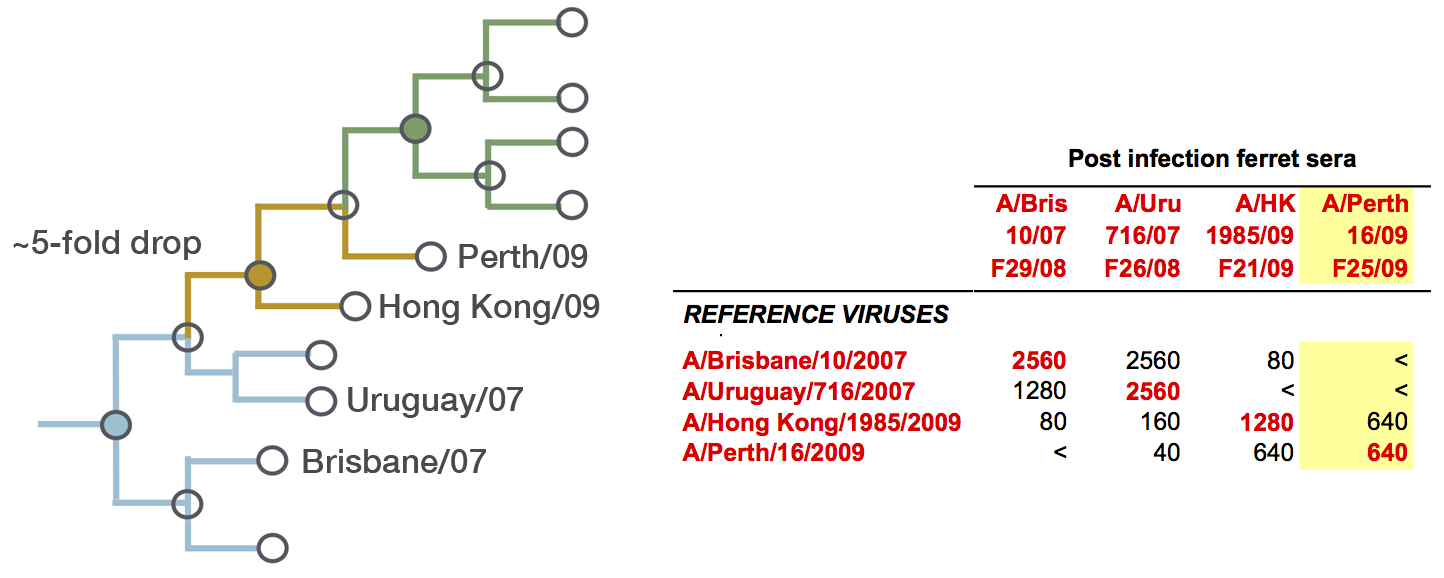
Predictor: calculate HI drop from ancestor,
drifted clades have high fitness
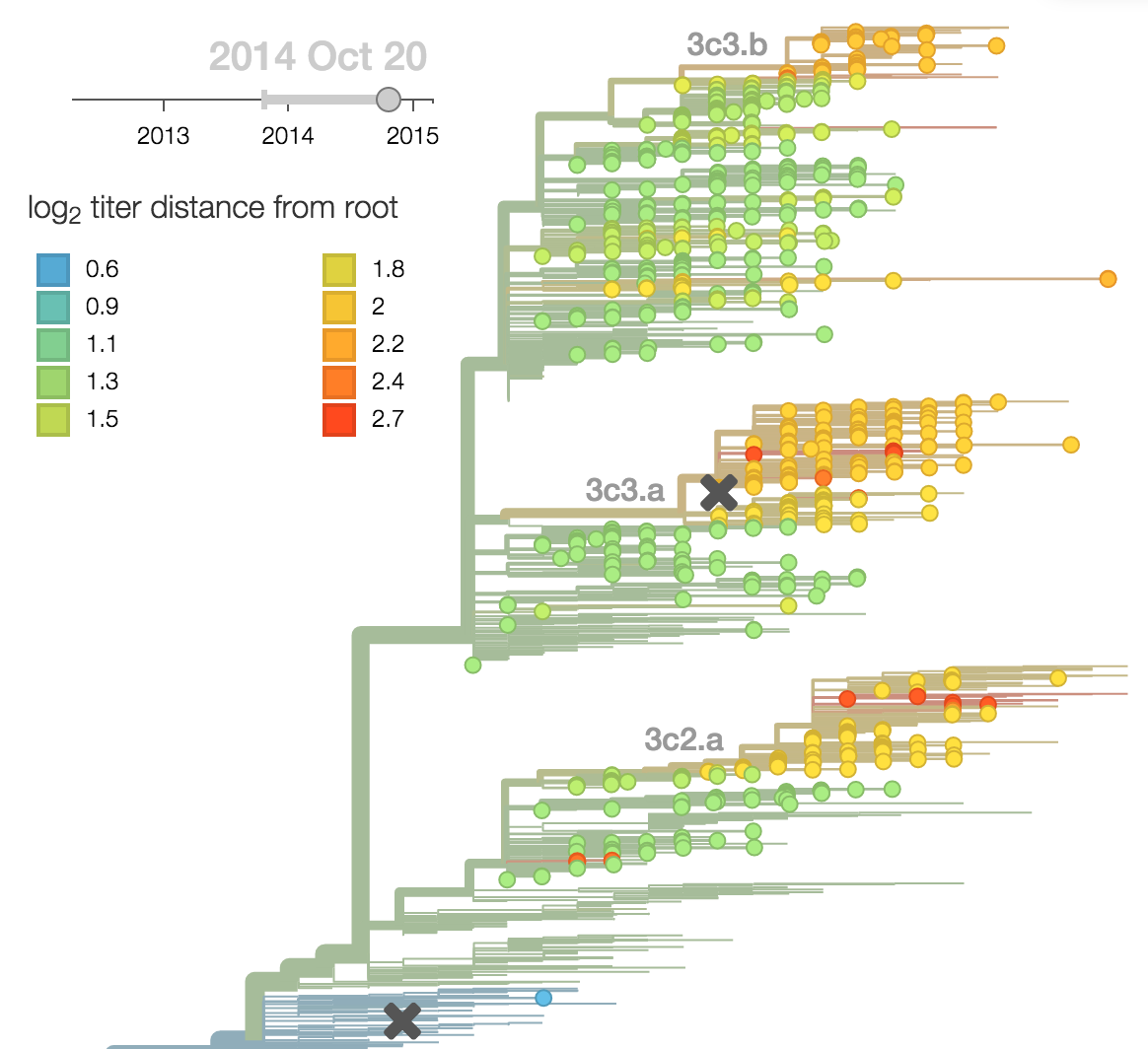
Predictor: project frequencies forward,
growing clades have high fitness
