Data rich phylodynamics
Trevor Bedford
Fred Hutchinson Cancer Center / Howard Hughes Medical Institute
31 Jan 2024
Science Meeting
HHMI
Slides at: bedford.io/talks
Genomic epidemiology during the COVID-19 pandemic
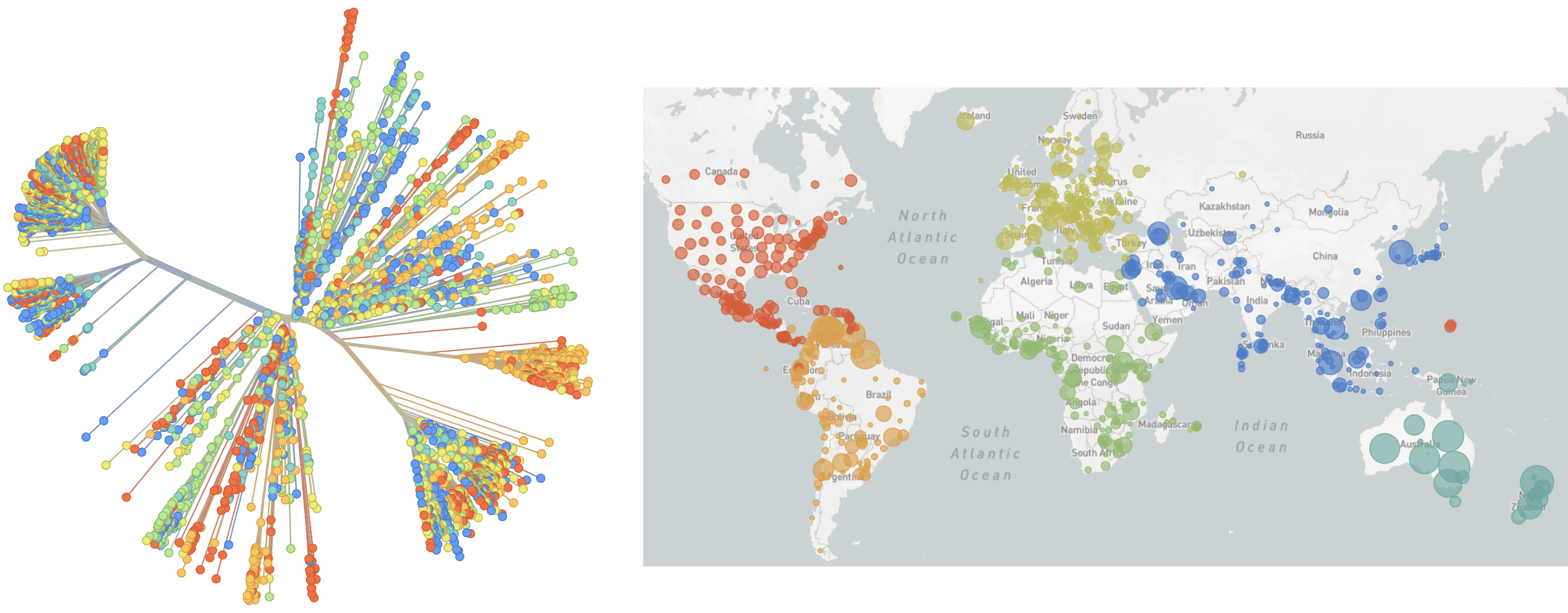
Over 16M SARS-CoV-2 genomes shared to GISAID and evolution tracked in real-time at nextstrain.org
![]() Richard Neher,
Richard Neher,
![]() Ivan Aksamentov,
Ivan Aksamentov,
![]() Kim Andrews,
Kim Andrews,
![]() Jennifer Chang
Jennifer Chang
![]() James Hadfield,
James Hadfield,
![]() Emma Hodcroft,
Emma Hodcroft,
![]() John Huddleston,
John Huddleston,
![]() Jover Lee,
Jover Lee,
![]() Victor Lin,
Victor Lin,
![]() Cornelius Roemer,
Cornelius Roemer,
![]() Thomas Sibley
