Genomic epidemiology of SARS-CoV-2
Trevor Bedford (@trvrb)
Oct 28, 2021
MCB532 Human Pathogenic Viruses
Sequencing to reconstruct pathogen spread
Epidemic process
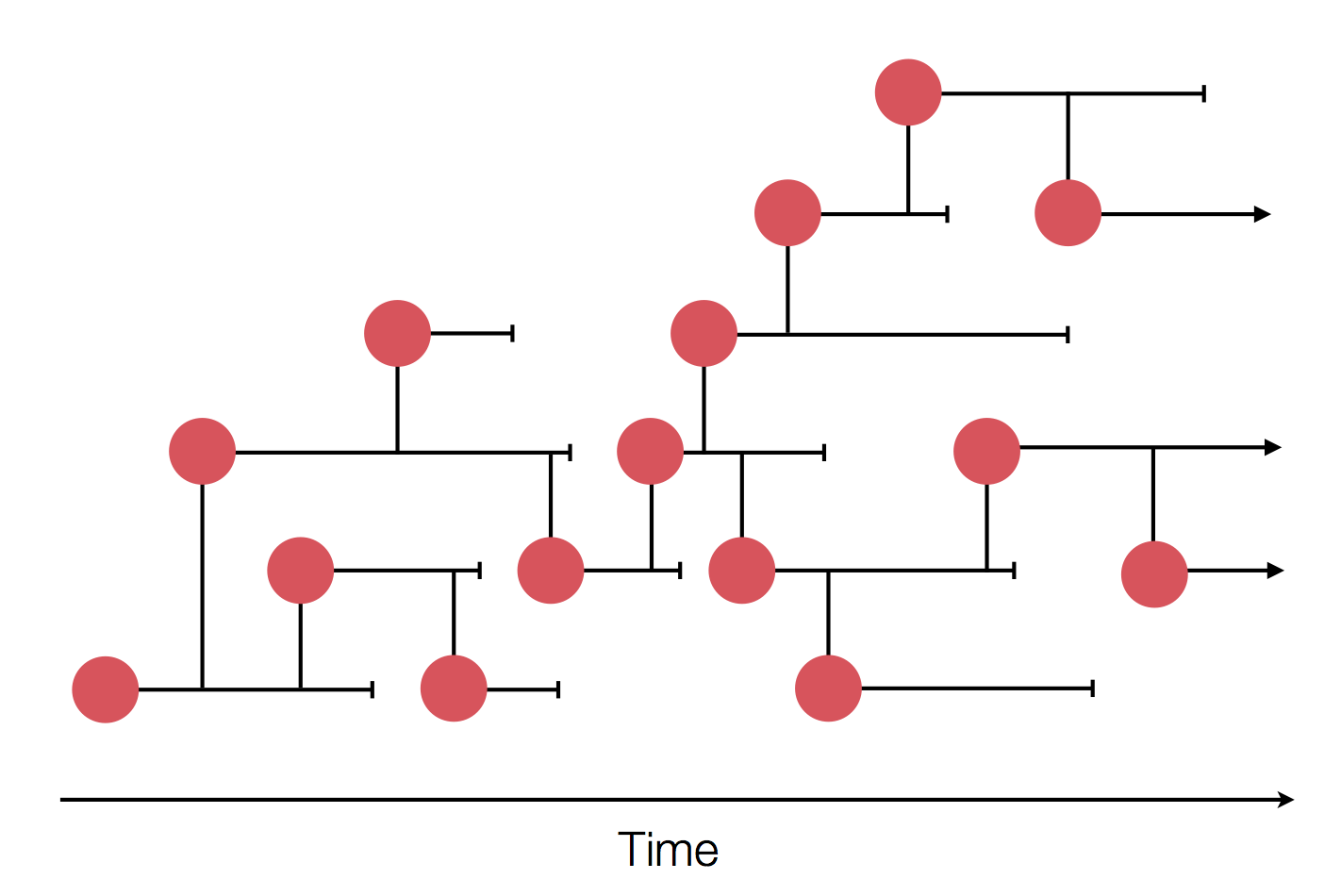
Sample some individuals
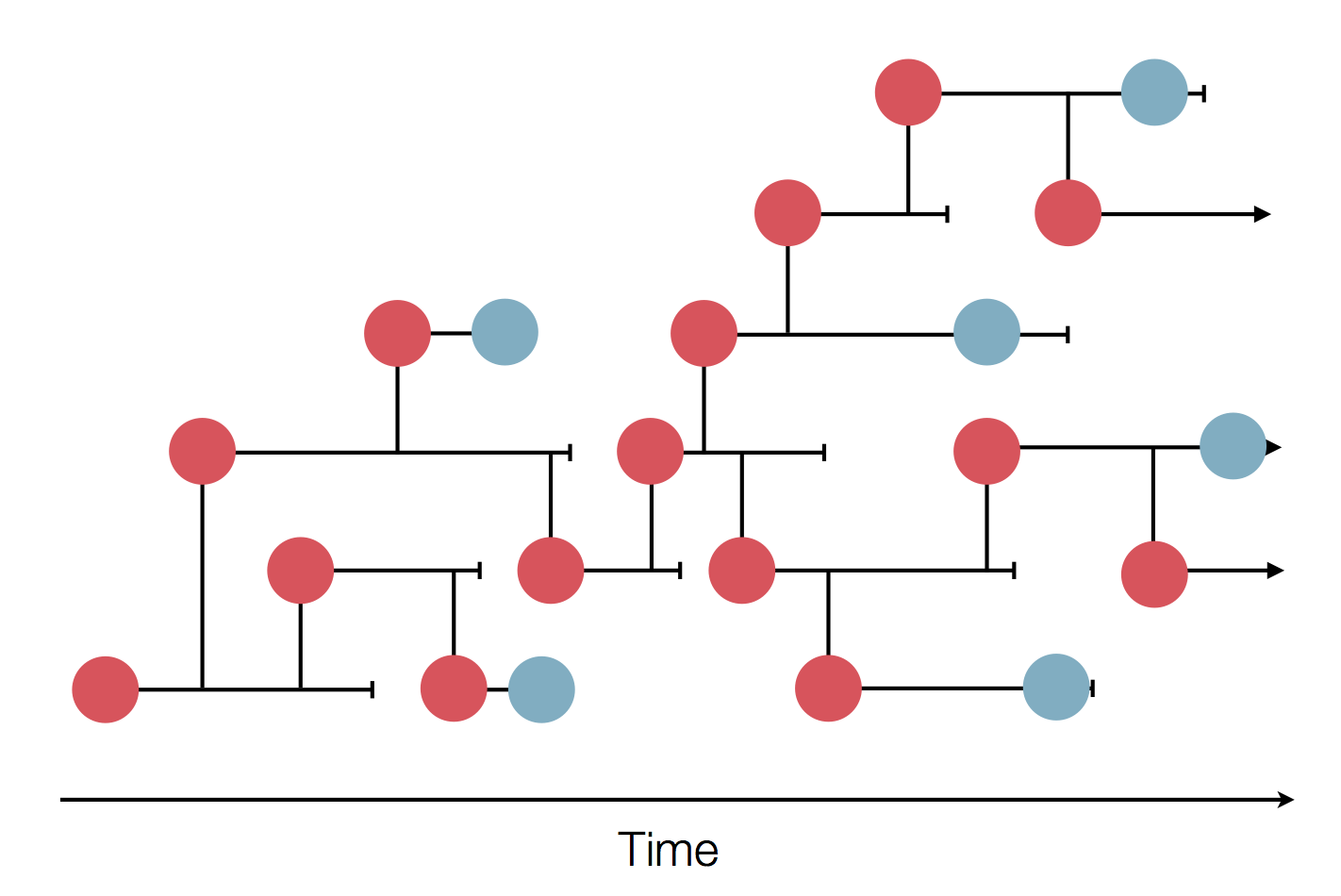
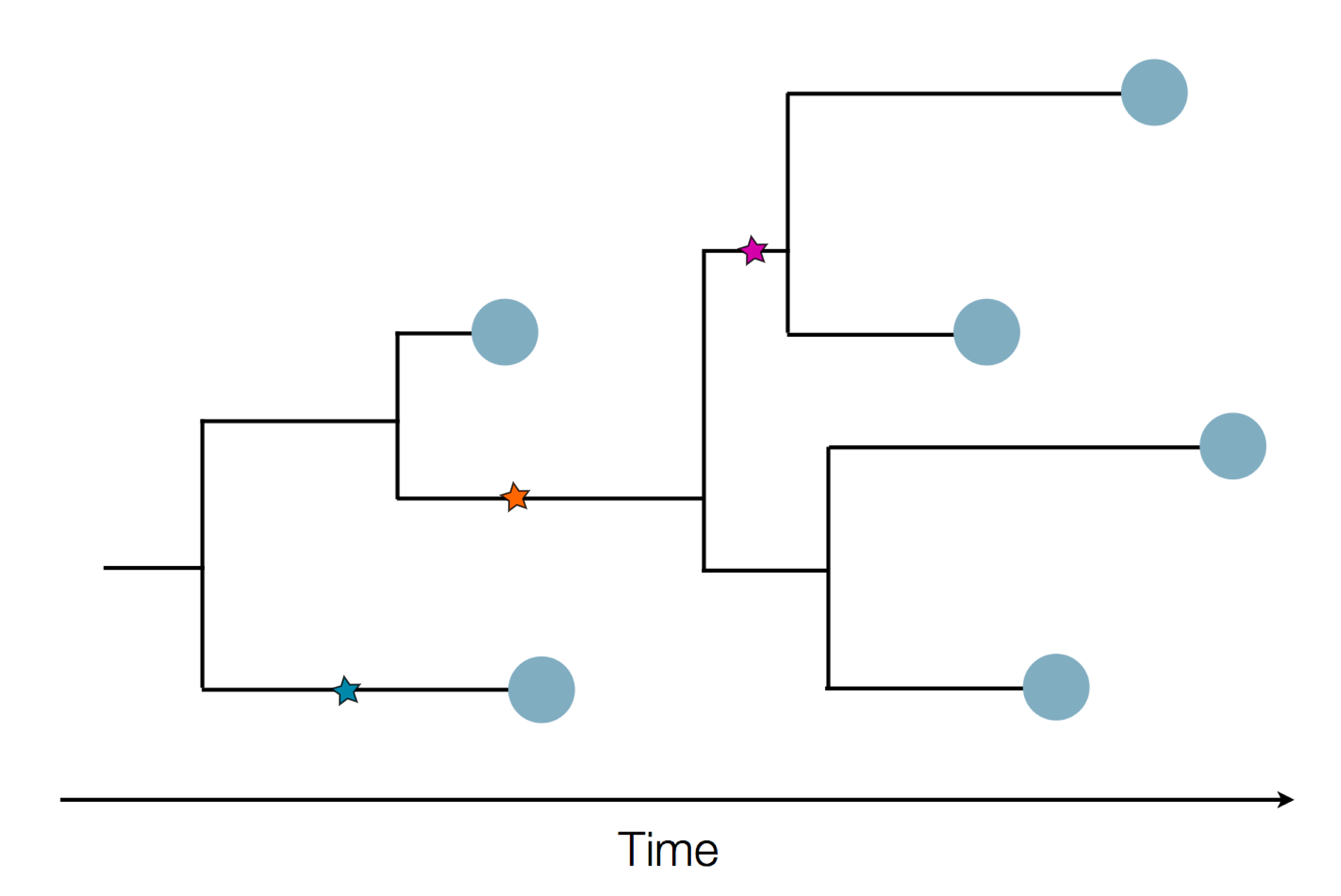
Sequence and determine phylogeny
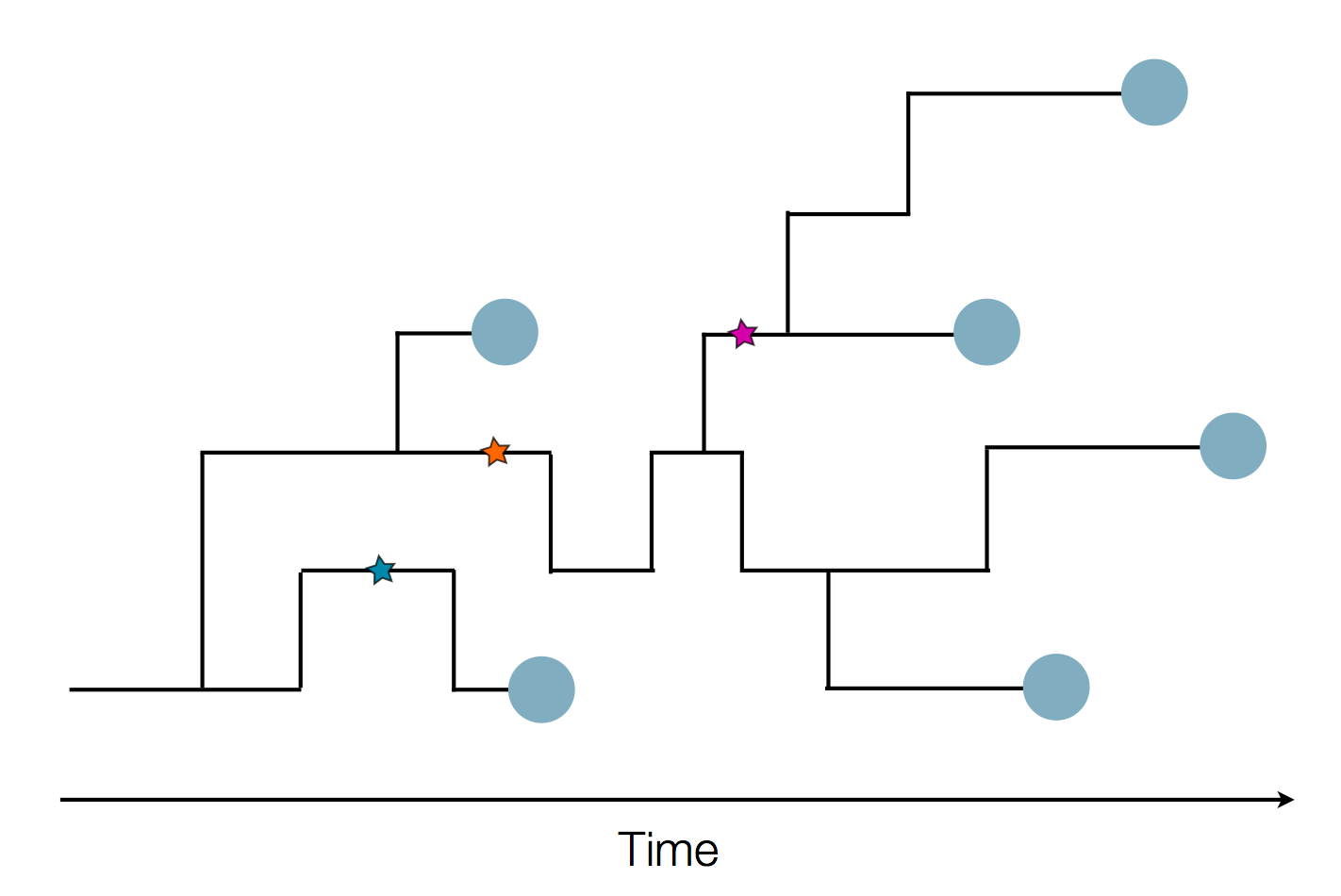
Sequence and determine phylogeny
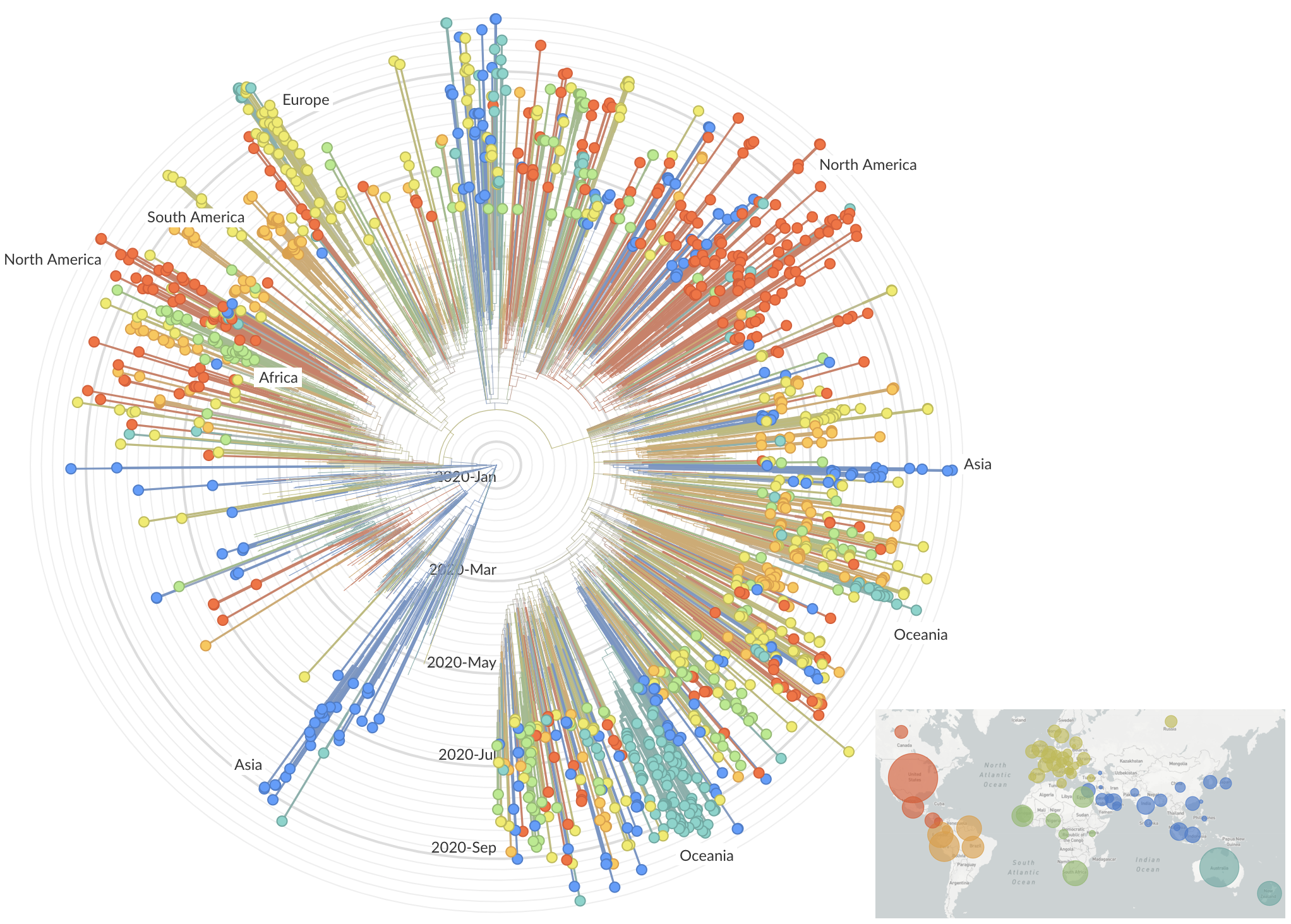
Genomic epidemiology during the COVID-19 pandemic