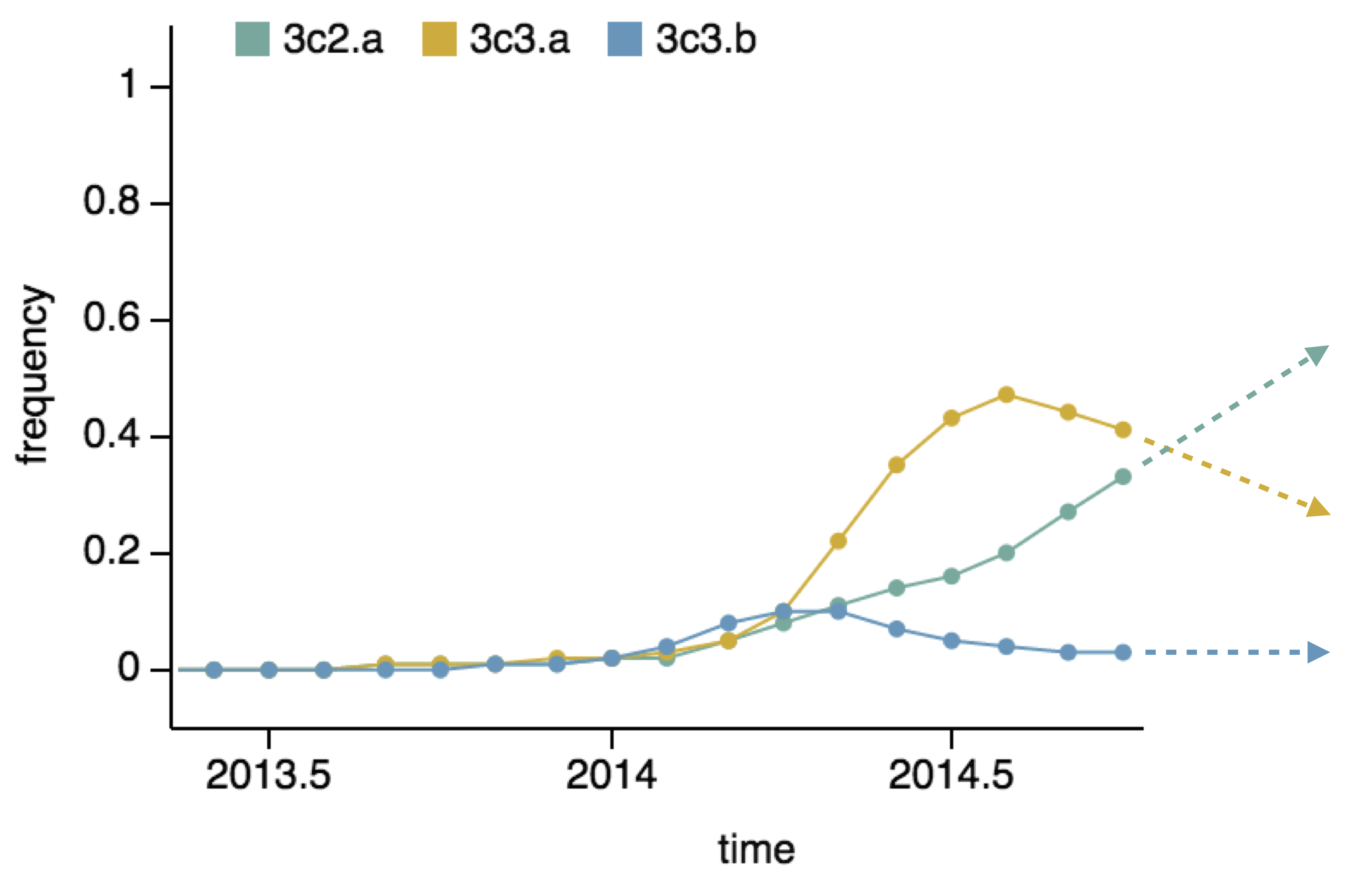
We predict fitness based on a simple formula
where the fitness $f$ of virus $i$ is estimated as
$$\hat{f}_i = \beta^\mathrm{HI} \, f_i^\mathrm{HI} + \beta^\mathrm{freq} \, f_i^\mathrm{freq}$$