Thomas Sibley
Three key insights that genomic epi provided during pandemic
- Rapid human-to-human spread in Wuhan beyond initial market outbreak
- Extensive local transmission while testing was rare
- Identification of variants of concern and mapping of increased transmission rates
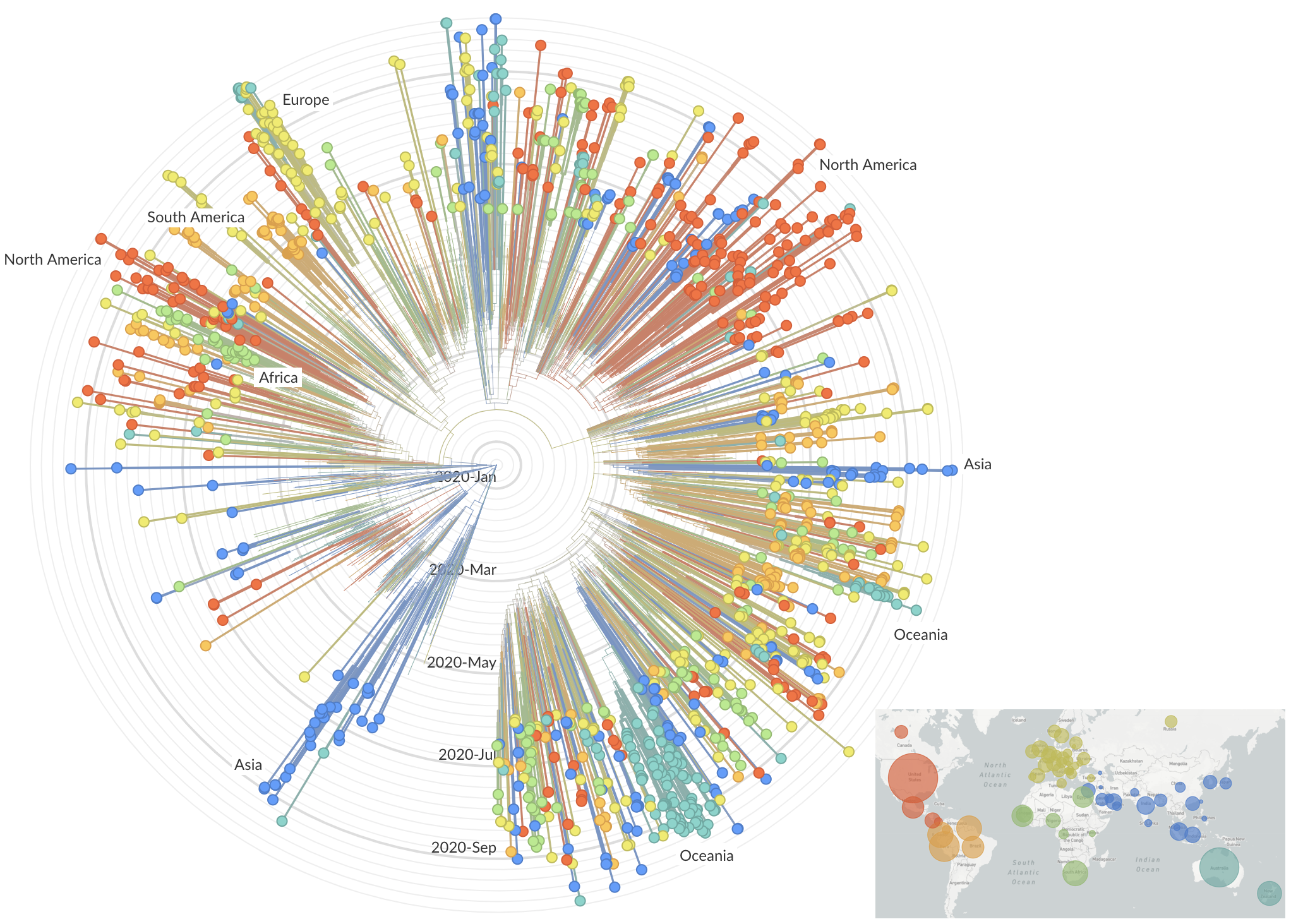
Jan 19: First 12 genomes from Wuhan (blue) and Bangkok (red) showed lack of genetic diversity
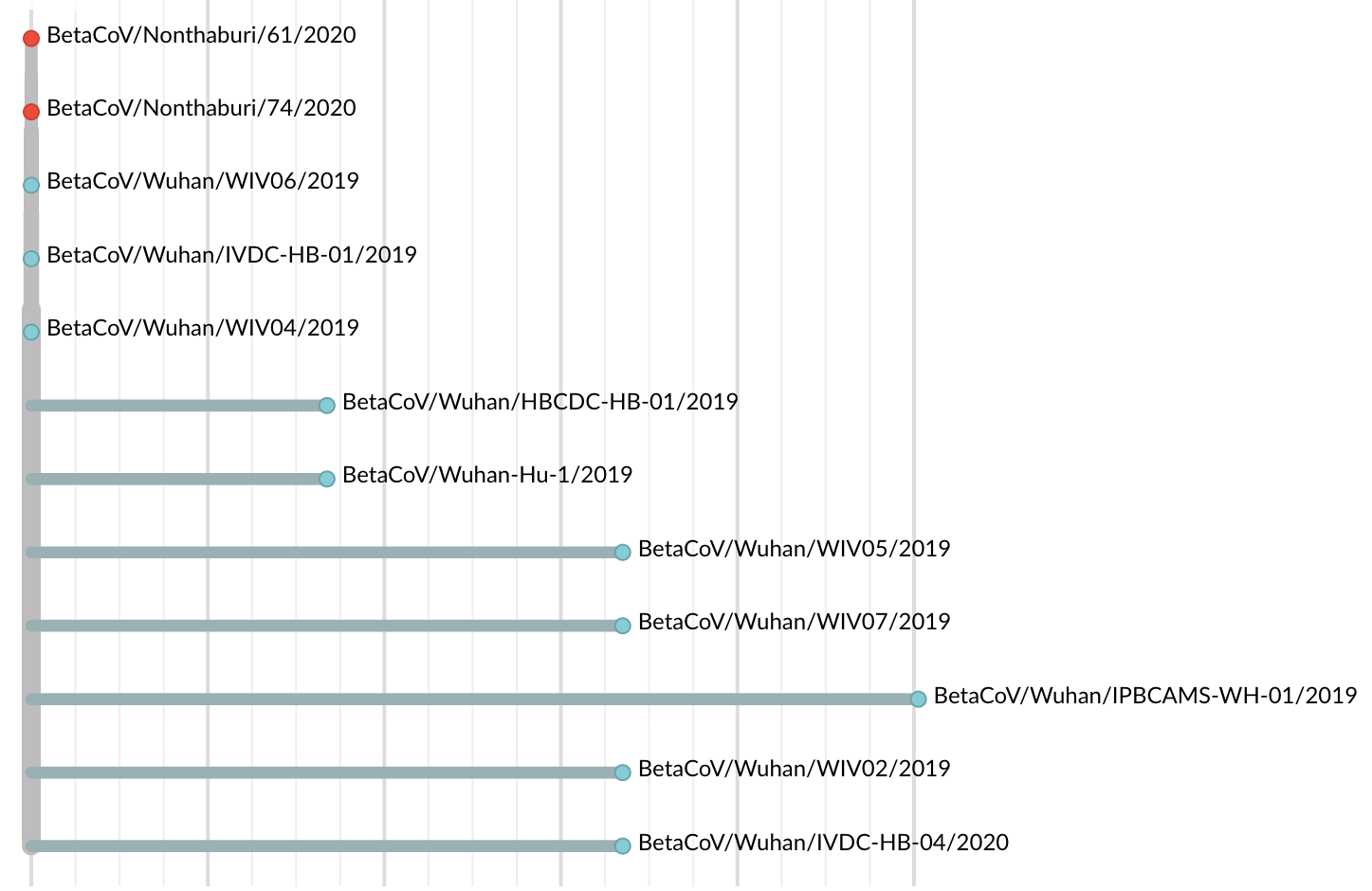
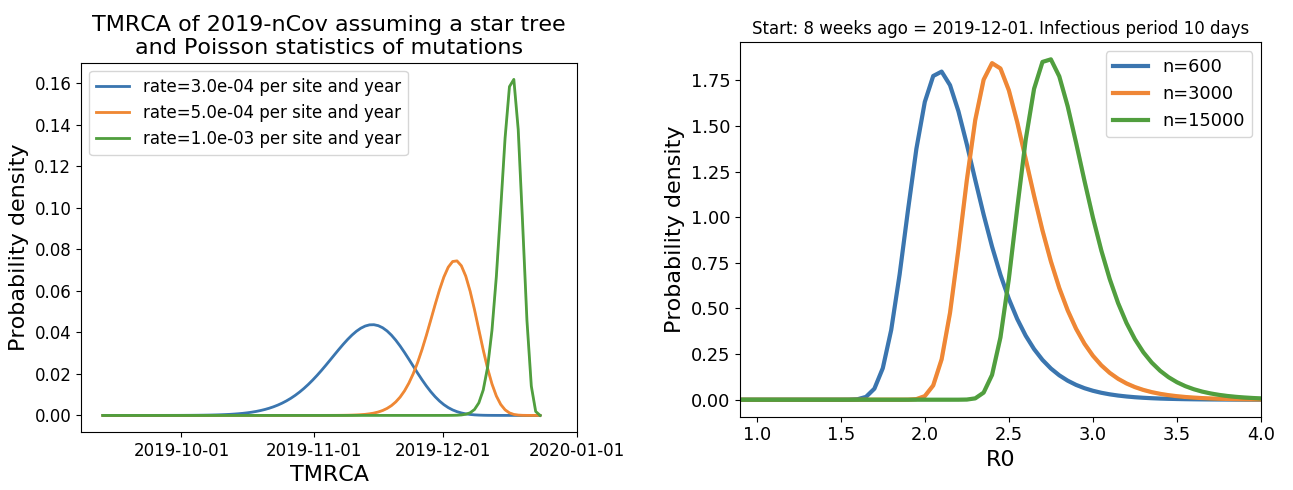
Jan 23: Introduction into the human population between Nov 15 and Dec 15 and subsequent rapid human-to-human spread
Epidemic in the USA was introduced from China in late Jan and from Europe during Feb
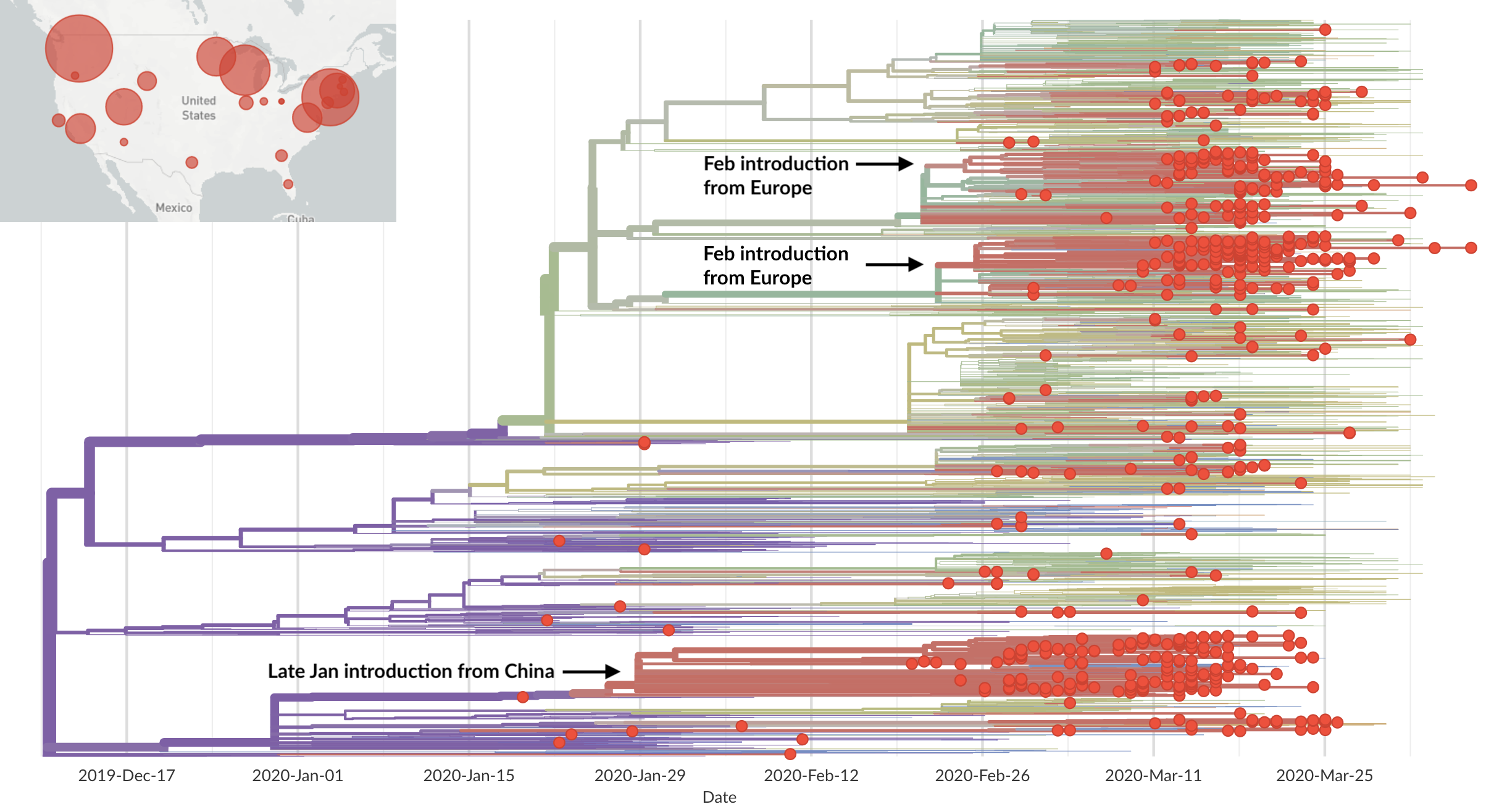