>4.6M SARS-CoV-2 genomes shared to GISAID with 630k genomes in Aug 2021 alone
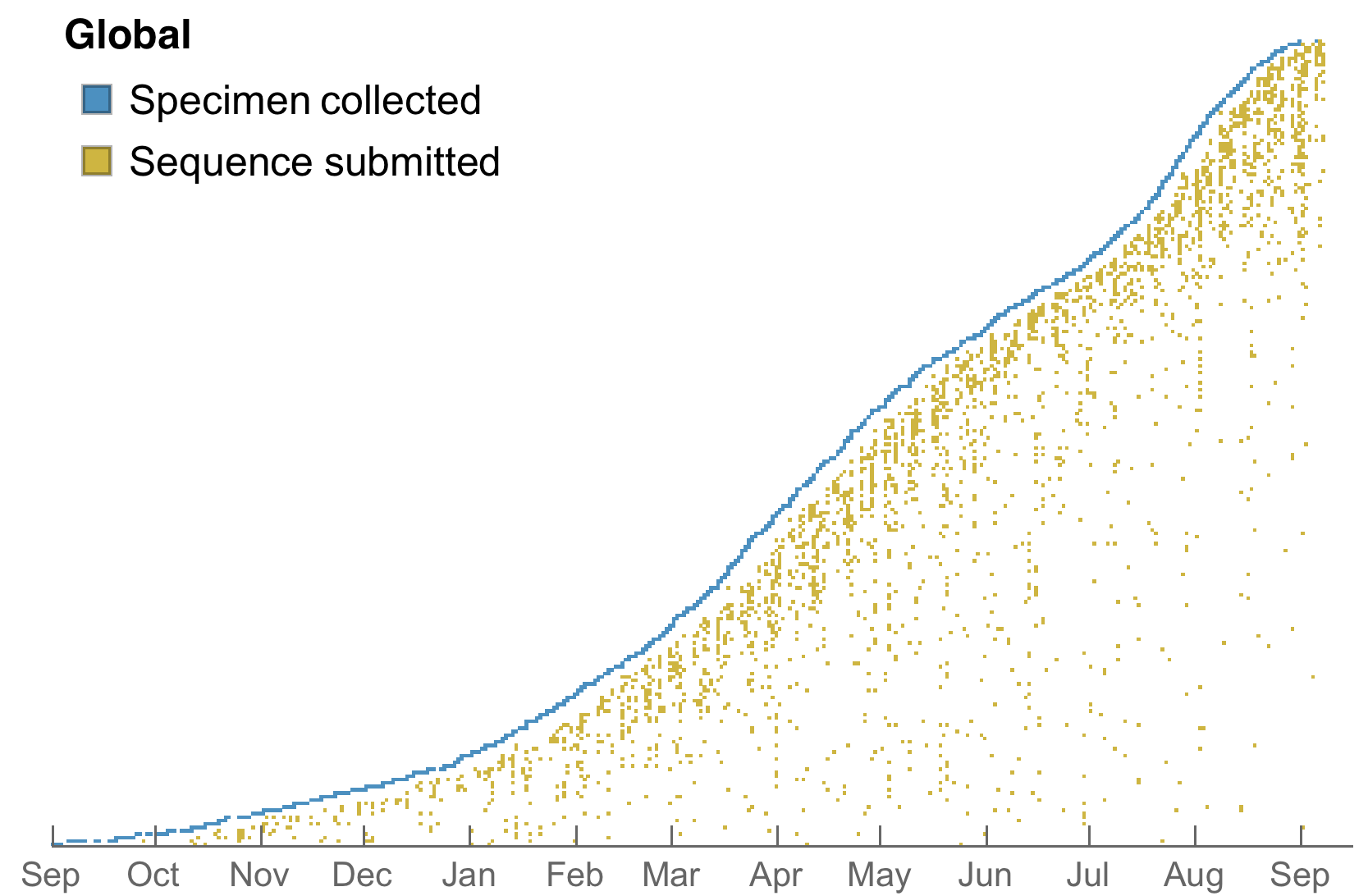
Data from gisaid.org
Three key insights that genomic epi provided during pandemic and concepts that underlie these insights
- Rapid human-to-human spread in Wuhan beyond initial market outbreak (molecular clocks)
- Extensive local transmission while testing was rare (phylogeography)
- Identification of variants of concern and mapping of increased transmission rates (adaptive evolution)