where $f_i^\mathrm{HI}$ measures antigenic drift via HI and $f_i^\mathrm{freq}$ measures clade growth/decline
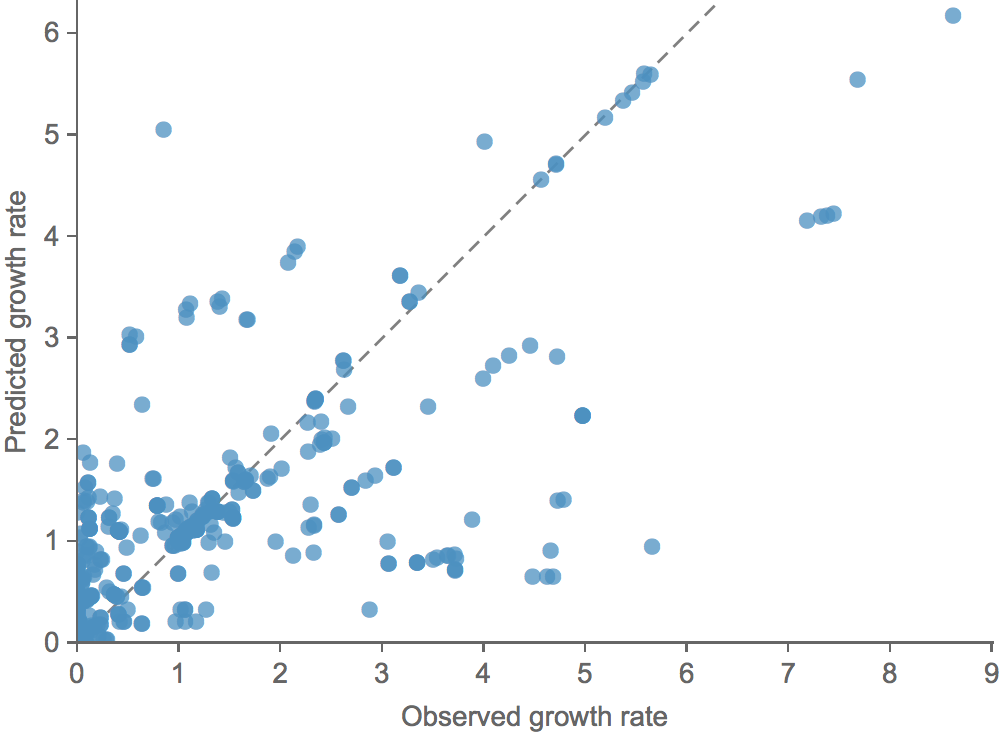
We learn coefficients and validate model based on previous 15 H3N2 seasons
Clade growth rate is well predicted (ρ = 0.66)