Early sequencing provided best estimate of extent of local outbreak
After initial wave, with mitigation
efforts and decreased travel,
regional clades emerge
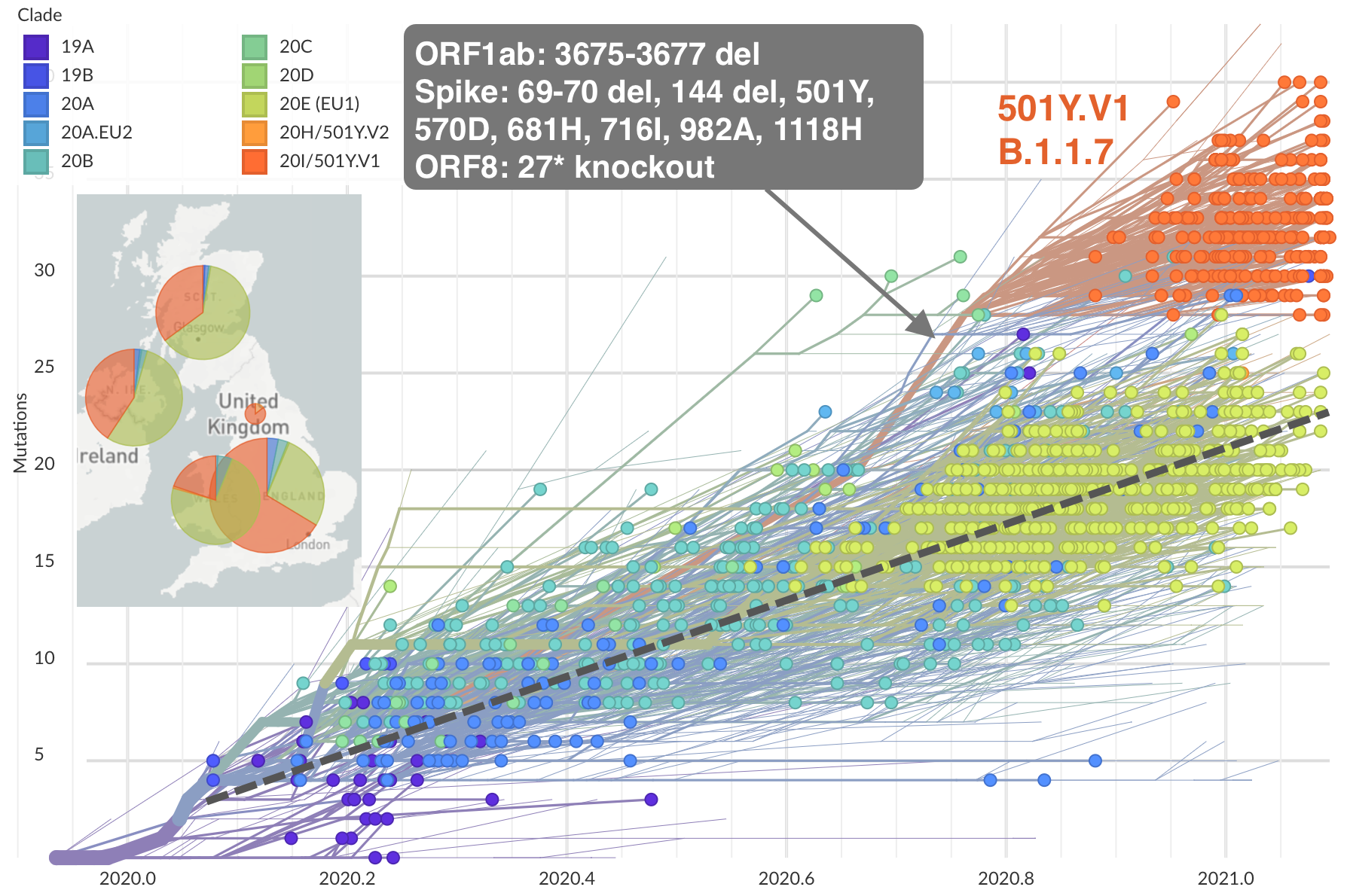
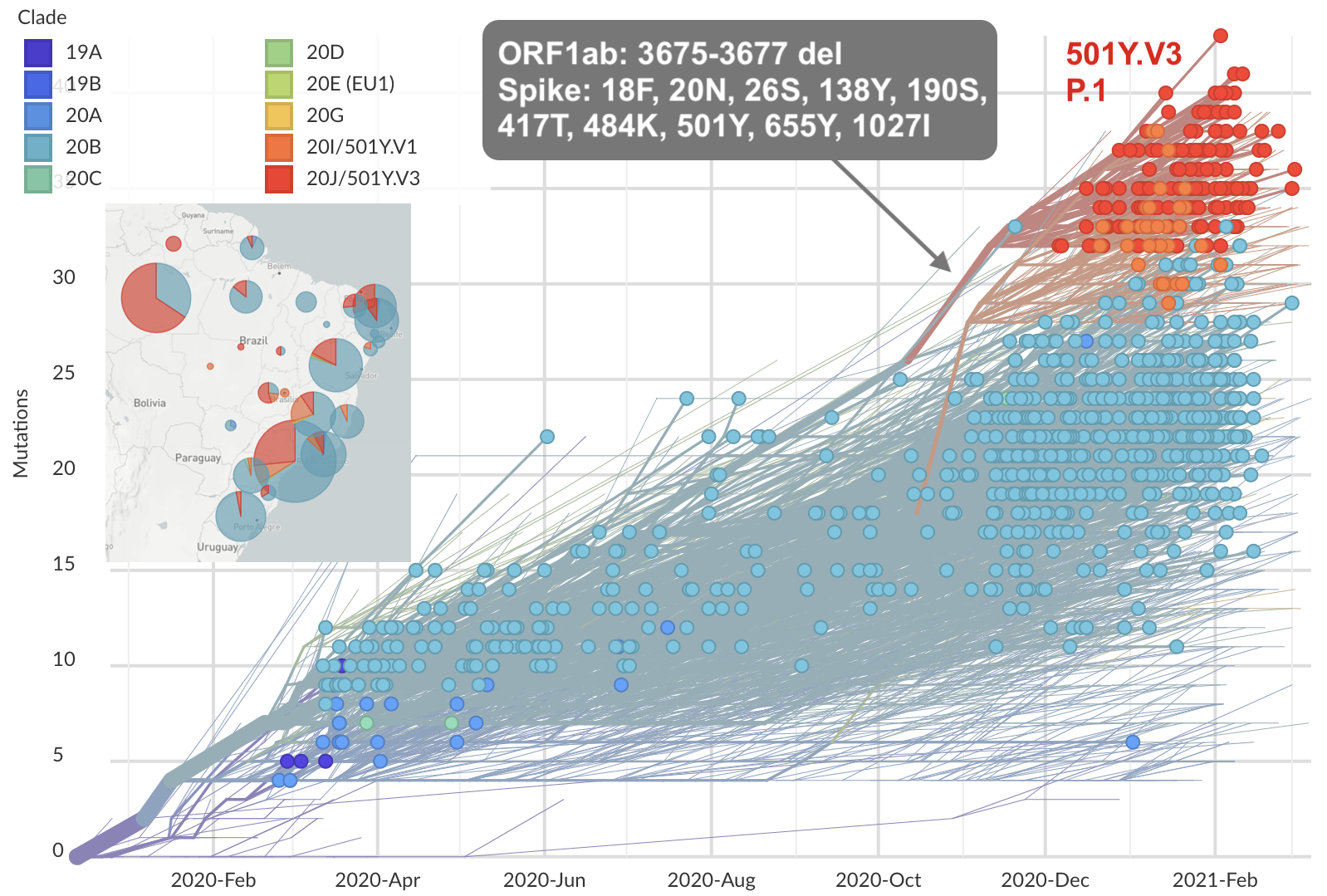
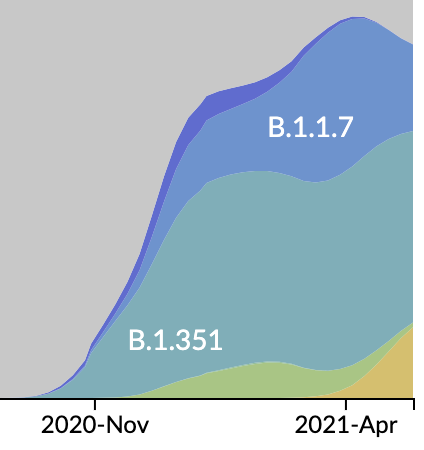
Emergence and spread of initial VOC viruses
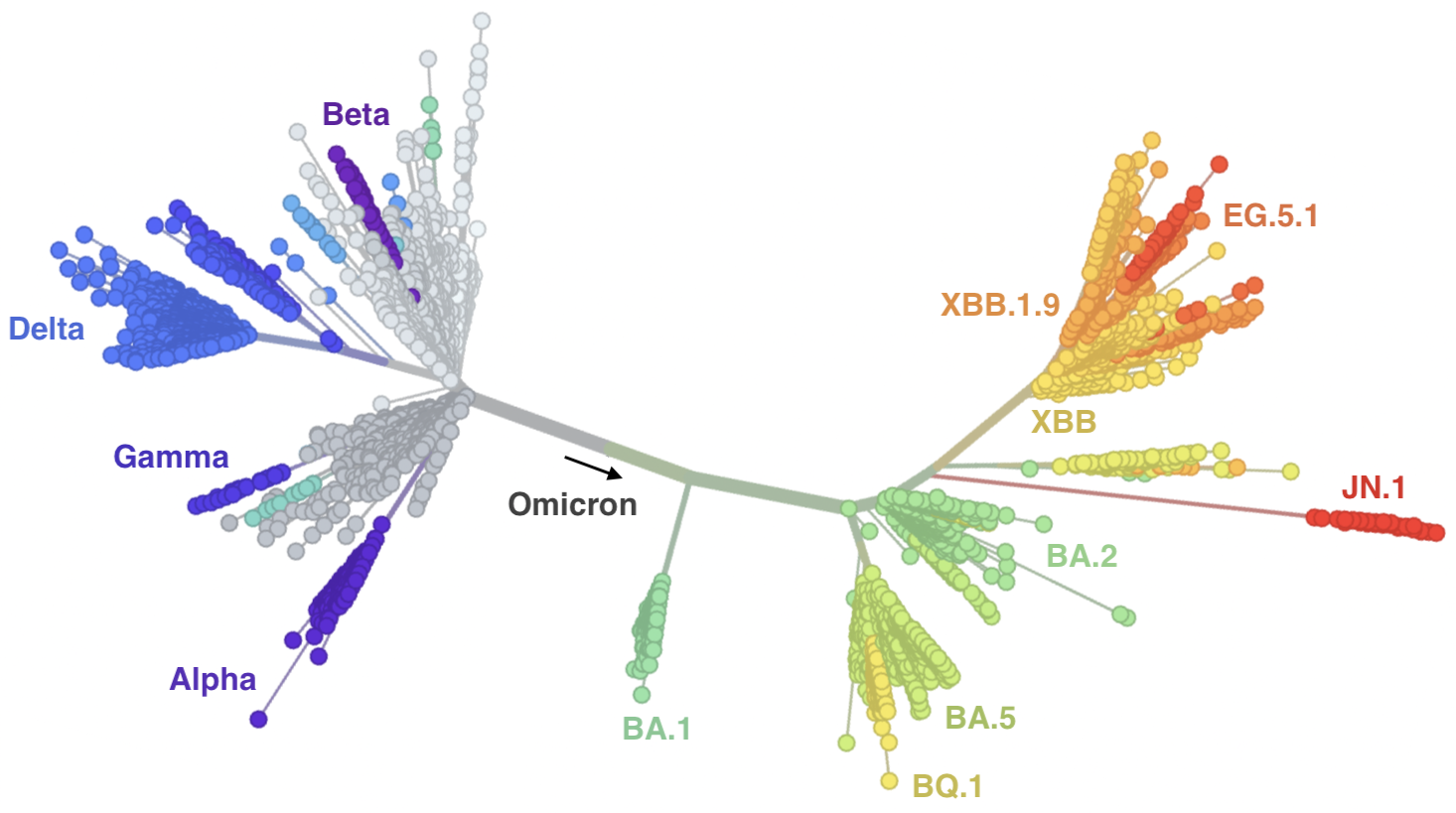
Continued evolution post-Omicron
Future of genomic epidemiology
The COVID-19 pandemic has pushed the field perhaps ~5 years into the future
| 2013-16 Ebola in West Africa | 29k confirmed cases | 1610 genomes |
| 2015-17 Zika in the Americas | 223k confirmed cases | 942 genomes |
| 2018-19 seasonal flu in US | 290k confirmed cases | 8864 genomes |