Molecular clocks and early human-to-human spread
Mutations tend to accumulate in a clock-like fashion
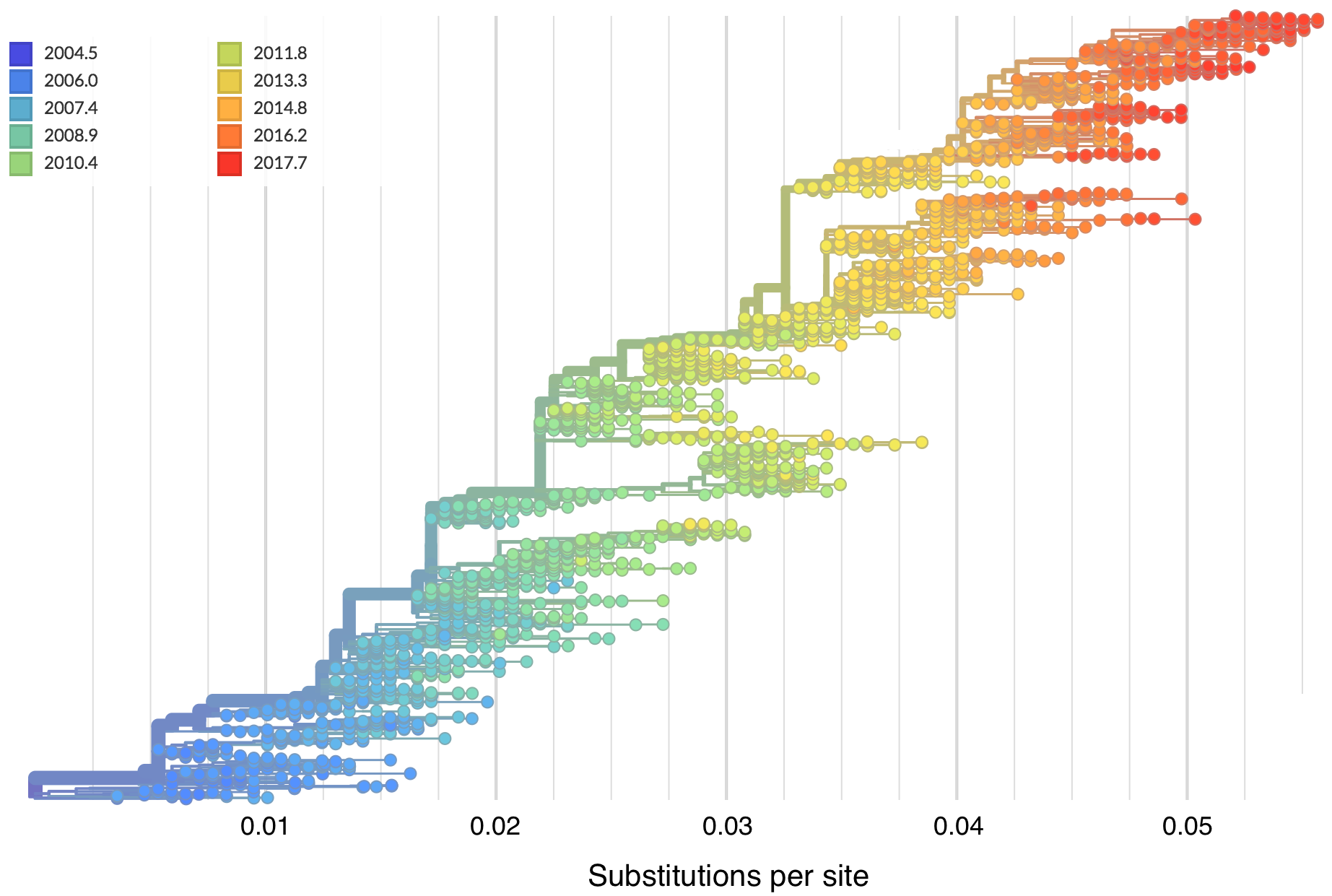
"Root-to-tip" plots show temporal signal
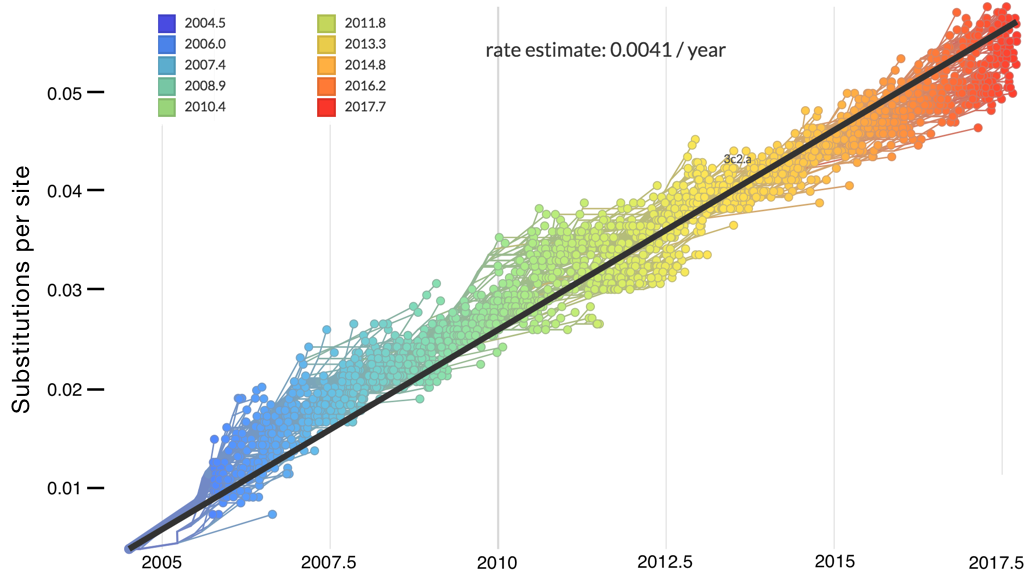
Allows conversion between branch length and time
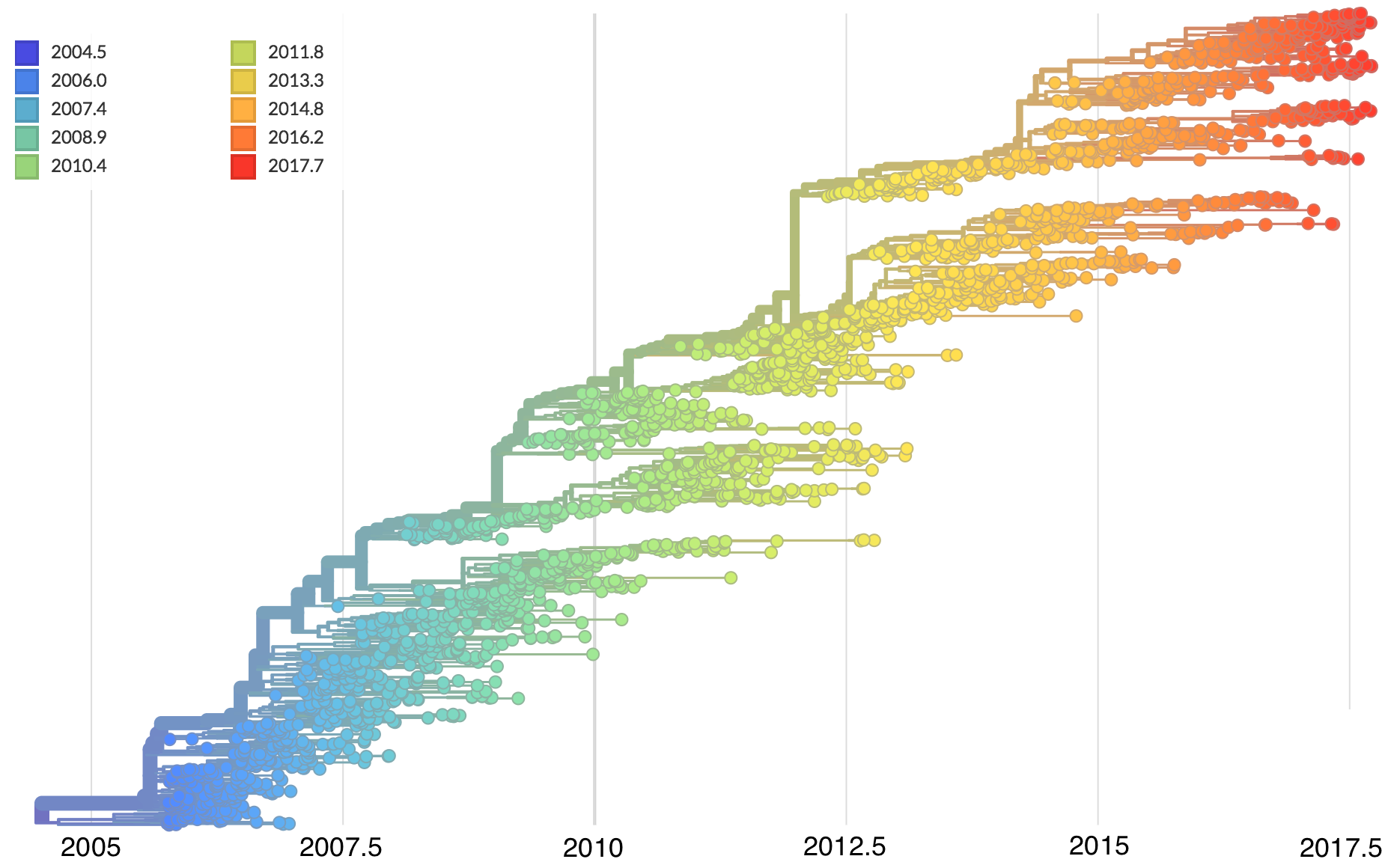
Jan 11: First five genomes from Wuhan showed a novel SARS-like coronavirus
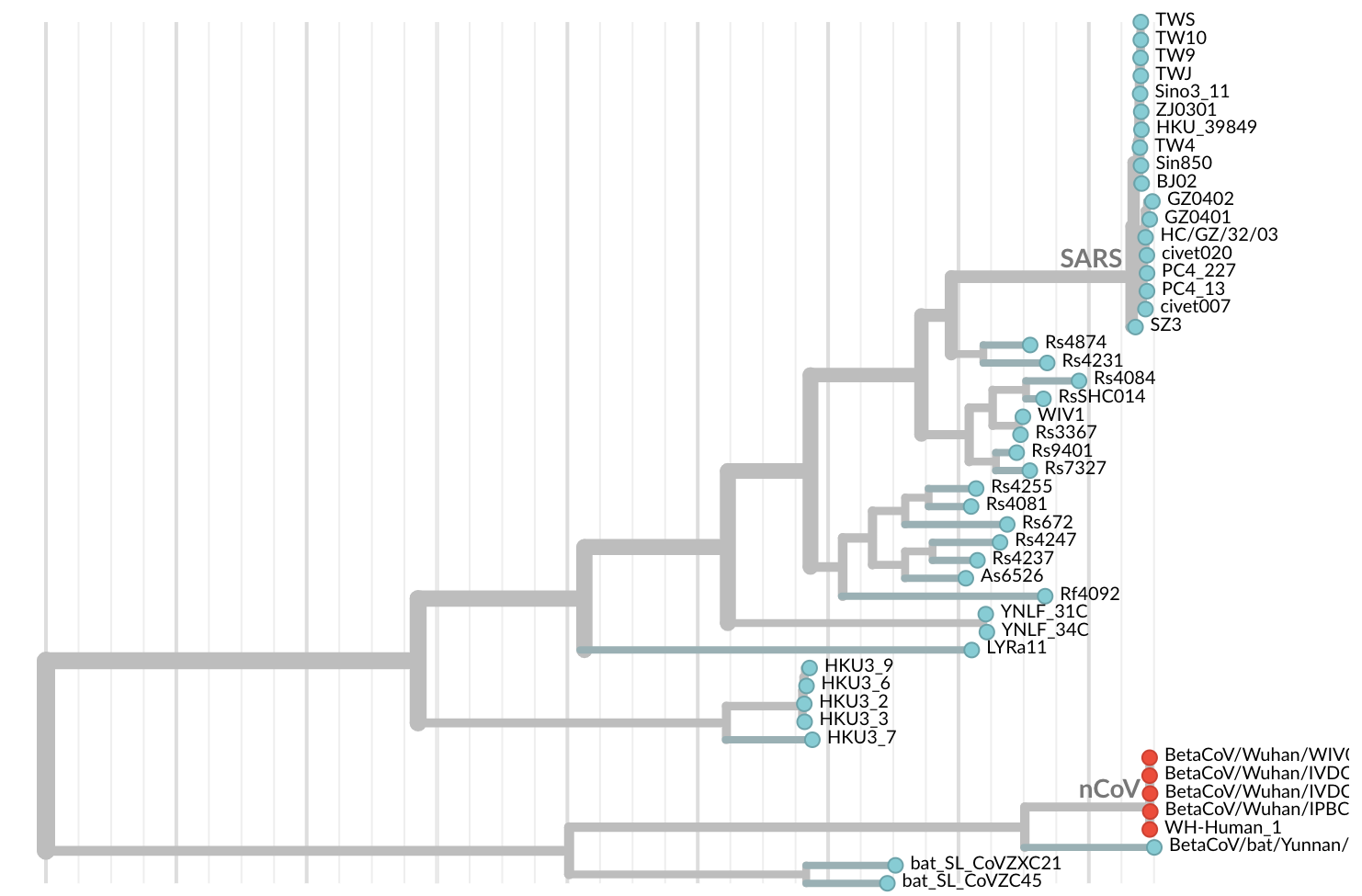