Growth vs decline correct in 84% of cases
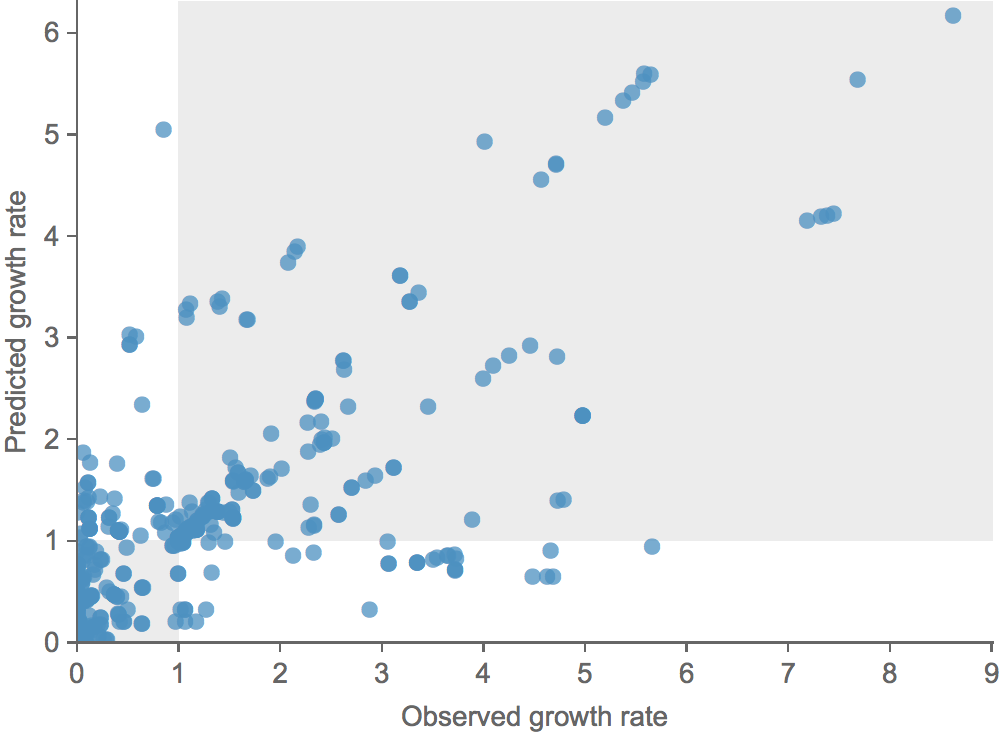
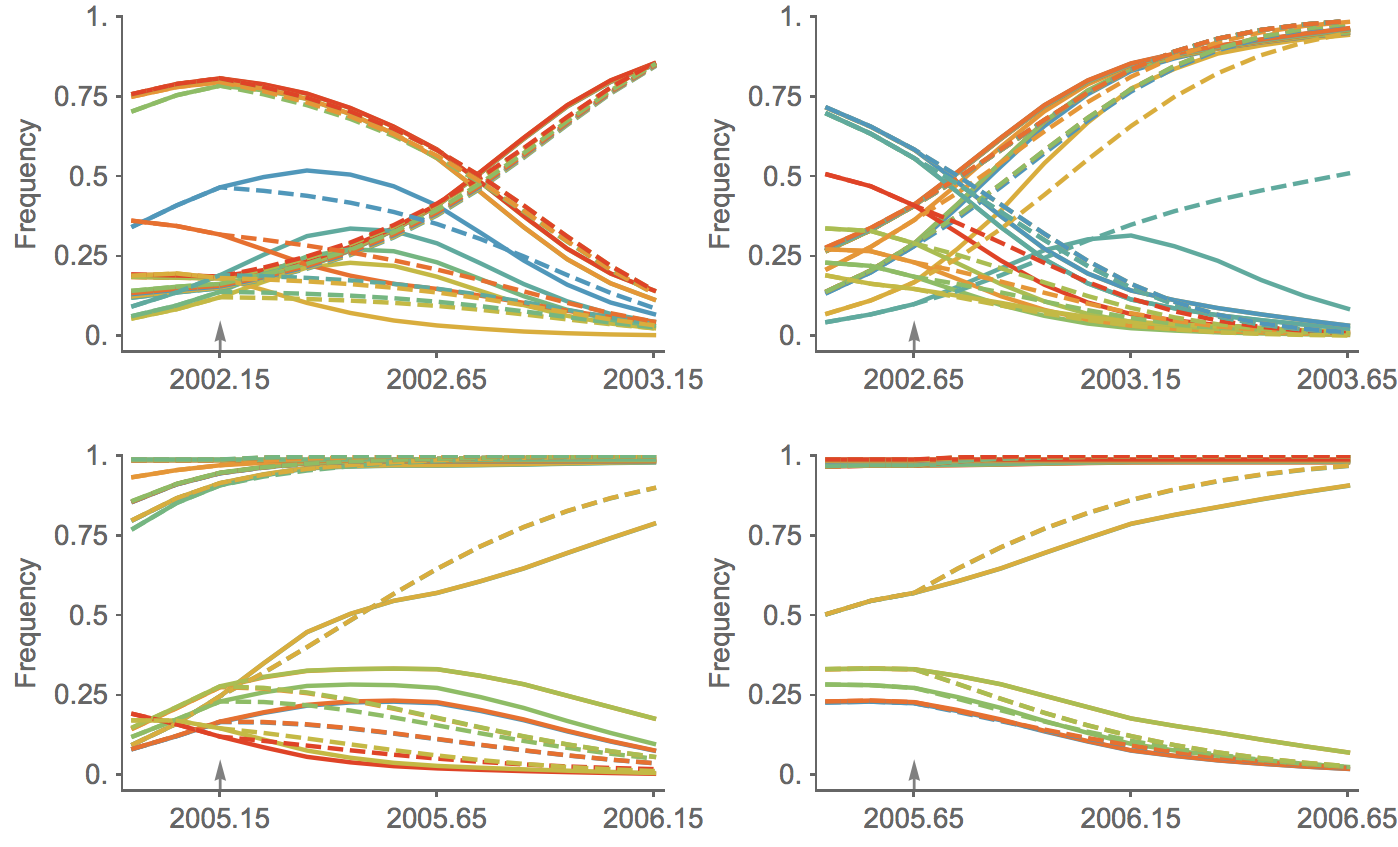
Trajectories show more detailed congruence
Trajectories show more detailed congruence
When does the forecast fail?