| 2020-22 COVID-19 pandemic | 732M confirmed cases | 14.5M genomes |

Scalable approaches to genomic epidemiology
- Genomic outbreak investigation: across pathogens with focus on spatial dynamics and recontructing spread
- Evolutionary forecasting: focus on seasonal influenza and SARS-CoV-2 for impact on vaccine strain selection
Genomic outbreak investigation
Phylogeography infers migration matrix via phylogeny
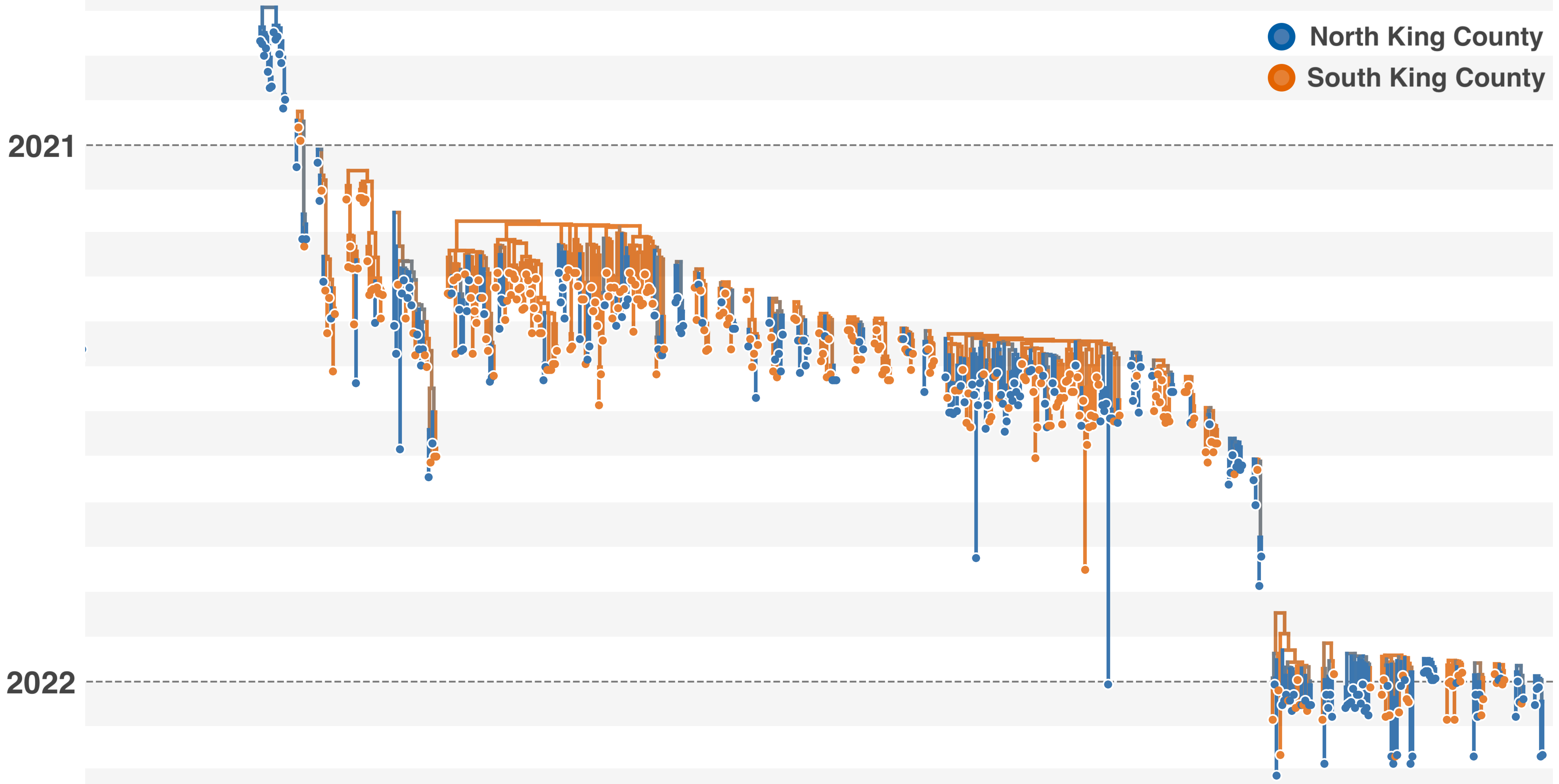
Phylogeography infers migration matrix via phylogeny
However, this approach faces significant issues with scalability and sampling bias
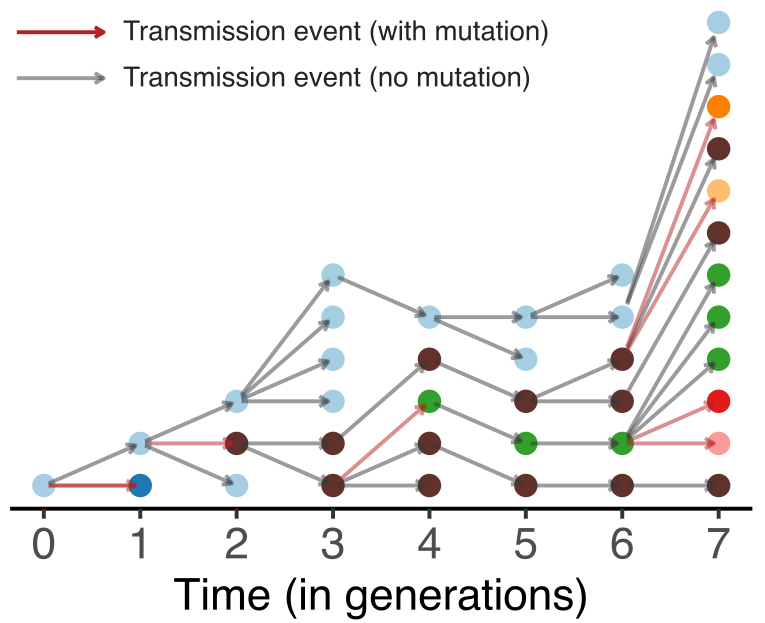
Model emergence, spread and dissolution of clusters of identical sequences
One mutation every ~13 days vs duration of infection of ~5 days