Jan 19: First 12 genomes from Wuhan and Bangkok showed lack of genetic diversity
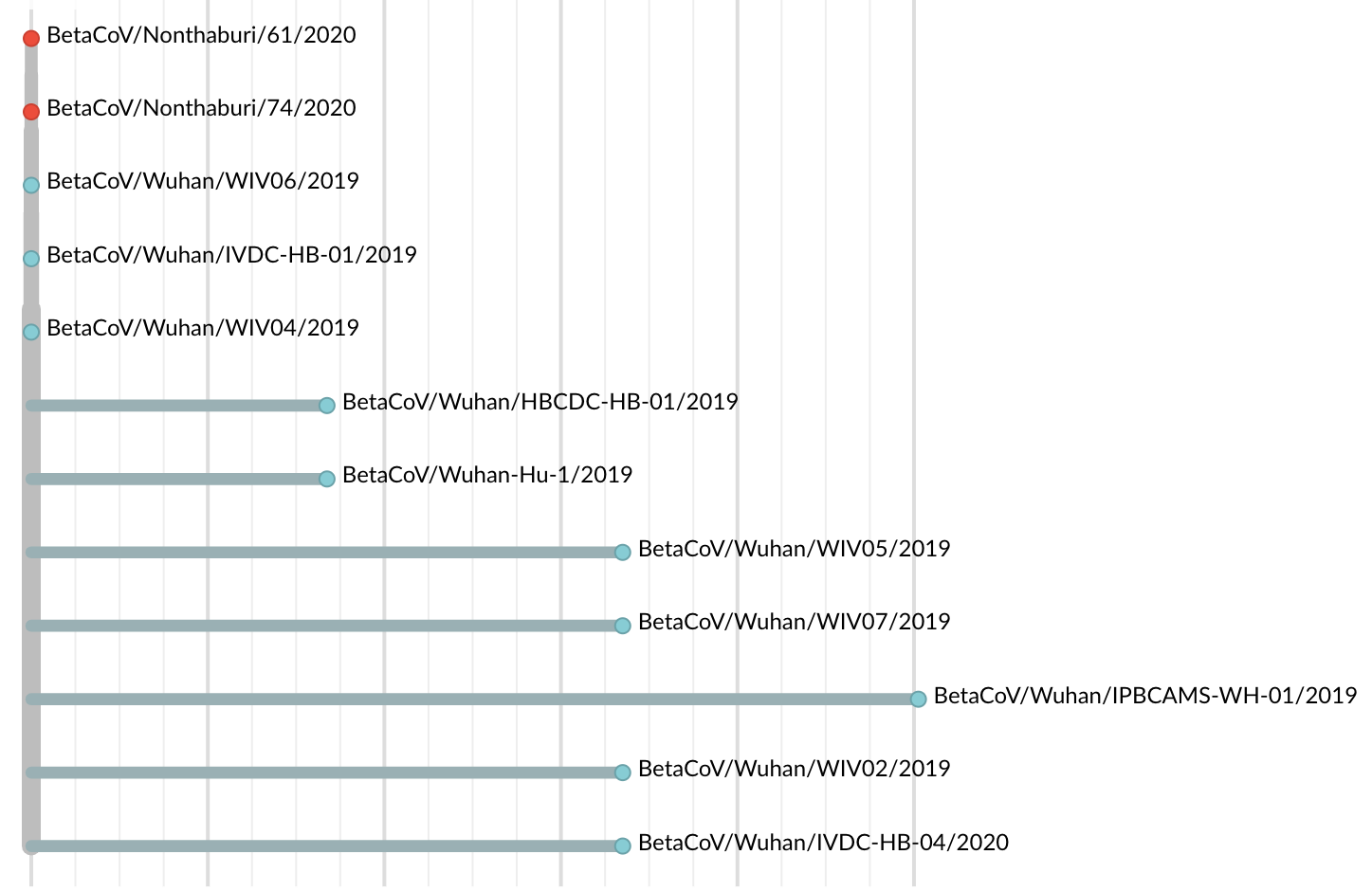
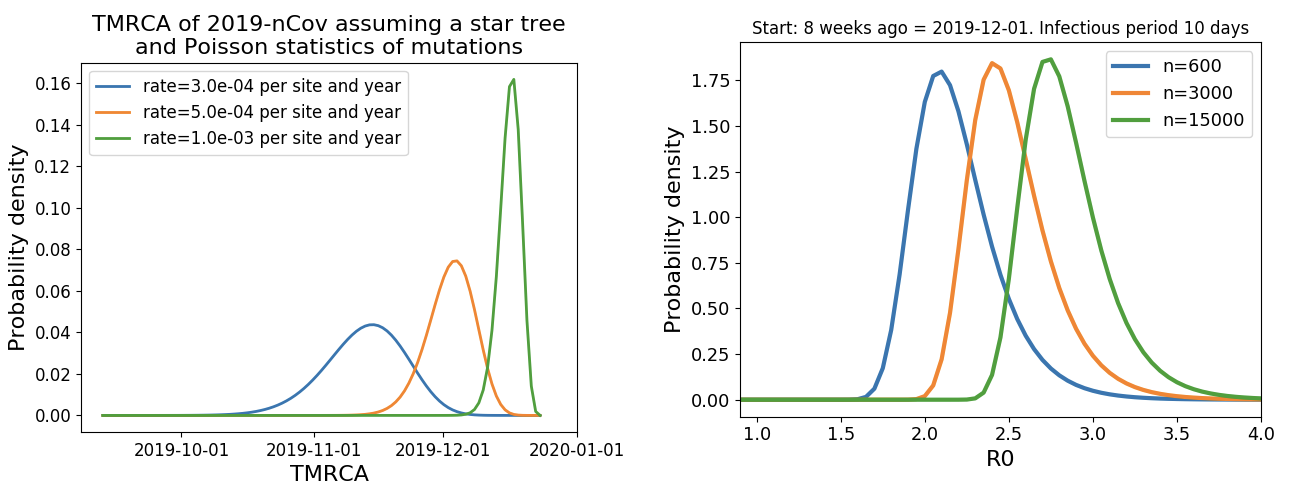
Single introduction into the human population between Nov 15 and Dec 15 and subsequent rapid human-to-human spread
Phylogeography and inferring local transmission
"Data" is a phylogeny and tip states
States include nucleotides, amino acids, geo locations, hosts, etc...
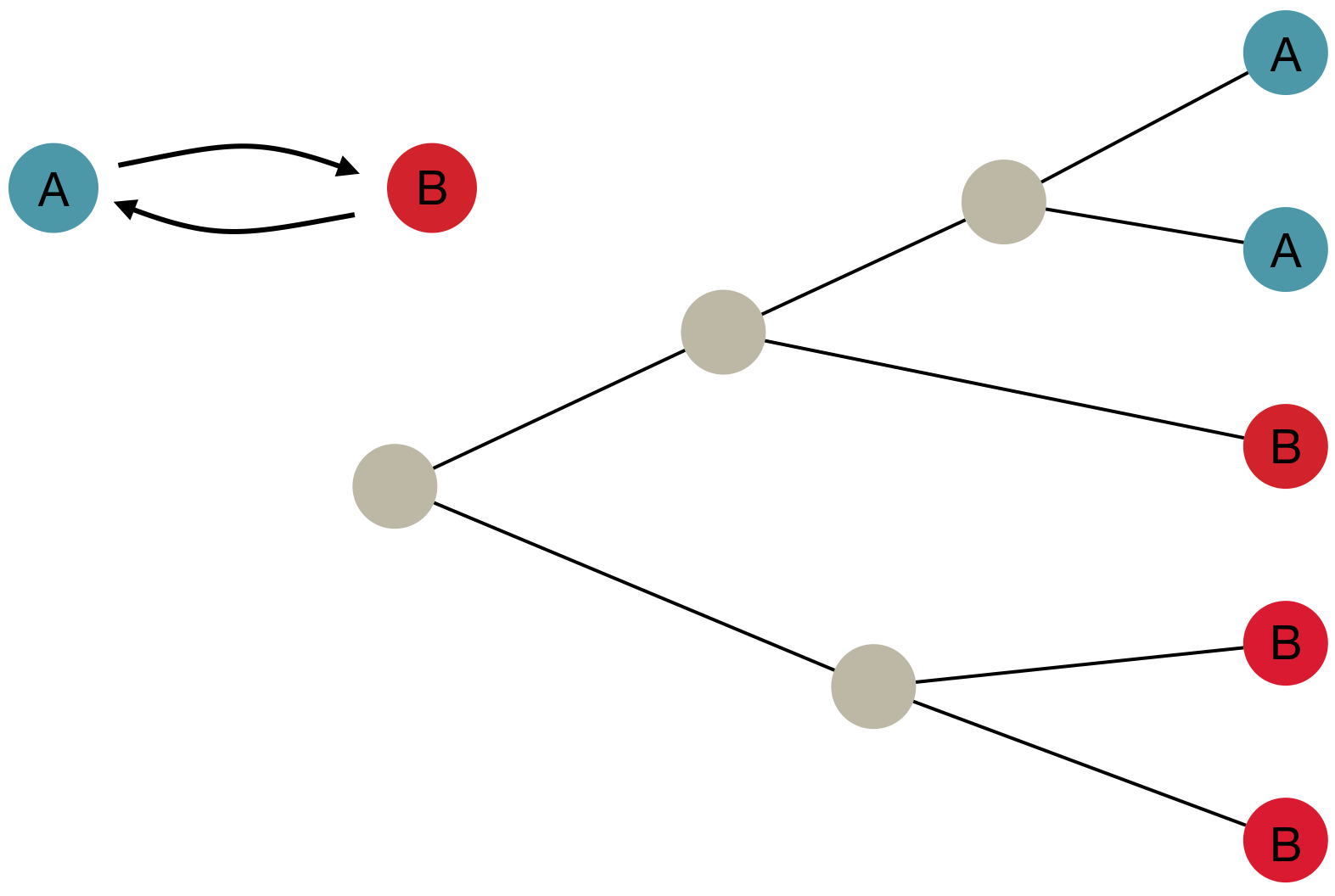
Model infers transition matrix and ancestral states