Emerging clades are difficult to forecast: little antigenic data and little evidence of "past performance"
Models work well for clades at >10%, but less well for clades <5%
New mutations difficult
Models can project forward from circulating strains, but cannot foresee the appearance of new mutations
Intrinsically limits the timescale of forecasting to ~1 year
Model is only as good as the data
Requires rapid shipping of samples, rapid sequencing and rapid antigenic characterization
Current situation
Further improvements to predictive modeling
- Extend to other seasonal viruses
- Forecast NA evolution
- Integrate neutralization (FRA) assay data
- Model effects of egg adaptation
- Incorporate an explicit geographic model
Real-time analyses are actionable and may inform influenza vaccine strain selection
More generally real-time analyses may be useful for other viruses
Zika
Zika's arrival and spread in the Americas
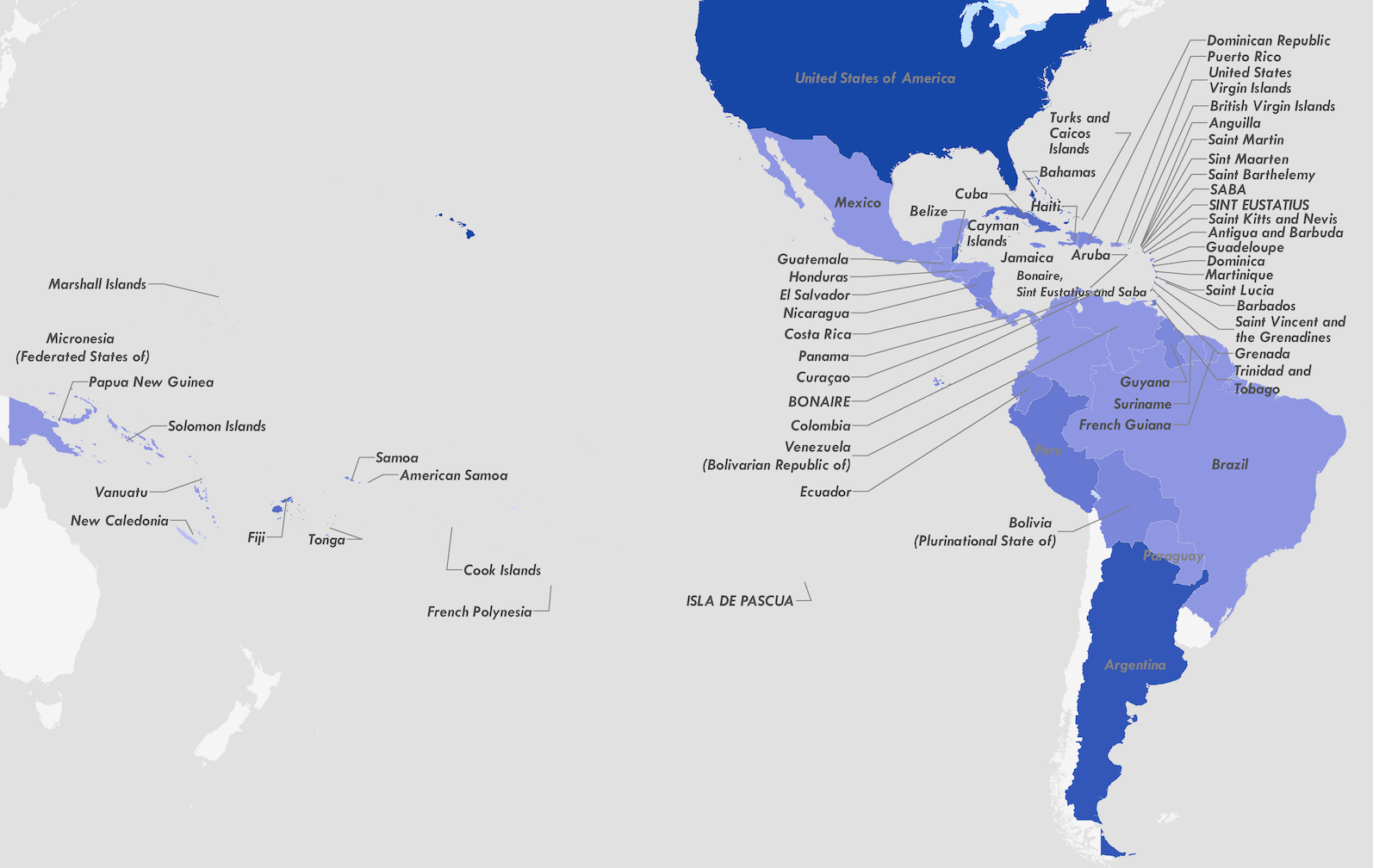
Establishment and cryptic transmission of Zika virus in Brazil and the Americas
with Nuno Faria, Nick Loman, Oli Pybus, Luiz Alcantara, Ester Sabino, Josh Quick,
![]() Alli Black,
Ingra Morales, Julien Thézé, Marcio Nunes, Jacqueline de Jesus,
Alli Black,
Ingra Morales, Julien Thézé, Marcio Nunes, Jacqueline de Jesus,
Marta Giovanetti, Moritz Kraemer, Sarah Hill and many others

Road trip through northeast Brazil to collect samples and sequence
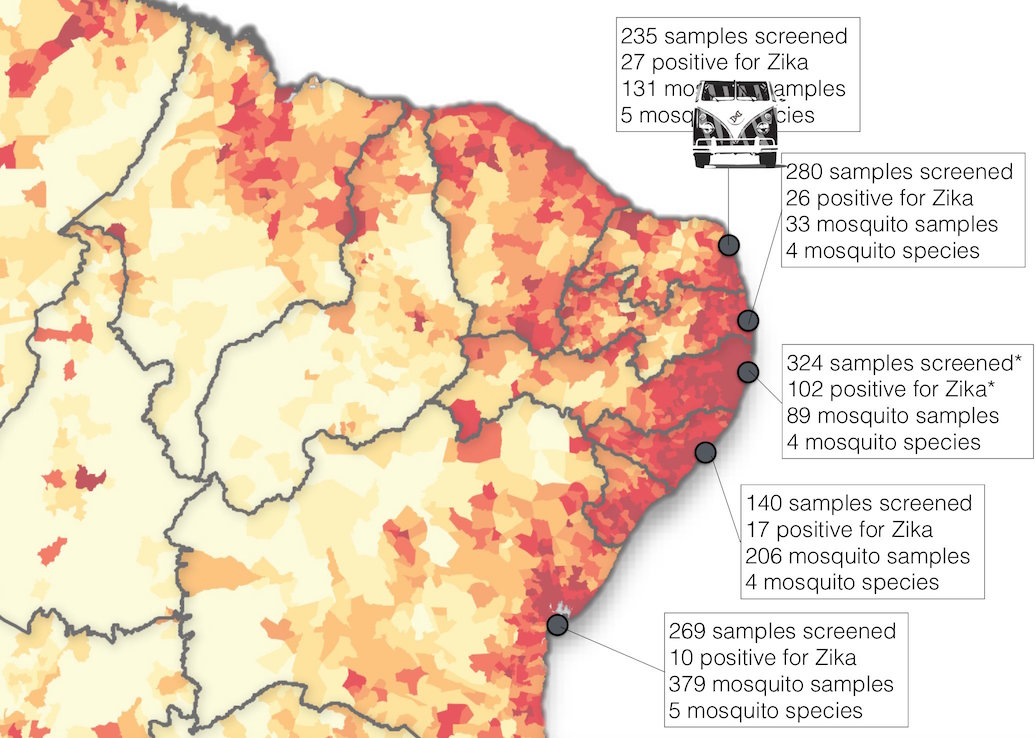
Case reports and diagnostics suggest initiation in northeast Brazil
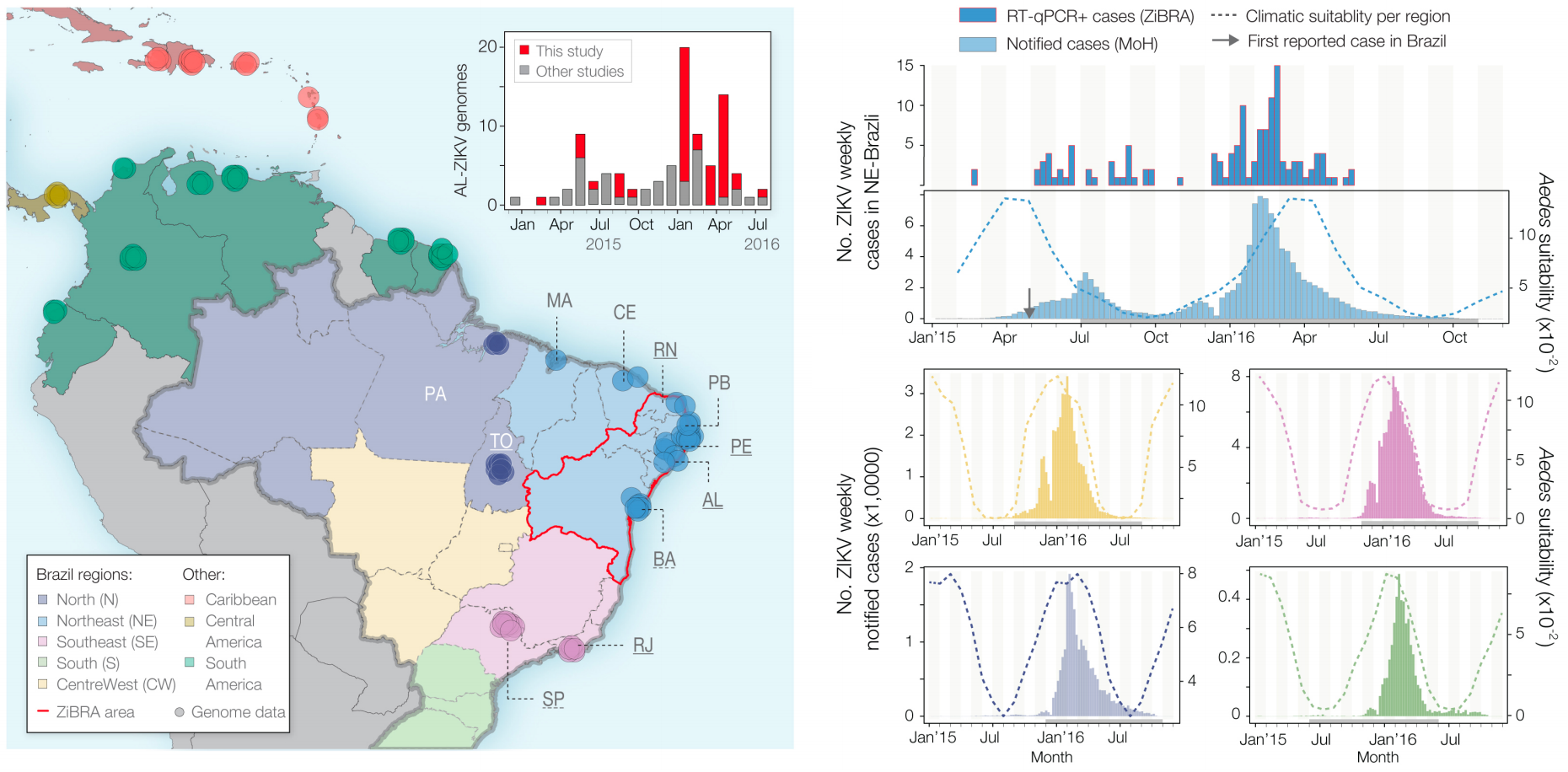
Phylogeny infers an origin in northeast Brazil