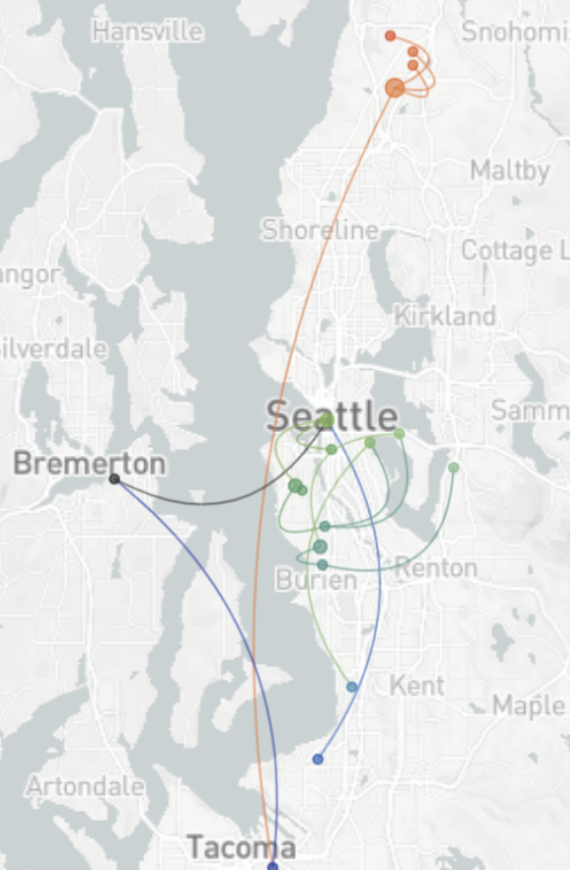
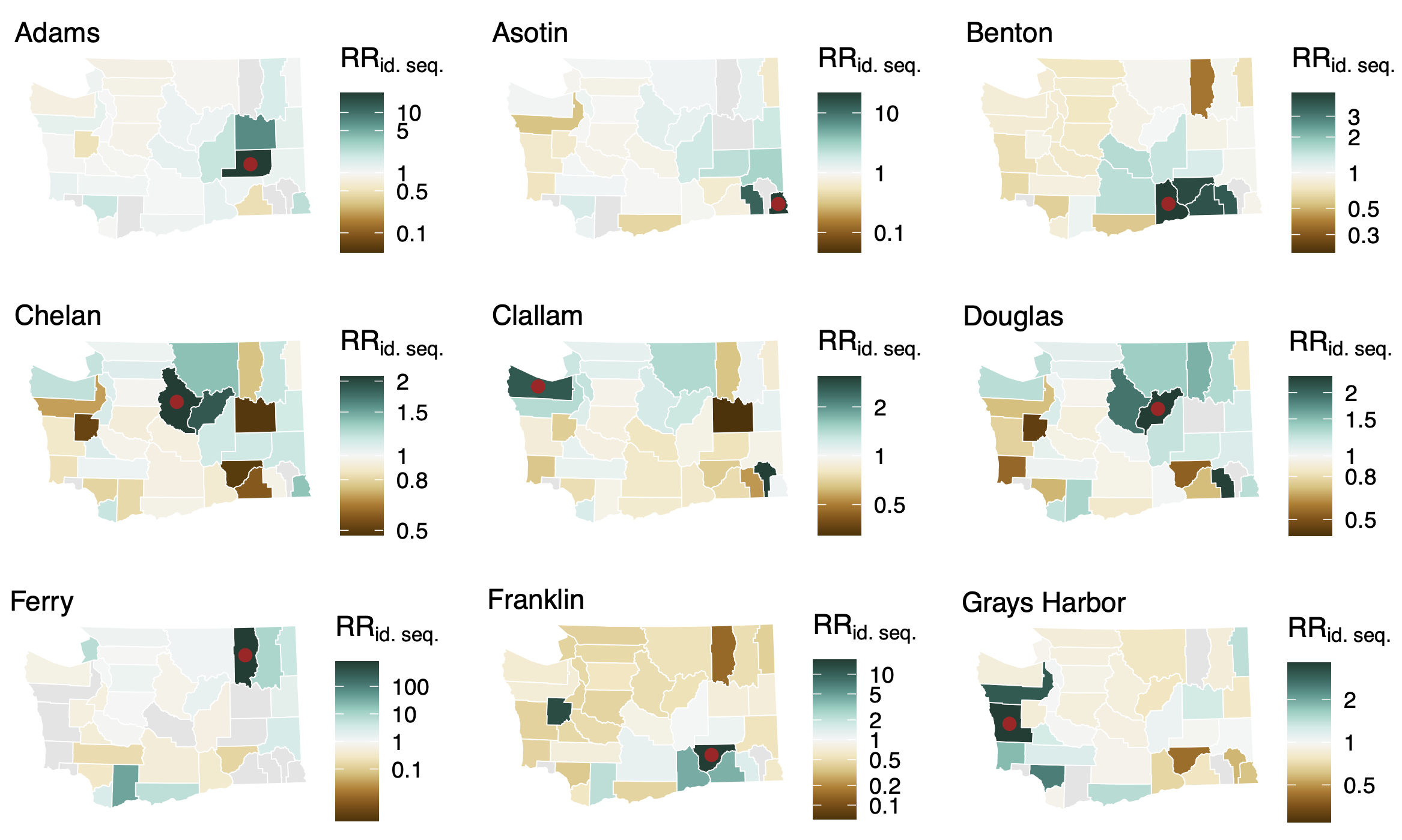
Spatial and social determinants of transmission from clusters of identical sequences
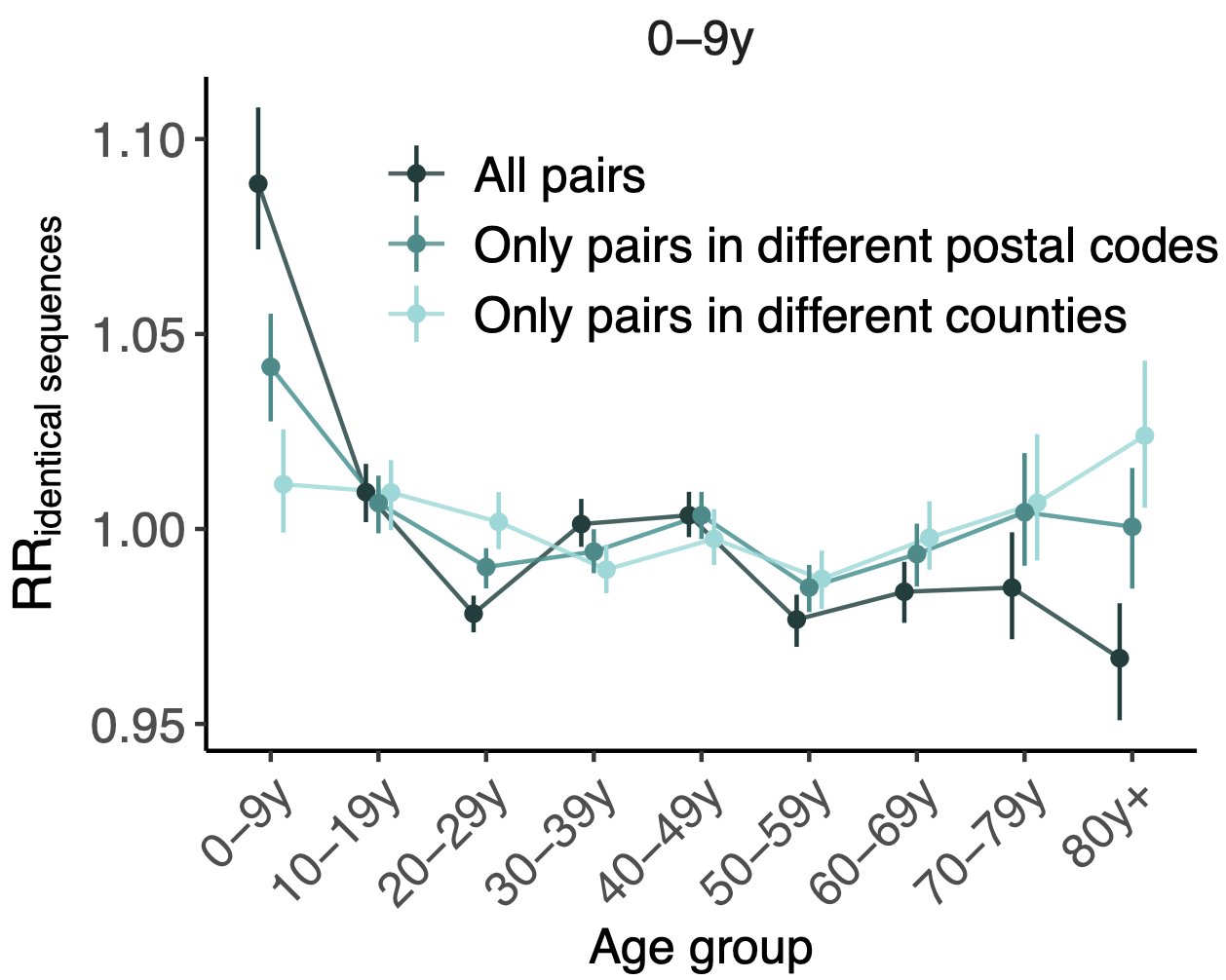
114k SARS-CoV-2 genomes from Washington State sentinel surveillance annotated with
geographic location and age
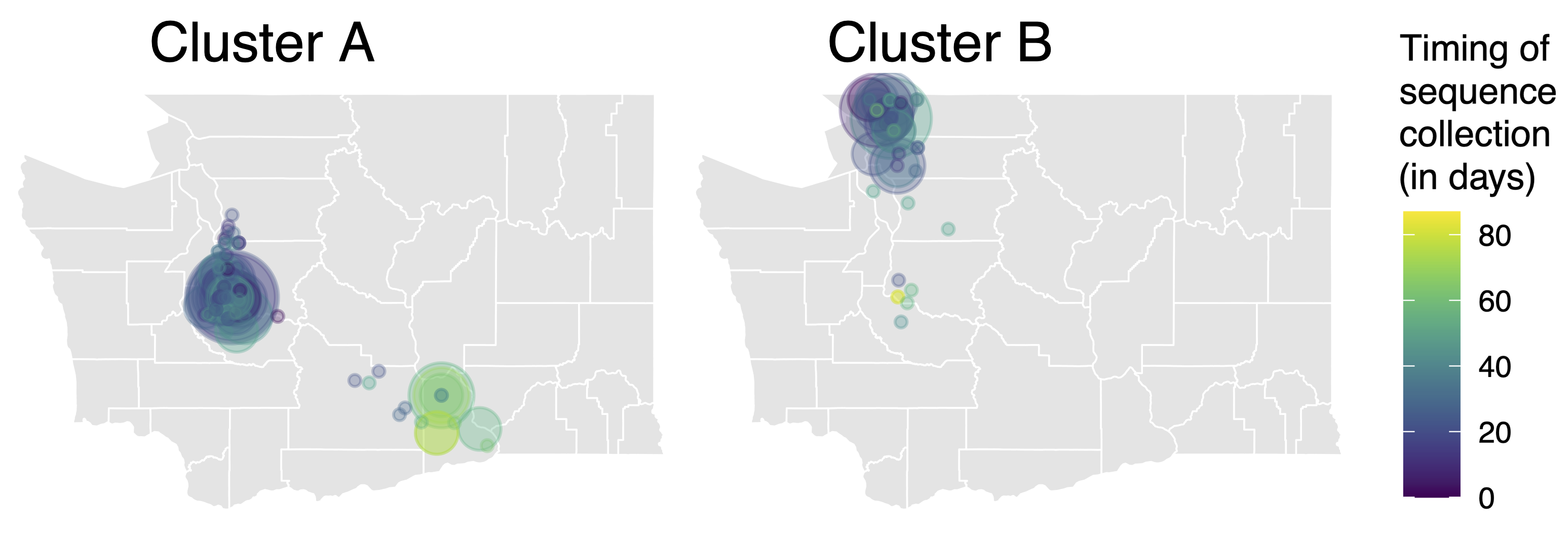
Pairs of identical sequences 5.1 times more likely to come from same county
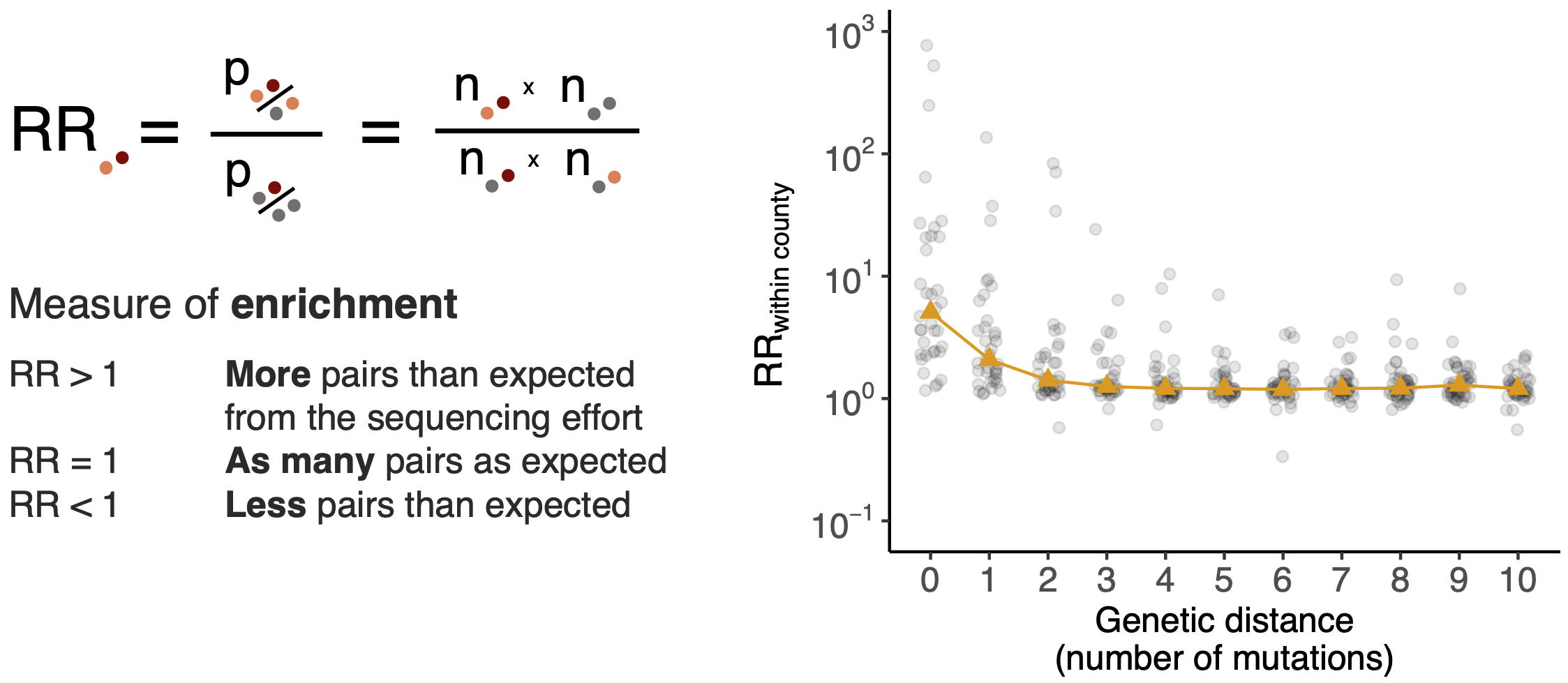
Between county enrichment corresponds well with geography
Between age group enrichment shows social mixing patterns
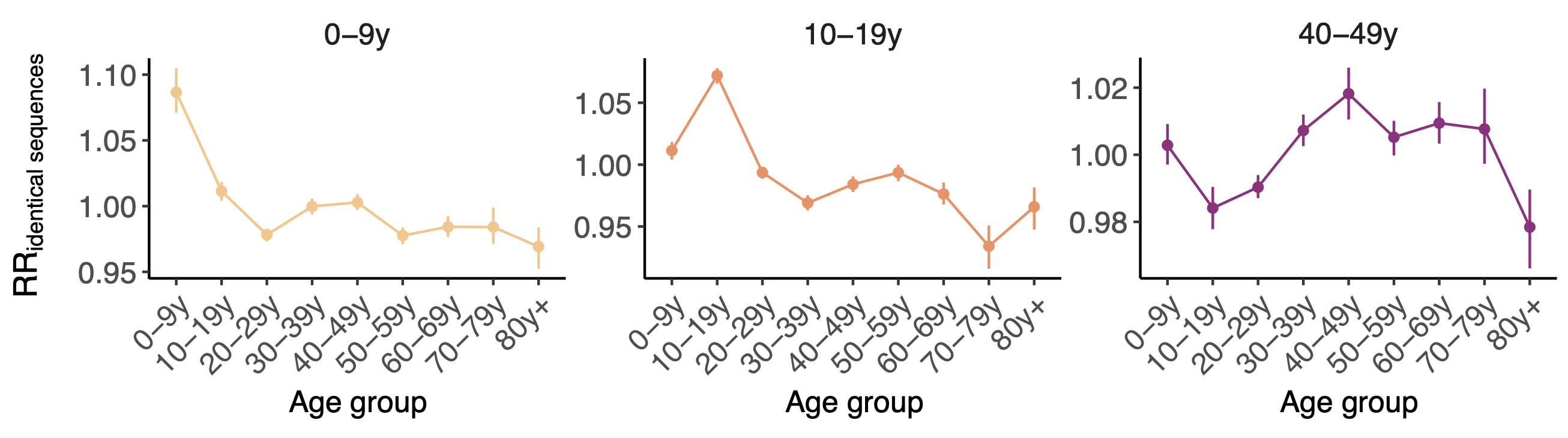
Signal for differential local vs long-distance age mixing
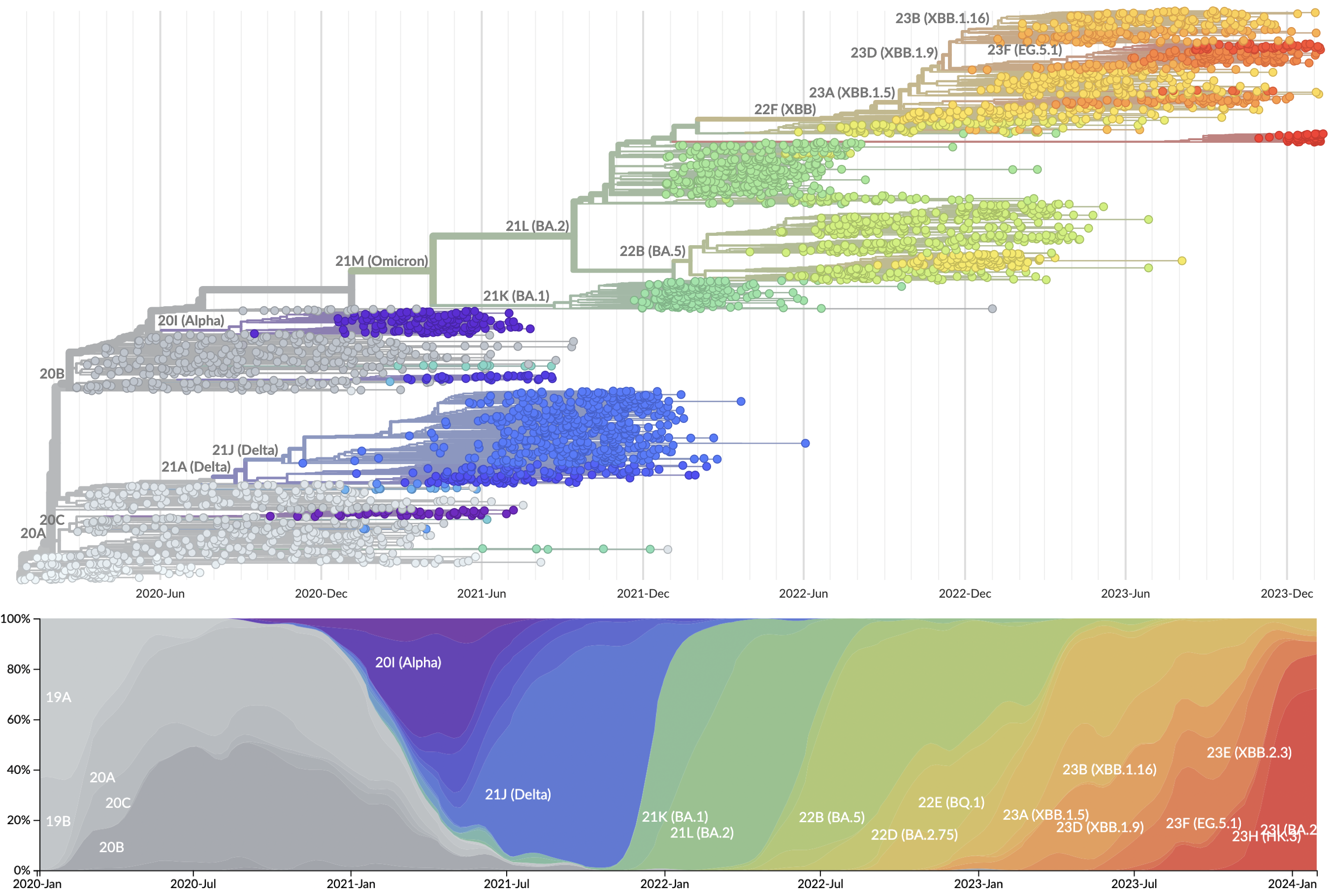