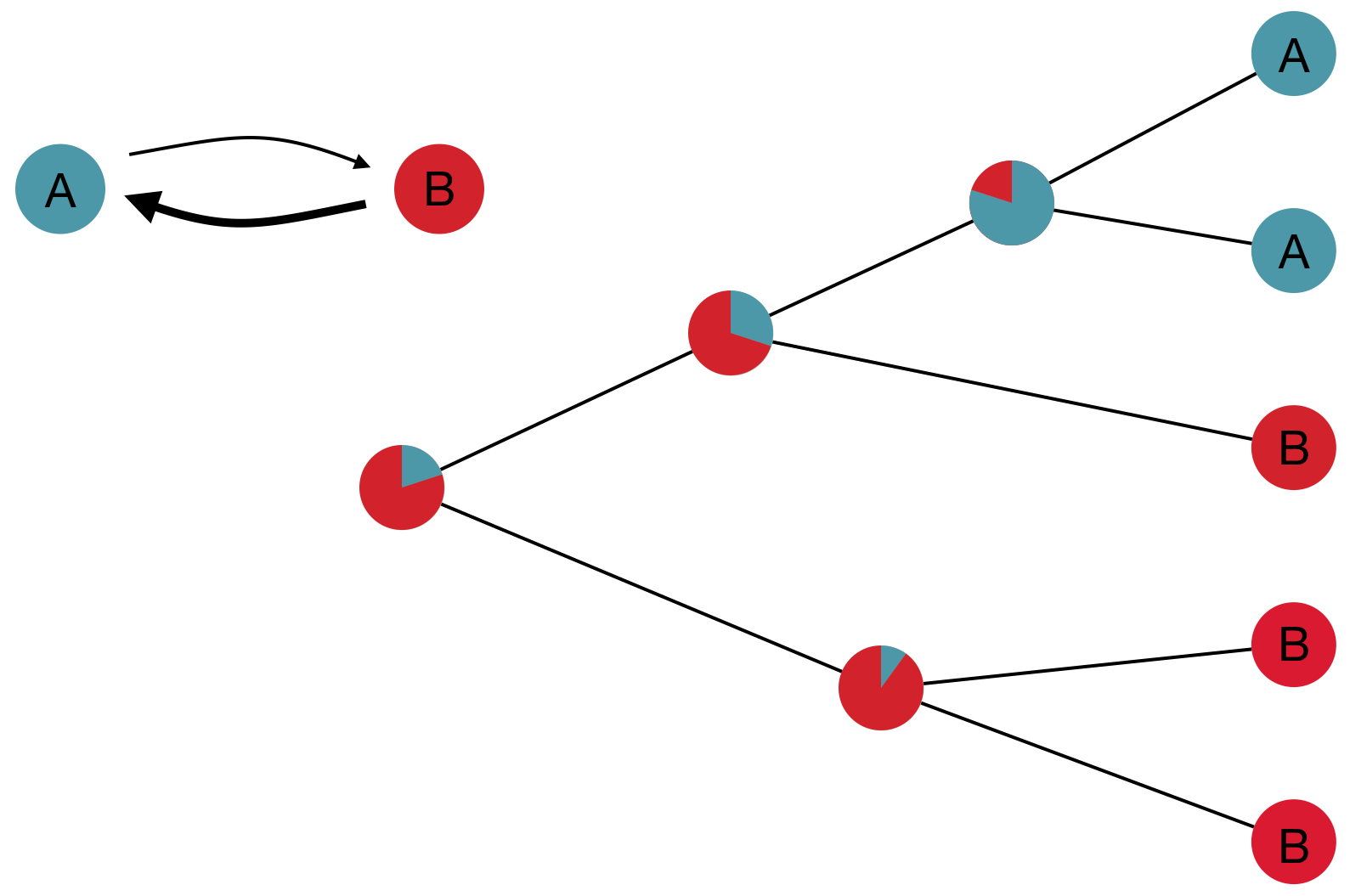
Rare transitions, short branches and many taxa increase confidence
Rapid global epidemic spread from China
Epidemic in the USA was introduced from China in late Jan and from Europe during Feb
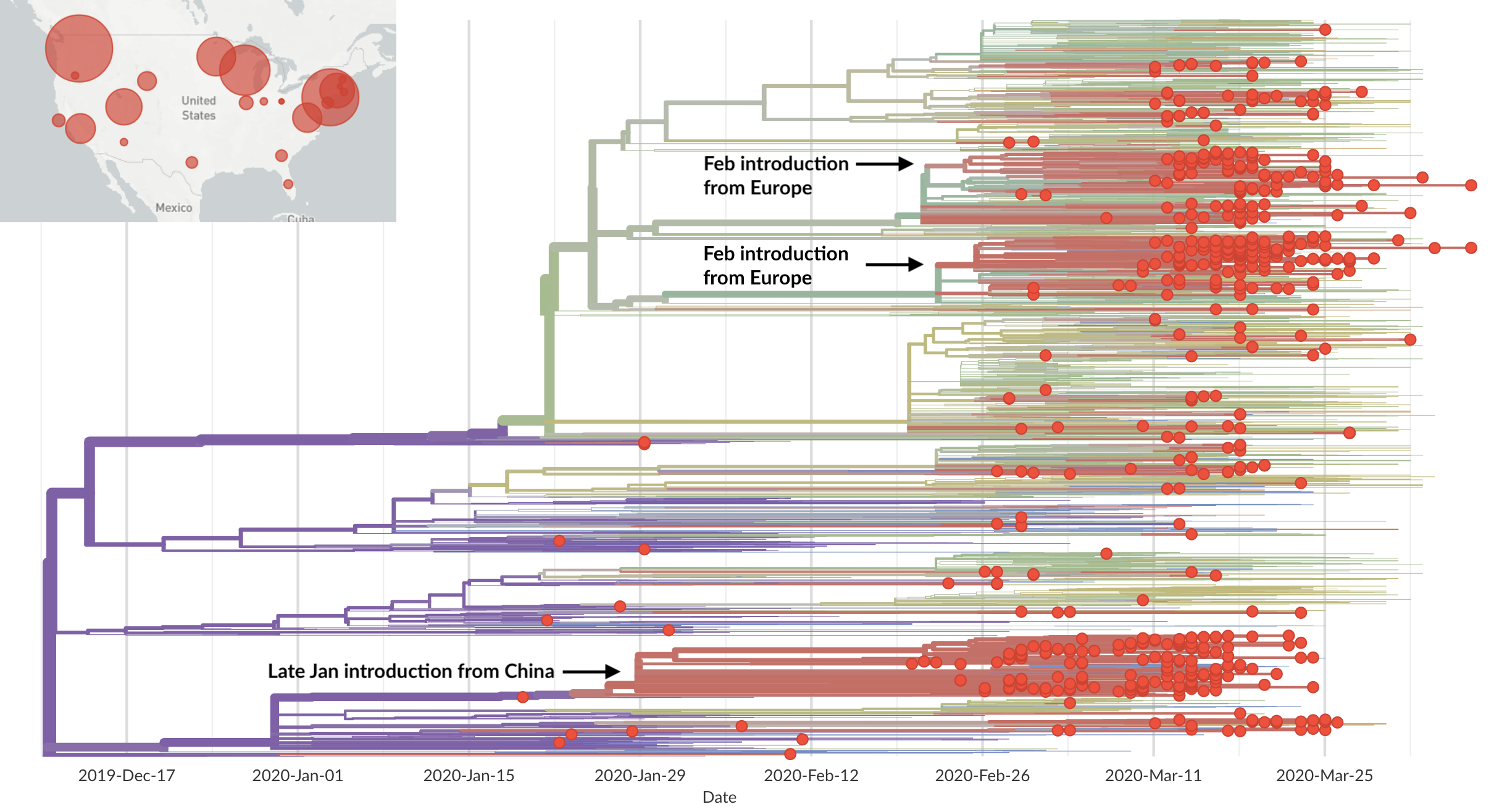
Seattle Flu Study detects local circulation and charts early epidemic in Washington State
Direct introduction from China ~Feb 1 responsible for the majority of the epidemic
Mitigation efforts visible in virus genetic diversity