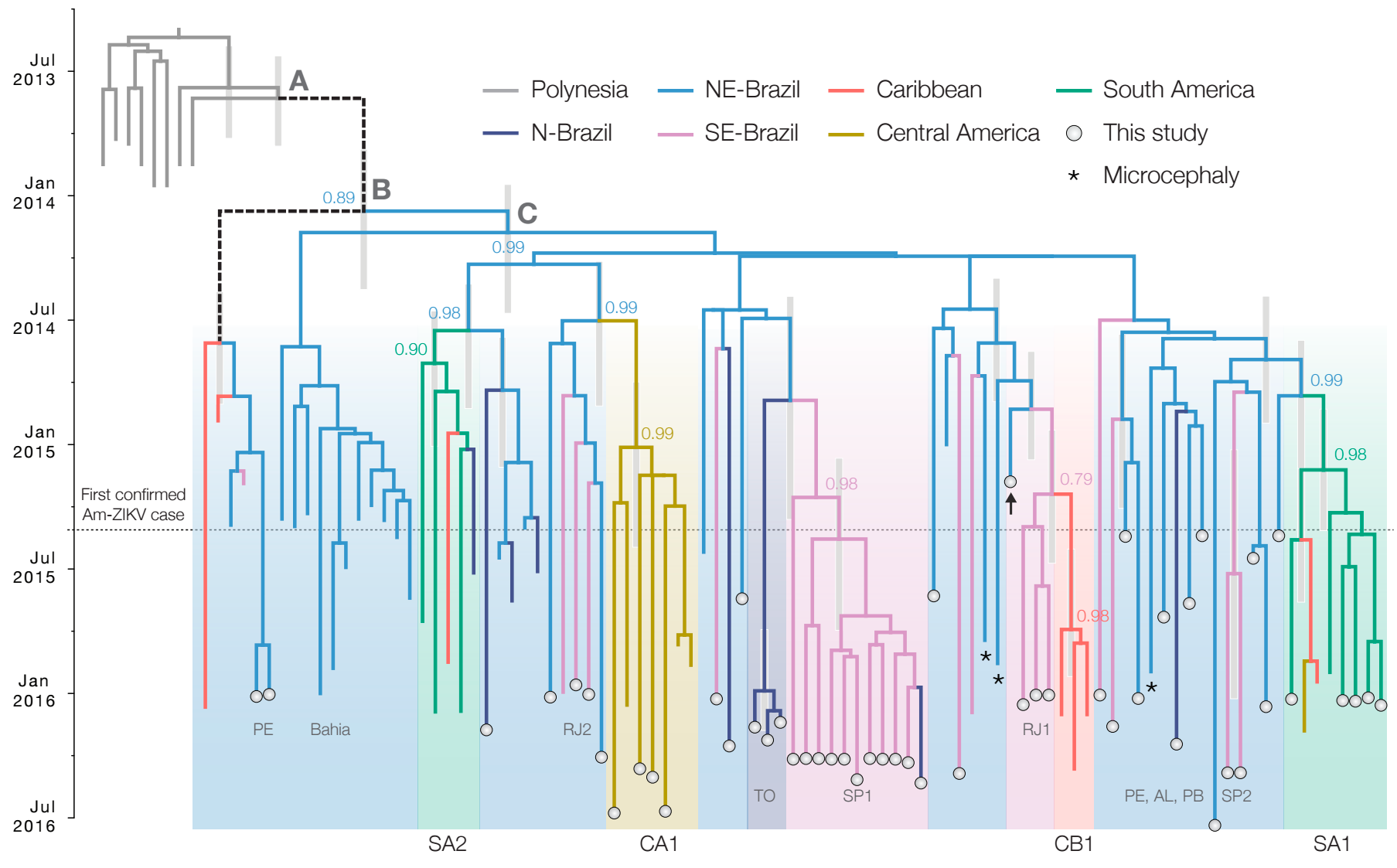
Important analyses, let's make them more rapid and more automated
nextstrain
Project to conduct real-time molecular epidemiology and evolutionary analysis of emerging epidemics
with
![]() Richard Neher,
Richard Neher,
![]() James Hadfield,
James Hadfield,
![]() Colin Megill,
Colin Megill,
![]() Sidney Bell,
Sidney Bell,
![]() Charlton Callender,
Charlton Callender,
![]() Barney Potter,
Barney Potter,
and ![]() John Huddleston
John Huddleston
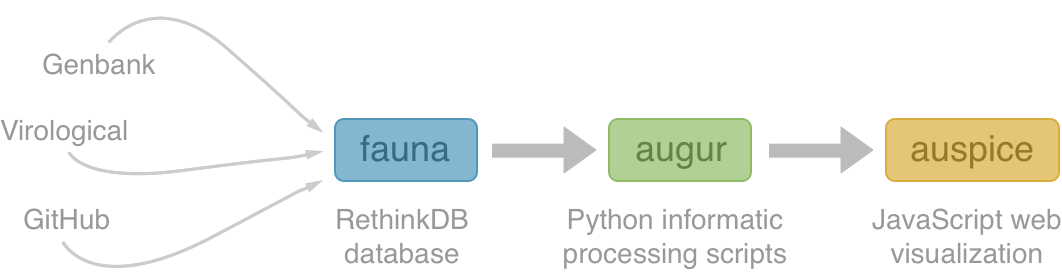
Nextstrain architecture
All code open source at github.com/nextstrain
nextstrain.org
Rapid on-the-ground sequencing by Ian Goodfellow, Matt Cotten and colleagues
Build out pipelines for different pathogens, improve databasing and lower bioinformatics bar
Acknowledgements
Bedford Lab:
![]() Alli Black,
Alli Black,
![]() Sidney Bell,
Sidney Bell,
![]() Gytis Dudas,
Gytis Dudas,
![]() John Huddleston,
John Huddleston,
![]() Barney Potter,
Barney Potter,
![]() James Hadfield,
James Hadfield,
![]() Louise Moncla
Louise Moncla
Influenza: WHO Global Influenza Surveillance Network, GISAID, Richard Neher, Colin Russell, Andrew Rambaut, Dave Wentworth, Becky Garten, Marta Łuksza, Michael Lässig
Zika: Nick Loman, Nuno Faria, Oli Pybus, Josh Quick, Kristian Andersen, Nathan Grubaugh, Jason Ladner, Gustavo Palacios, Sharon Isern, Gytis Dudas, Alli Black, Barney Potter, Esther Ellis, Louise Moncla, Diana Rojas
Nextstrain: Richard Neher, James Hadfield, Colin Megill, Sidney Bell, Charlton Callender, Barney Potter, John Huddleston, Emma Hodcroft