Evolutionary Forecasting
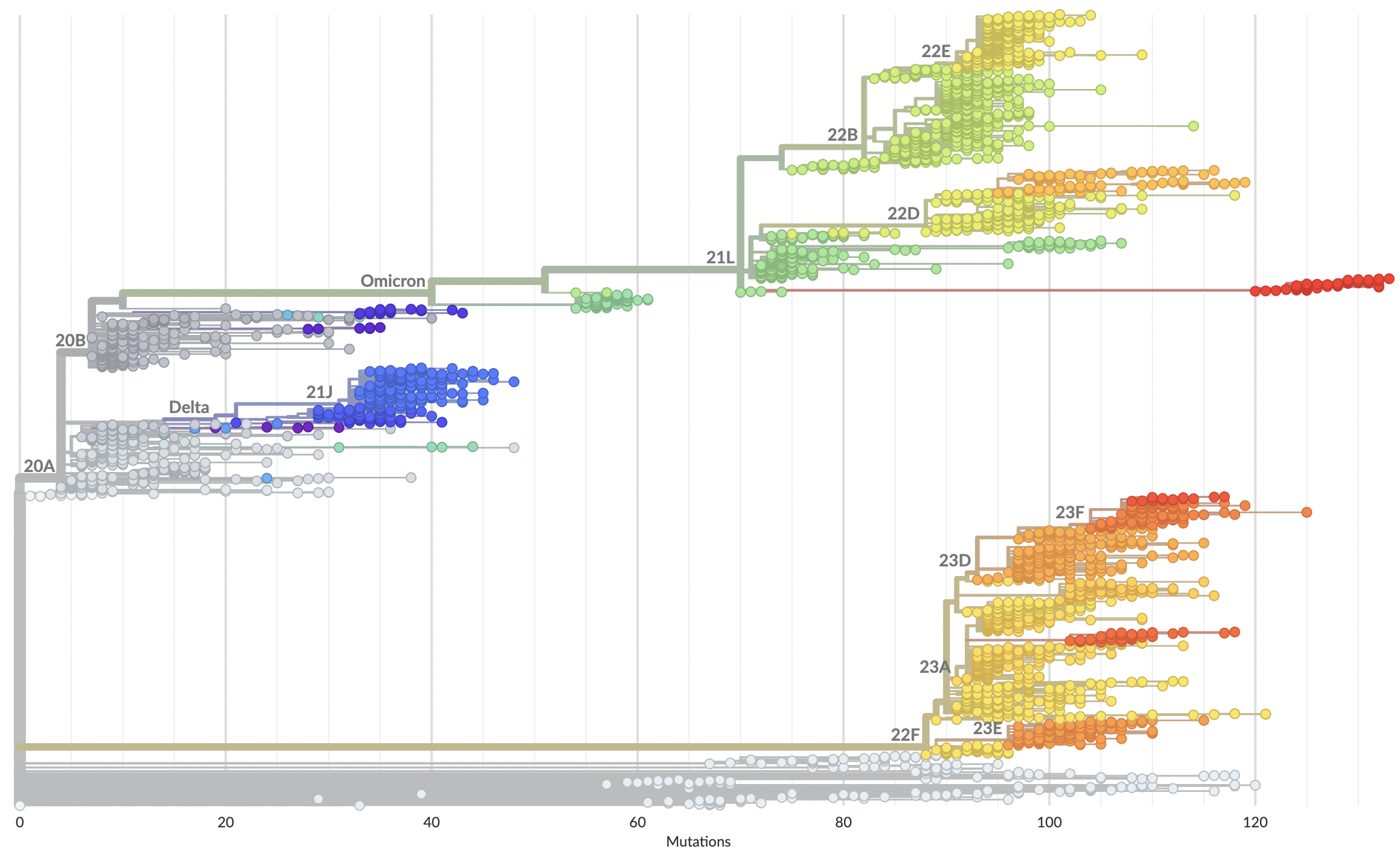
Recent advances
- Granular nomenclature and rapid classification
- Detailed frequency data allowing multinomial logistic regression
Currently 2857 Pango lineages and samples can be rapidly assigned lineages in Nextclade or UShER
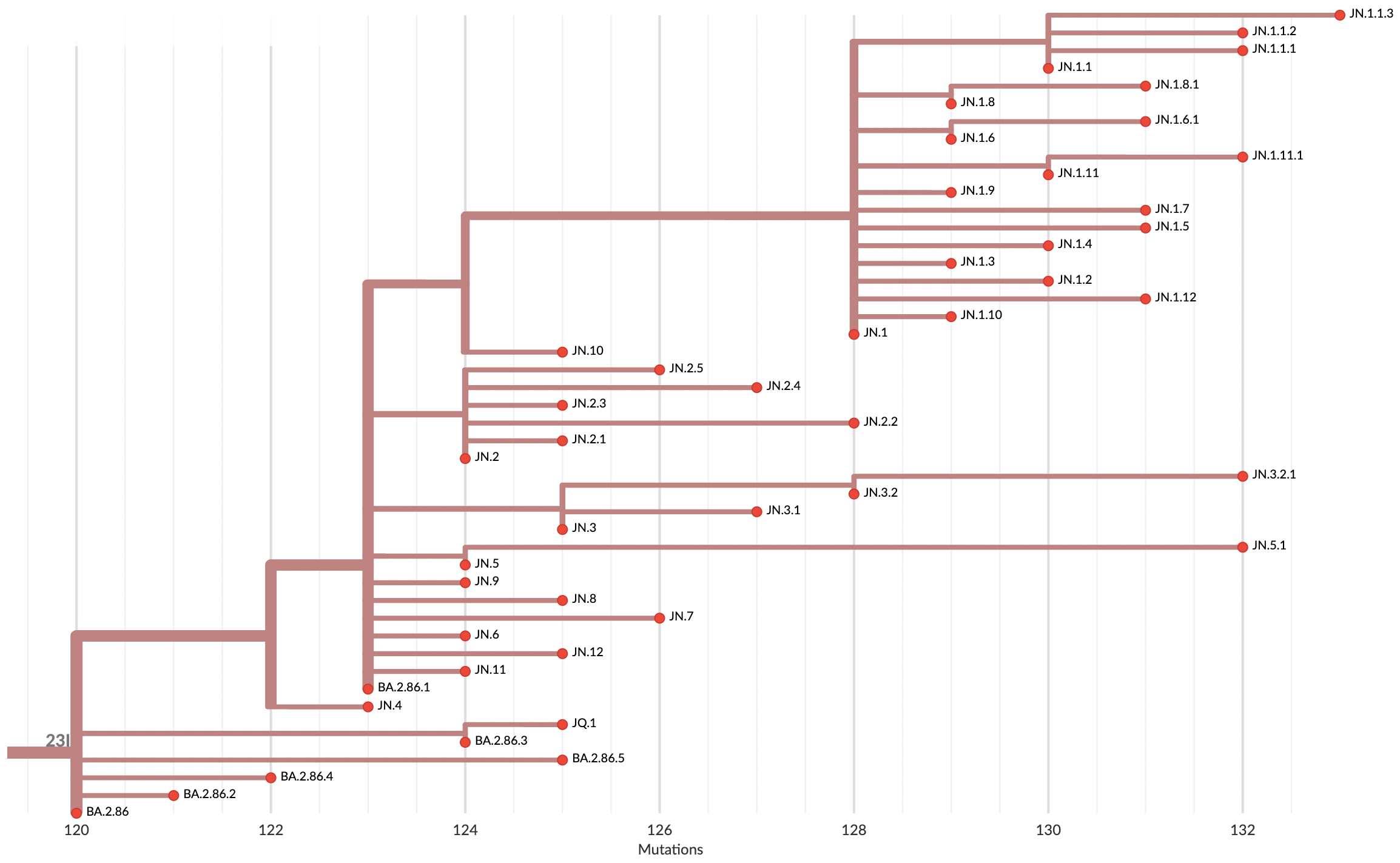
Variants that are just 1-2 mutations different will get a label
Multinomial logistic regression
Multinomial logistic regression across $n$ variants models the frequency $x$ of variant $i$ at time $t$ as
$$x_i(t) = \frac{p_i \, \mathrm{exp}(f_i \, t)}{\sum_{1 \le j \le n} p_j \, \mathrm{exp}(f_j \, t) }$$
estimating parameters for the initial frequency $p$ and the growth rate or fitness $f$.