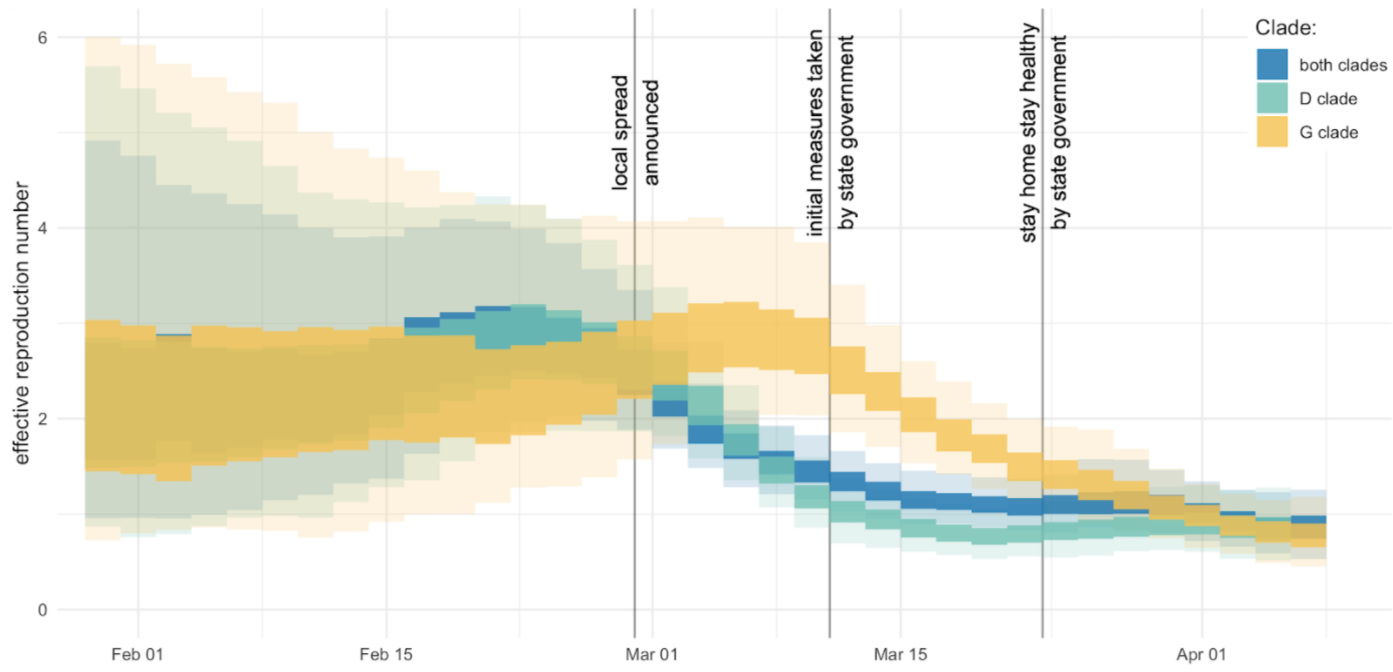
Similar stories from elsewhere in the world, particularly when genomic sequencing is strong relative to case-based surveillance
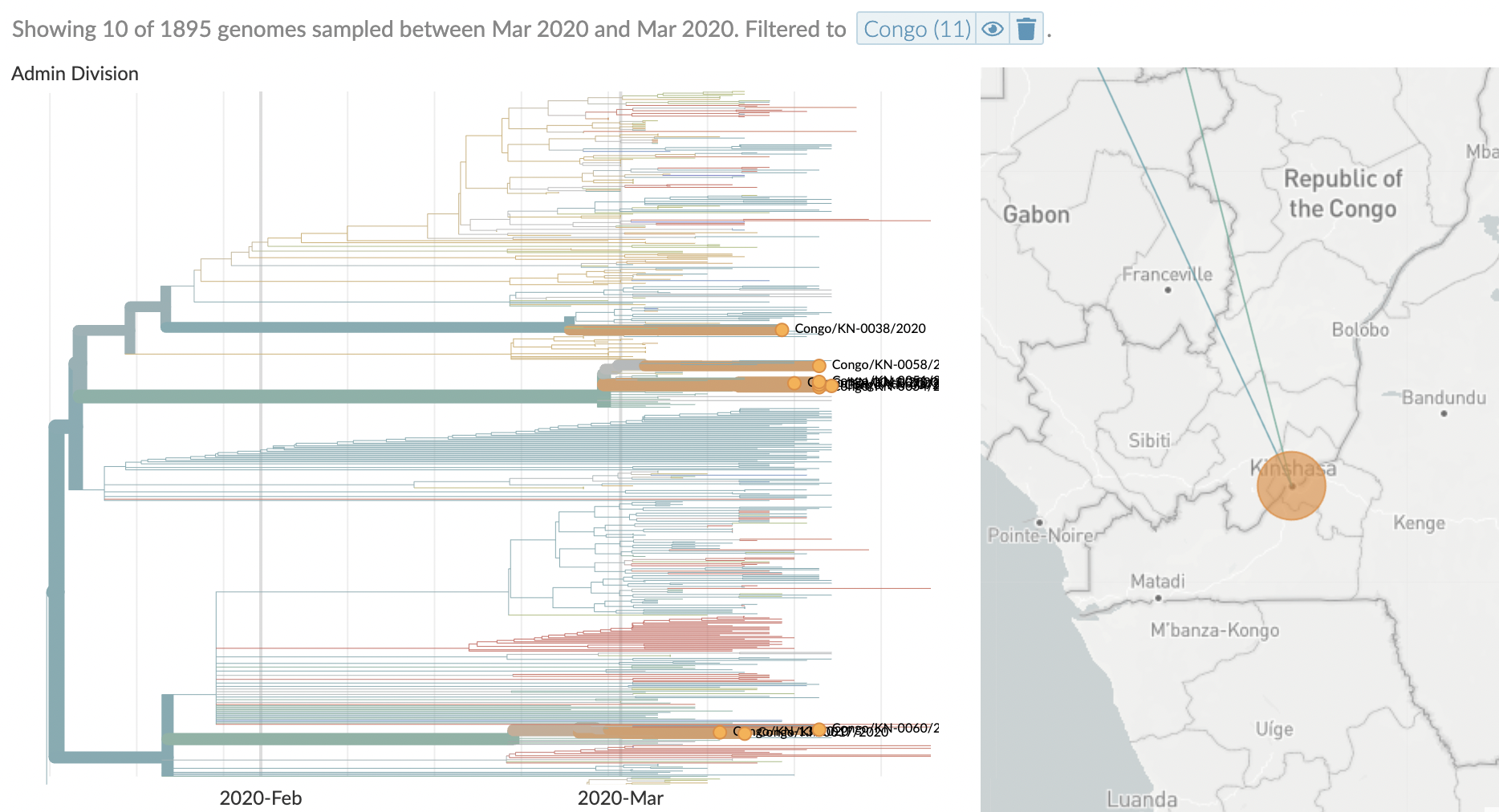
After initial wave, with mitigation
efforts and decreased travel,
regional clades emerge
Adaptive evolution and emergence and spread of variant of concern viruses
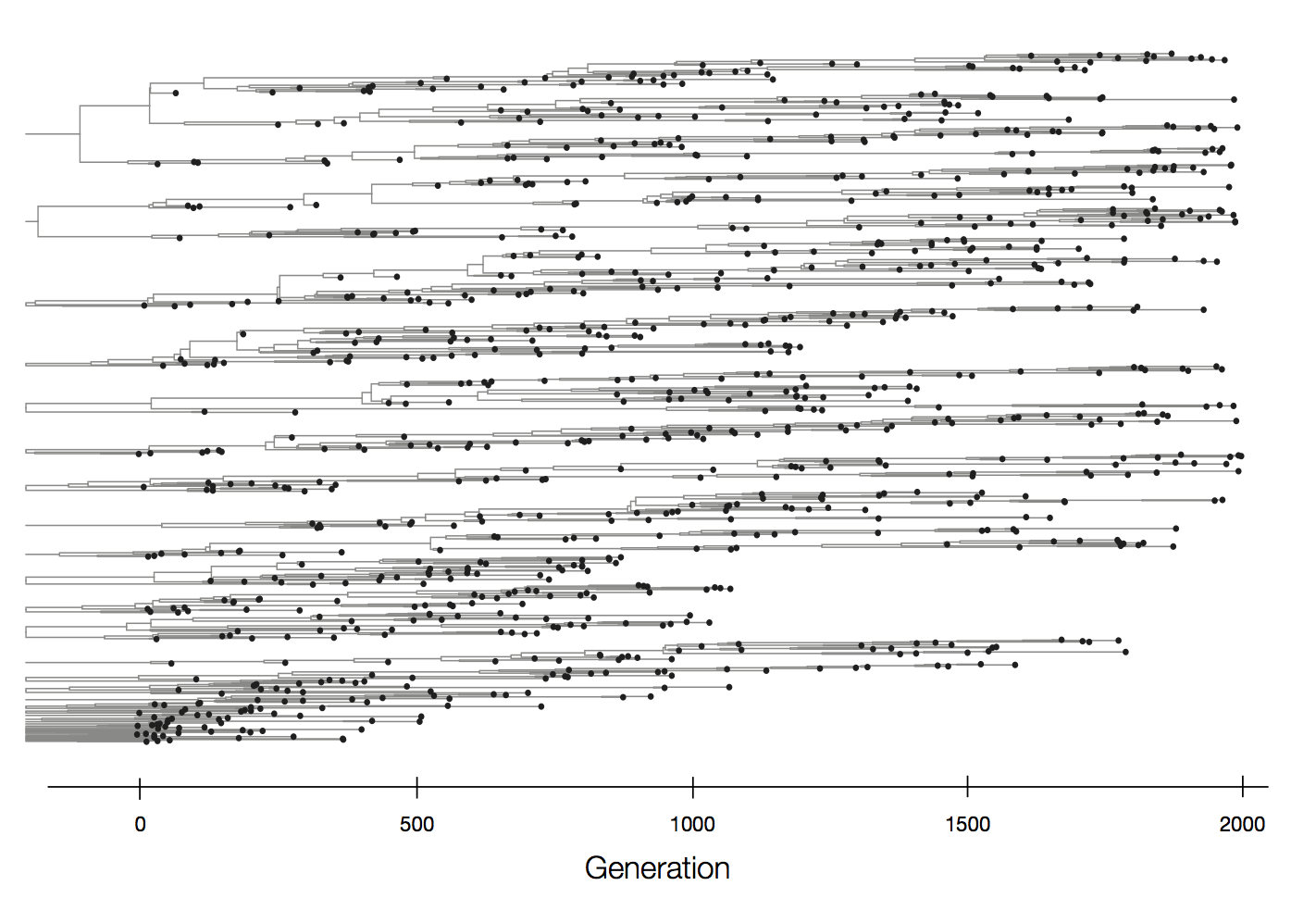
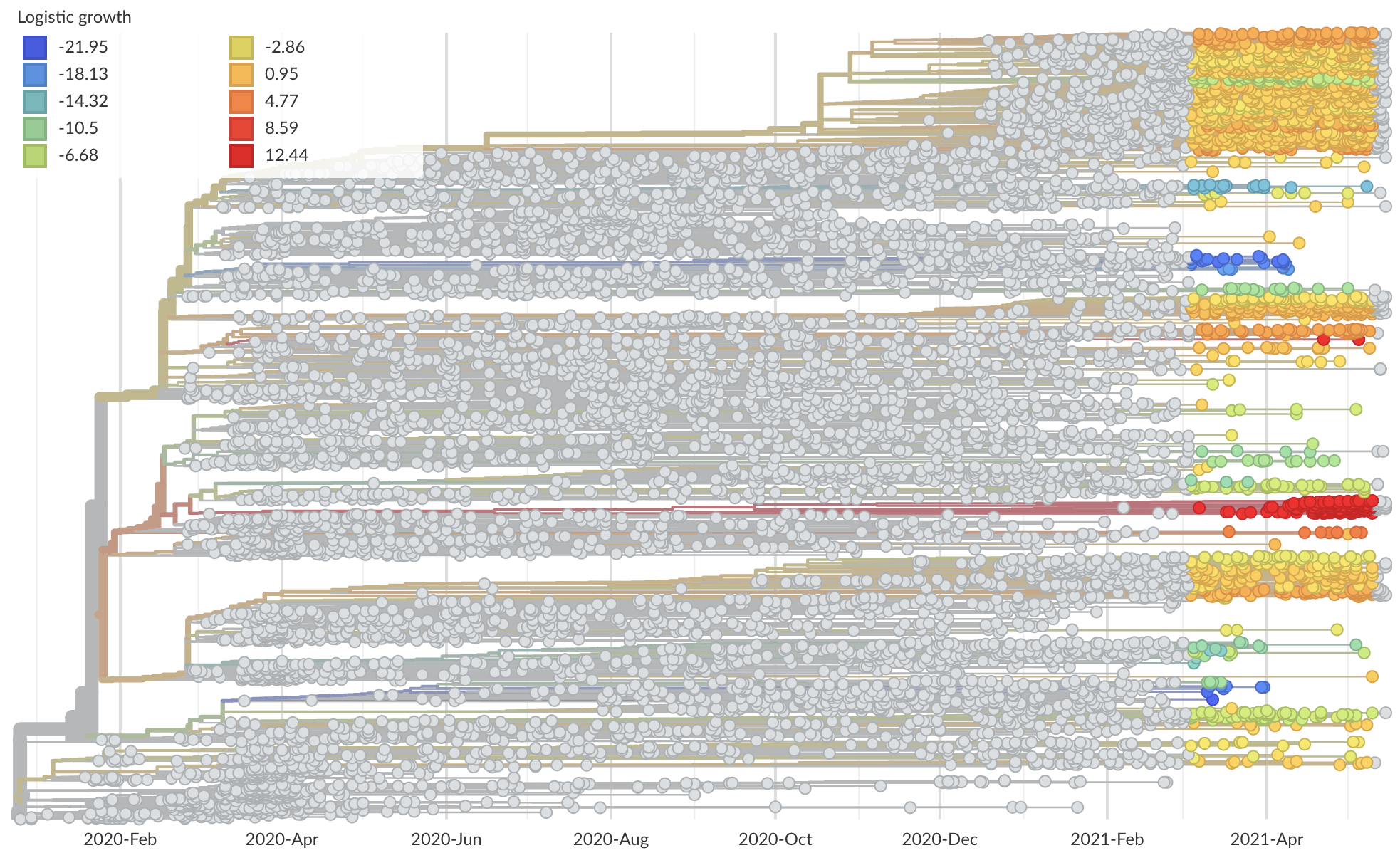
Mutations that increase viral fitness will increase in frequency
- This distorts coalescent branching patterns
- This increases observed rate of mutations at selected sites
Neutral population dynamics
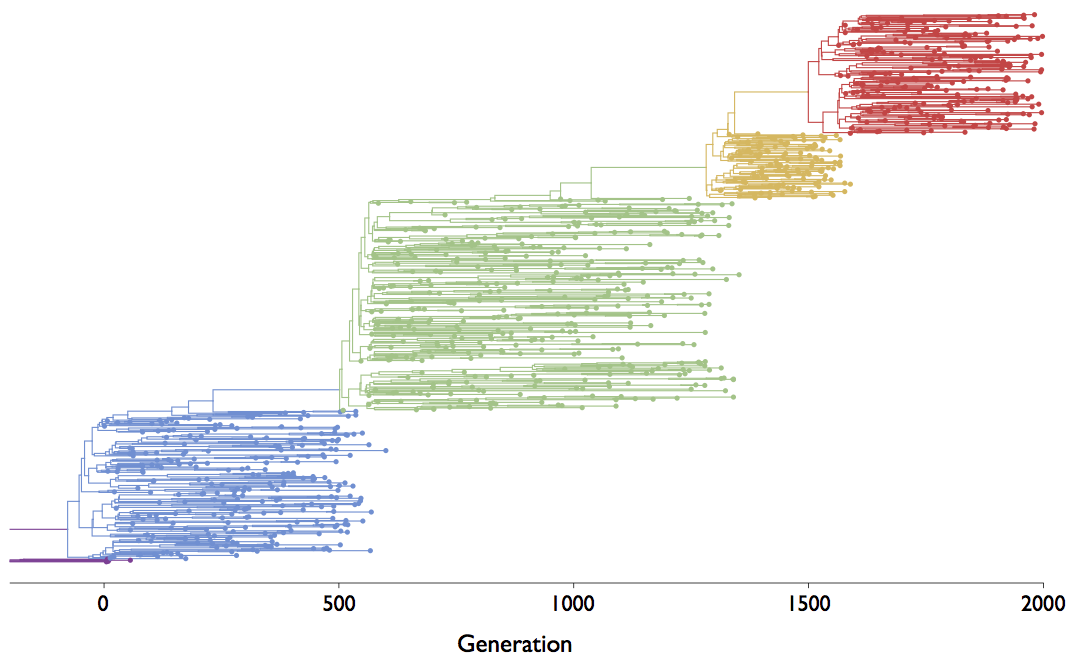
Episodic positive selection shows selective sweeps
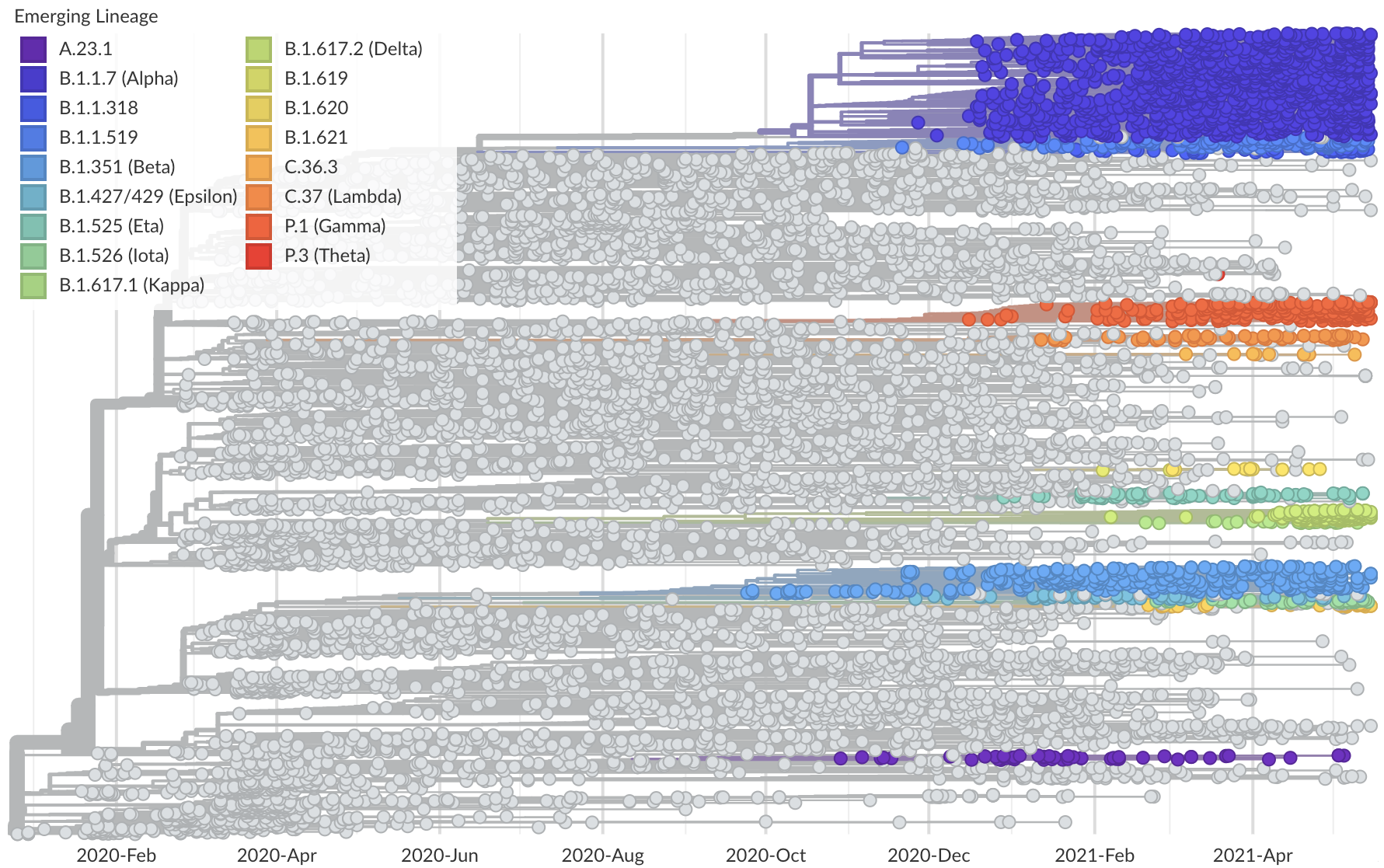
Spread of VOC / VOI lineages across the world
Delta is outcompeting other variants and is on track to sweep
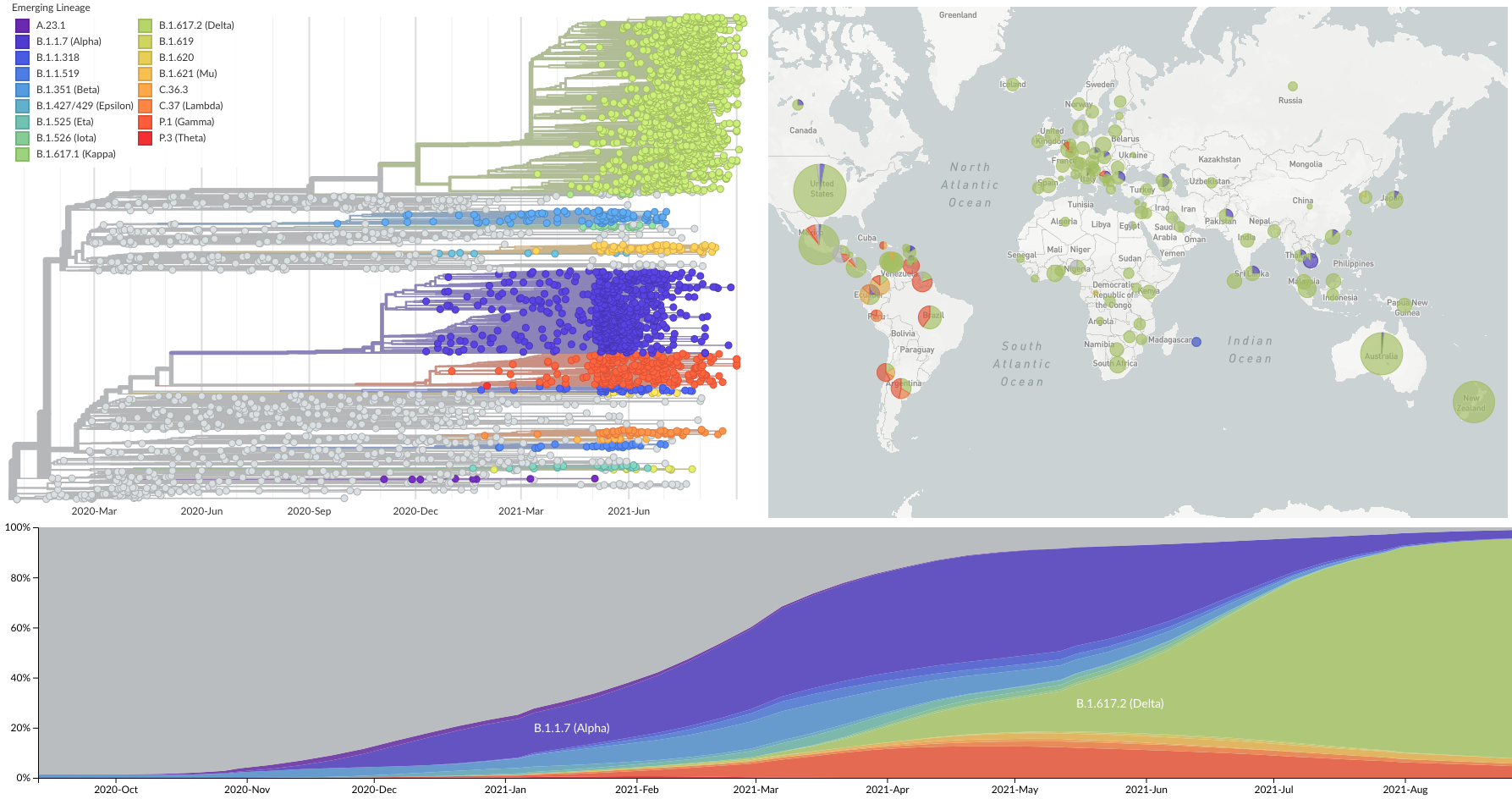