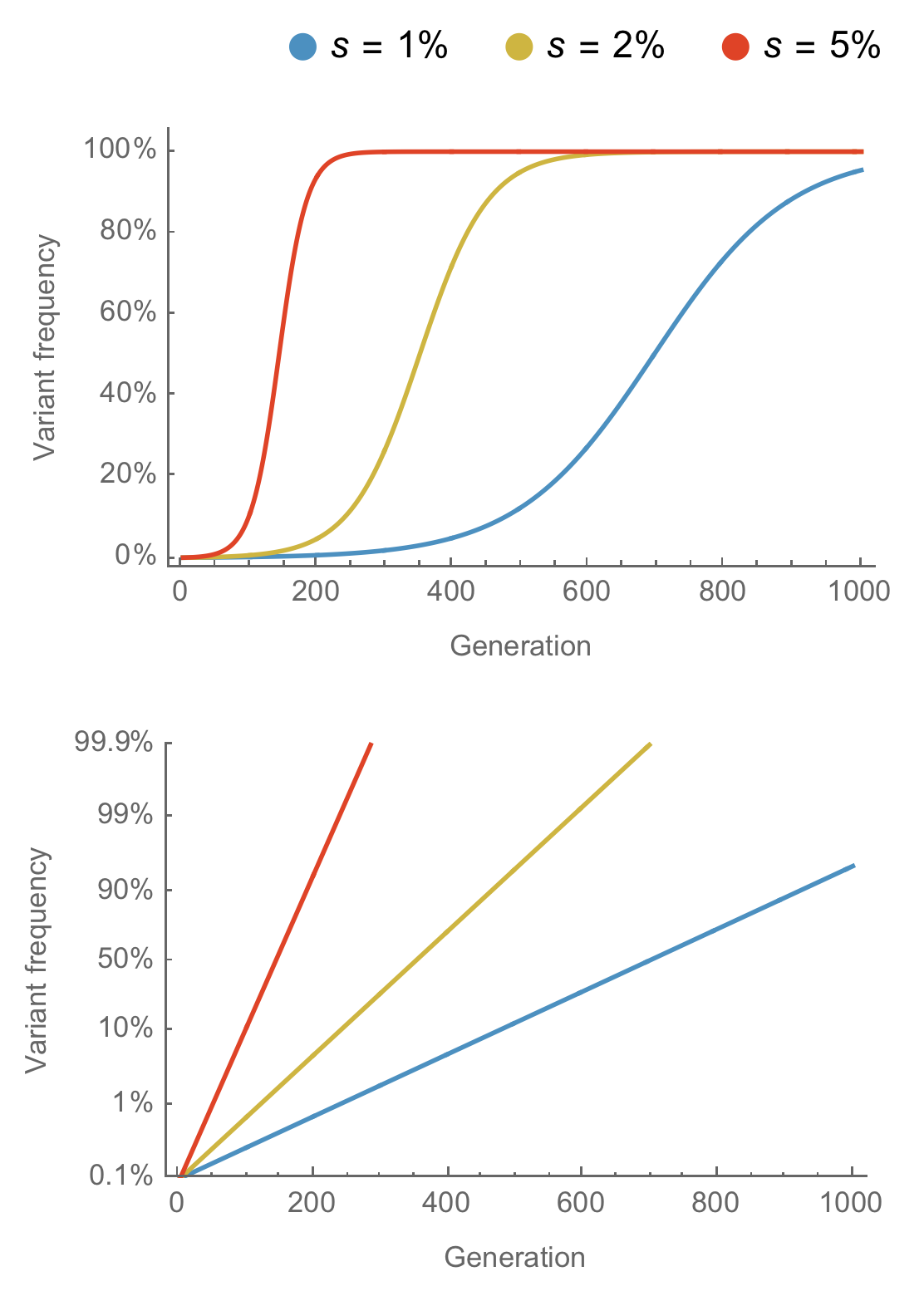
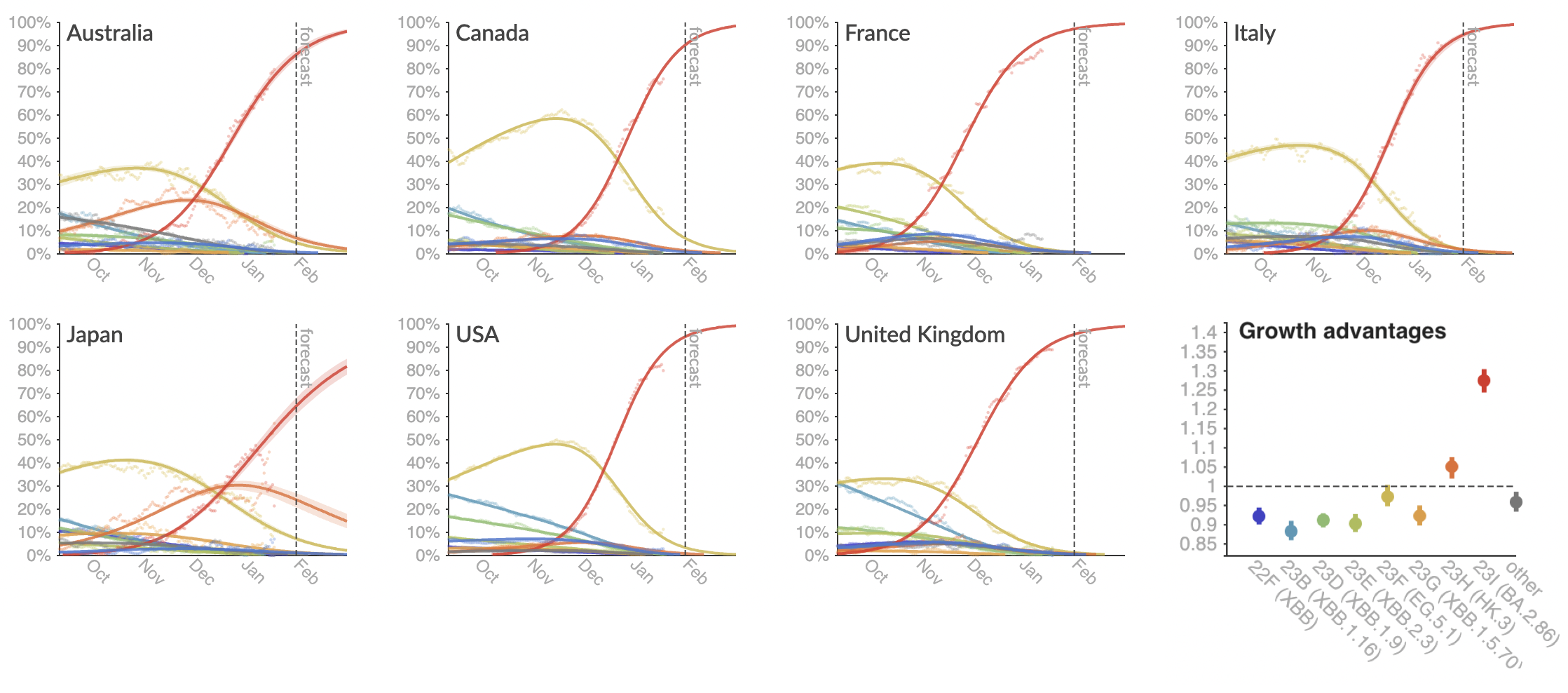
Provide continuously updated clade and lineage forecasts
The global sweep of JN.1 is now largely complete
Growth advantage readily identifiable while lineage is still rare
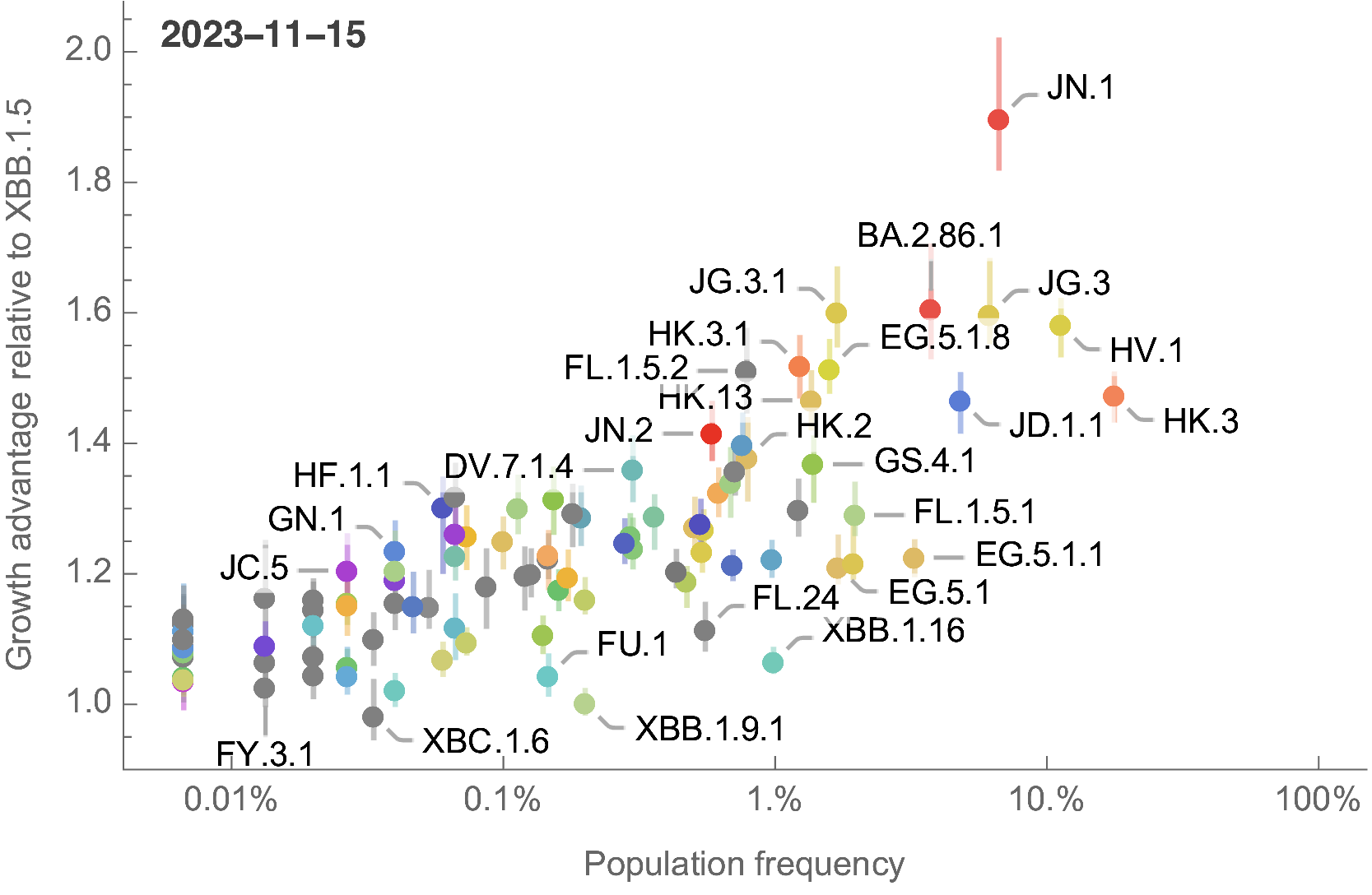
Growth advantage readily identifiable while lineage is still rare
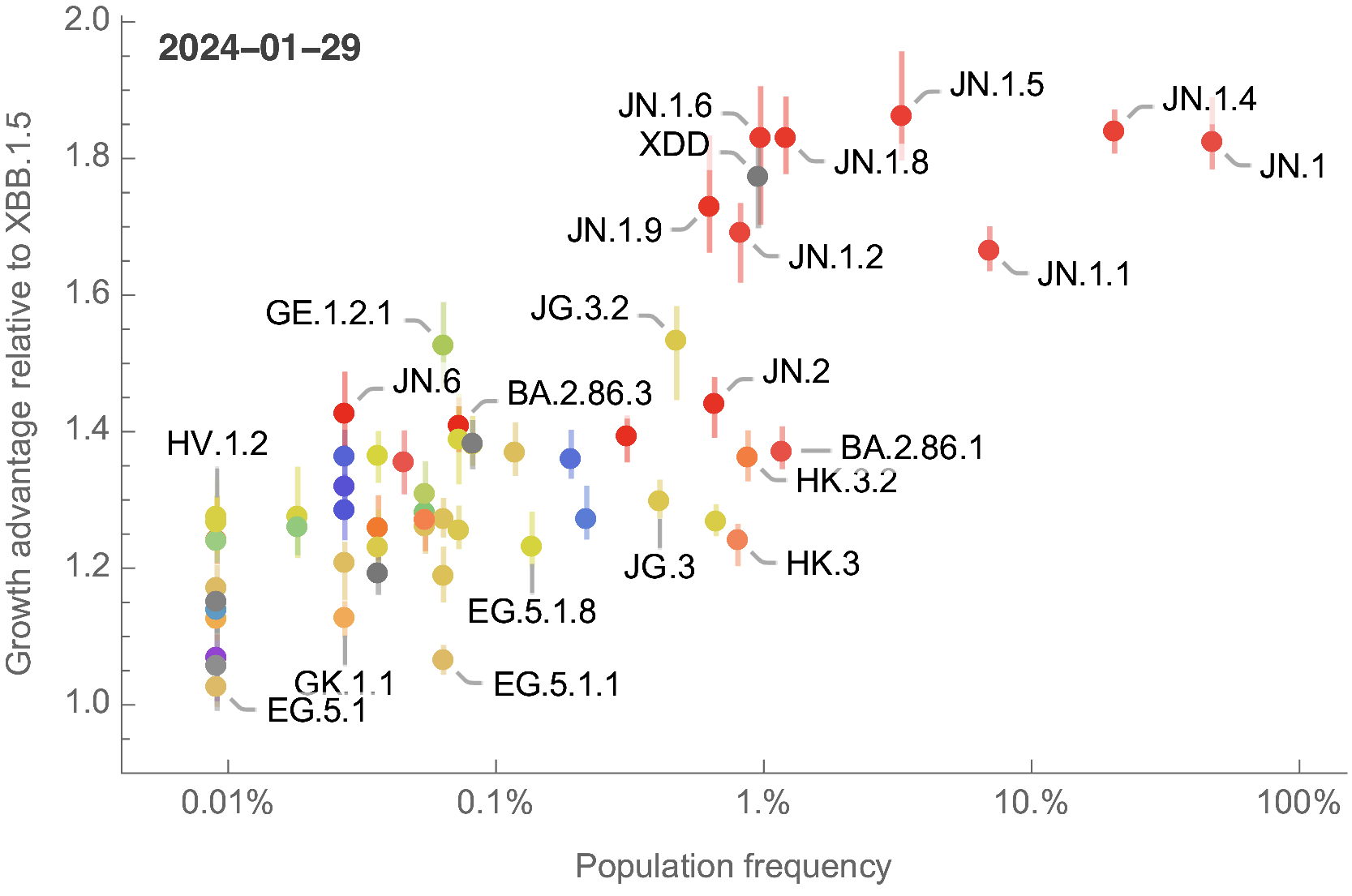
Ongoing work to lengthen prediction horizon by incorporating high-throughput experimental measurements of ACE2 binding and immune escape
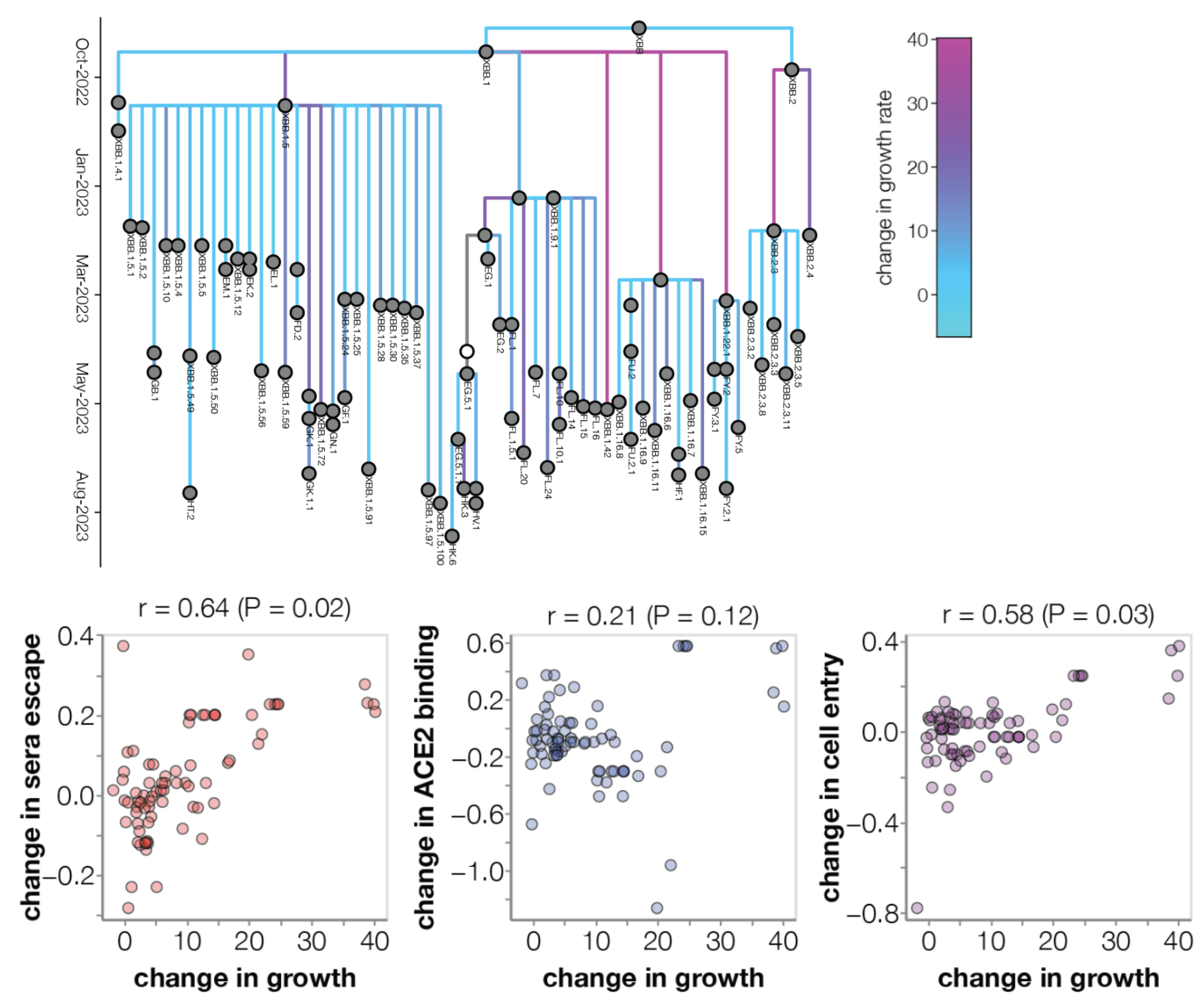
Lay ground-work for near ubiquitous pathogen sequencing
Acknowledgements
SARS-CoV-2 genomic epi: Data producers from all over the world and GISAID
Nextstrain: Richard Neher, Ivan Aksamentov, Kim Andrews, Jennifer Chang, James Hadfield, Emma Hodcroft, John Huddleston, Jover Lee, Victor Lin, Cornelius Roemer, Thomas Sibley
Determinants of transmission: Cécile Tran Kiem, Amanda Perofsky, Miguel Paredes, Lauren Frisbie, Allison Black, Cécile Viboud
Evolutionary forecasting: Marlin Figgins, Jover Lee, James Hadfield, John Huddleston, Cornelius Roemer, Richard Neher
Bedford Lab:
![]() John Huddleston,
John Huddleston,
![]() James Hadfield,
James Hadfield,
![]() Katie Kistler,
Katie Kistler,
![]() Thomas Sibley,
Thomas Sibley,
![]() Jover Lee,
Jover Lee,
![]() Cassia Wagner,
Cassia Wagner,
![]() Miguel Paredes,
Miguel Paredes,
![]() Nicola Müller,
Nicola Müller,
![]() Marlin Figgins,
Marlin Figgins,
![]() Victor Lin,
Victor Lin,
![]() Jennifer Chang,
Jennifer Chang,
![]() Eslam Abousamra,
Eslam Abousamra,
![]() Nashwa Ahmed,
Nashwa Ahmed,
![]() Cécile Tran Kiem,
Cécile Tran Kiem,
![]() Kim Andrews
Kim Andrews