Phylogeny of 10k genomes equitably sampled in space and time
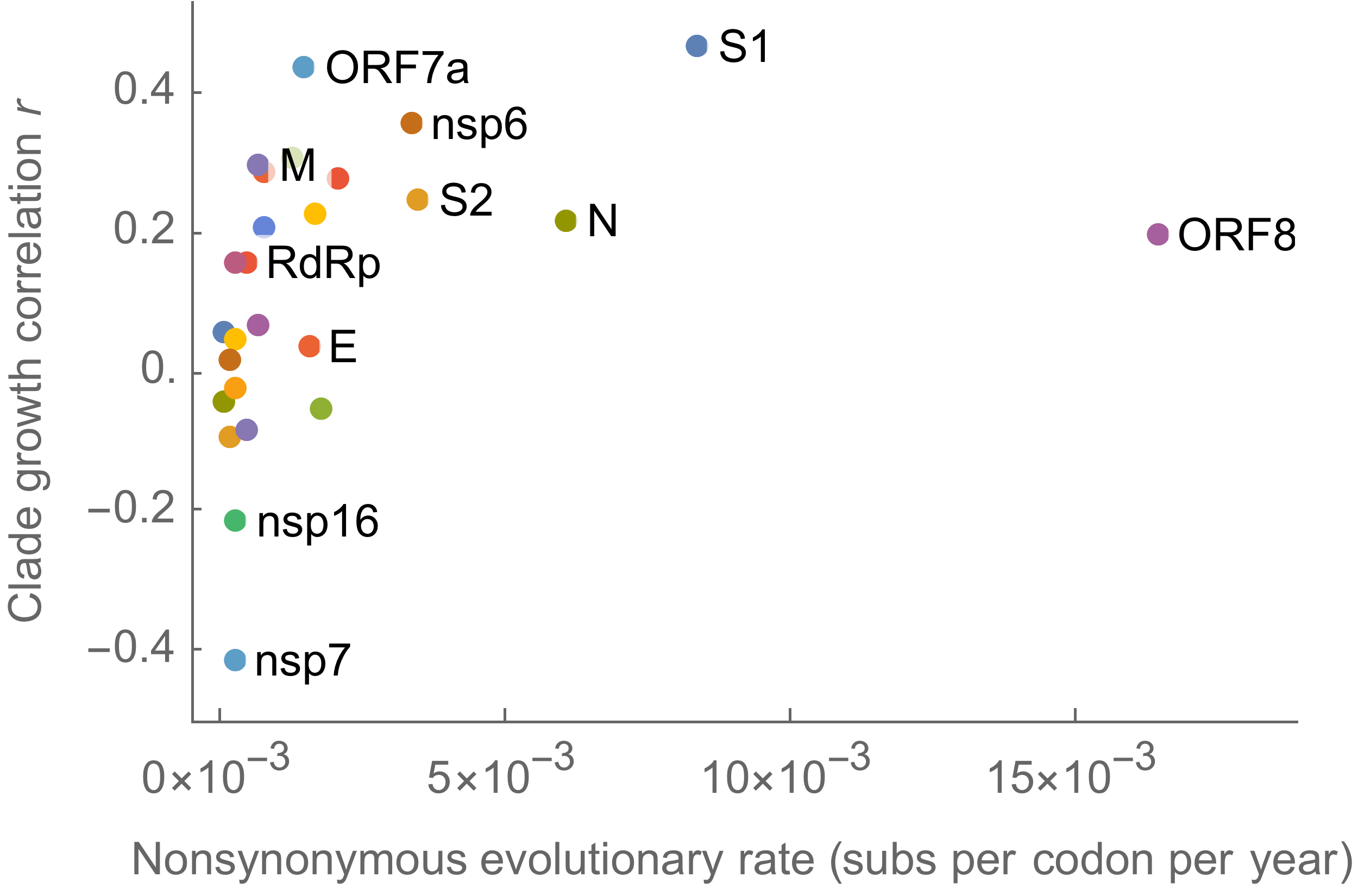
Measure clade growth as a proxy for viral fitness
Clades with more S1 nonsynonymous mutations grow faster
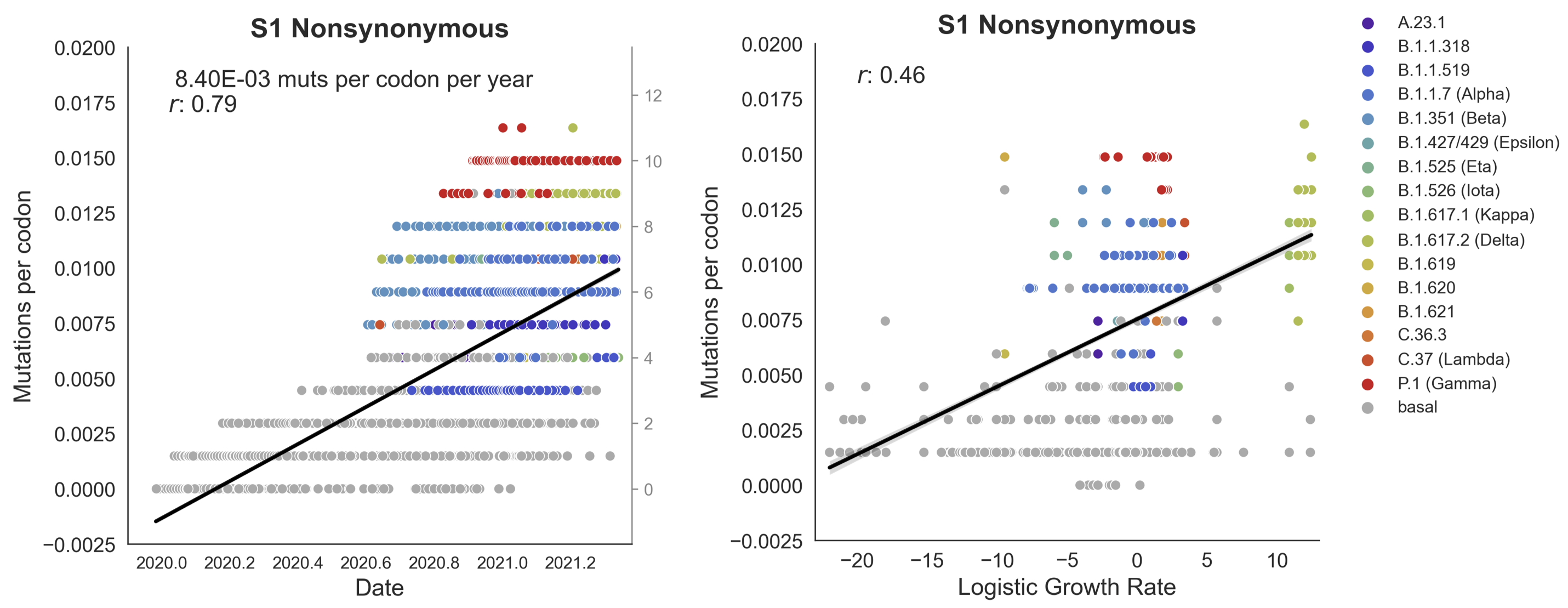
S1 is quickly evolving and highly correlated with clade growth
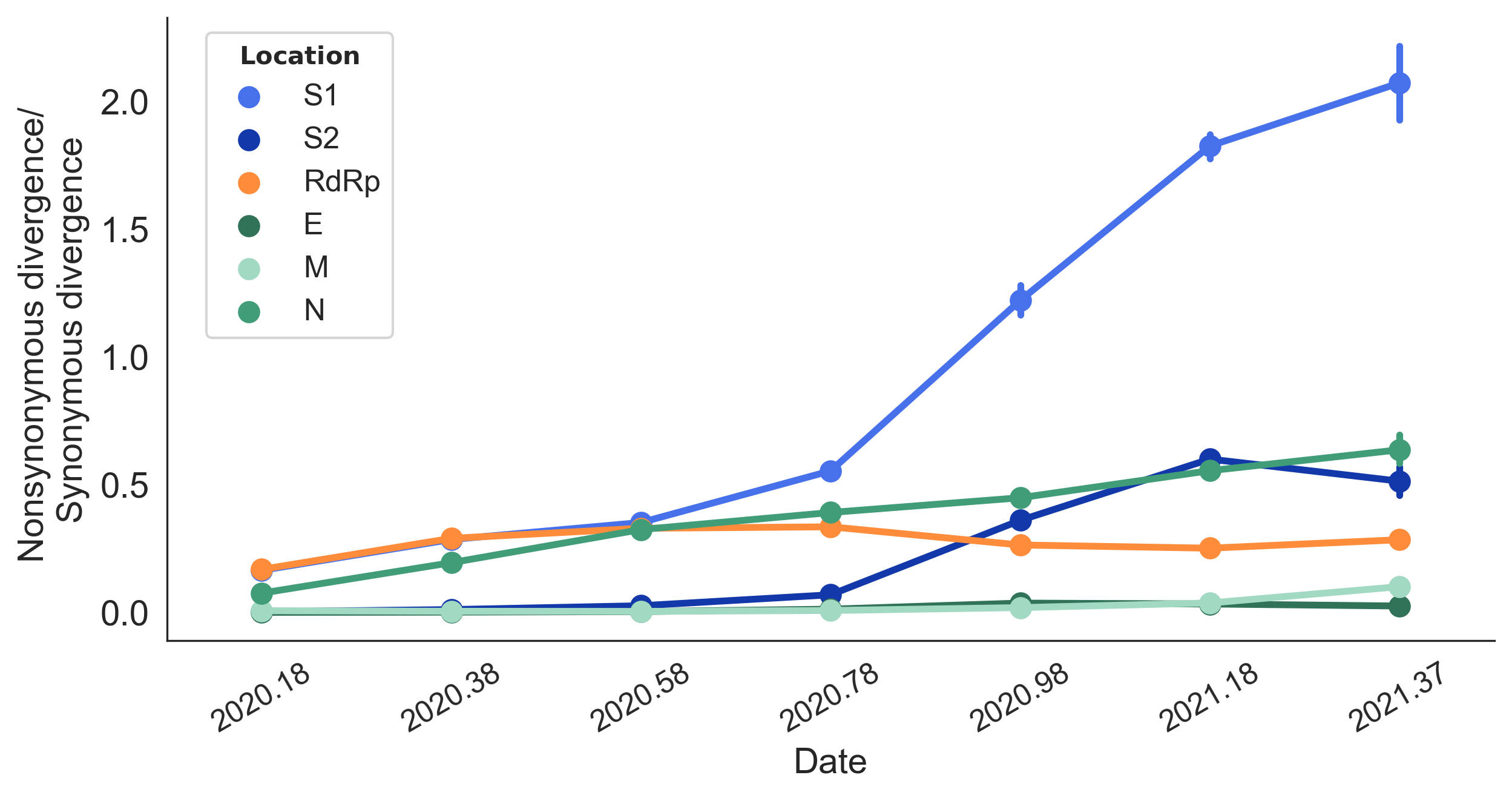
dN/dS through time further highlights adaptive evolution
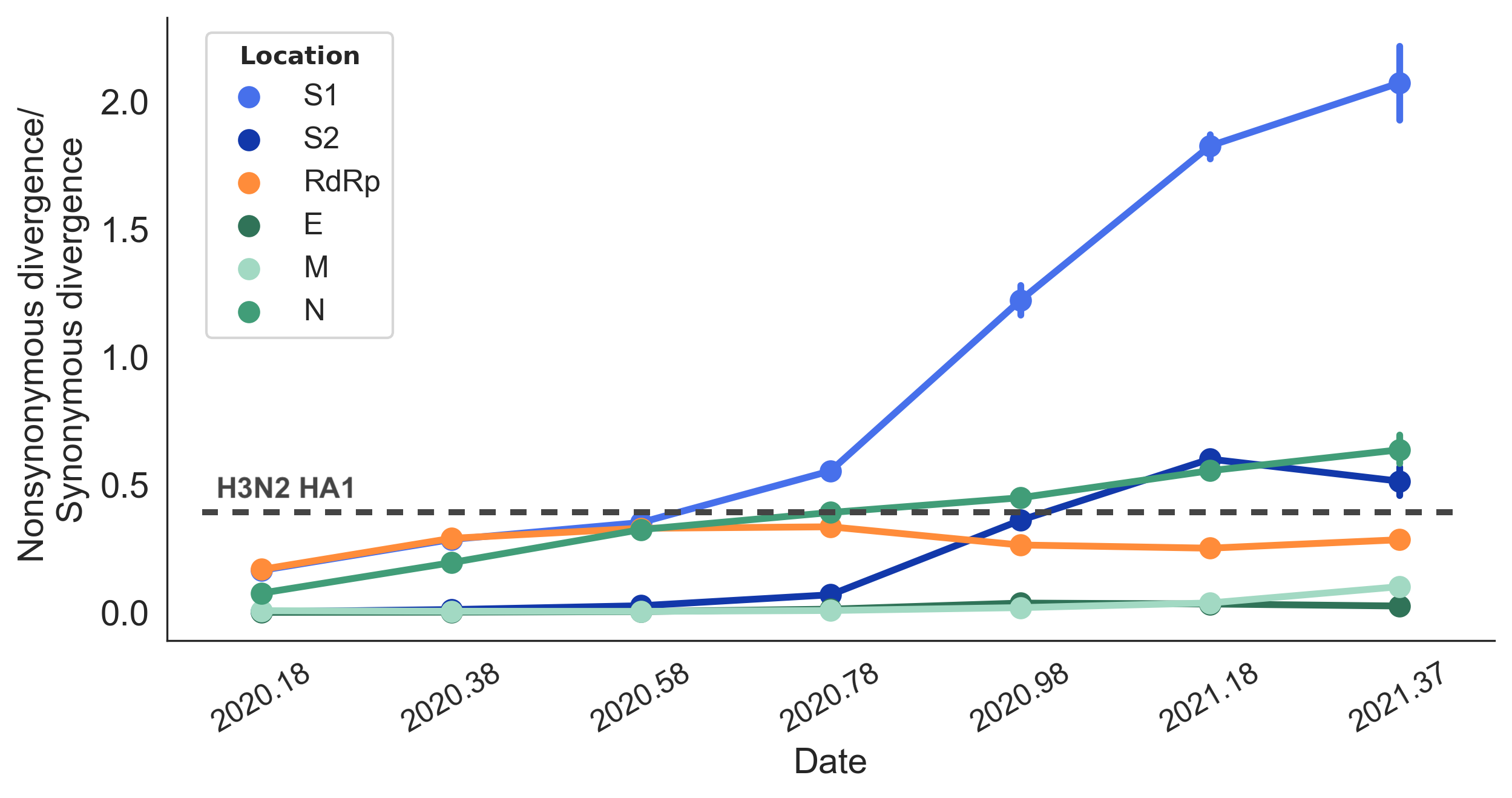
Rapid pace of adaptive evolution relative to H3N2 influenza
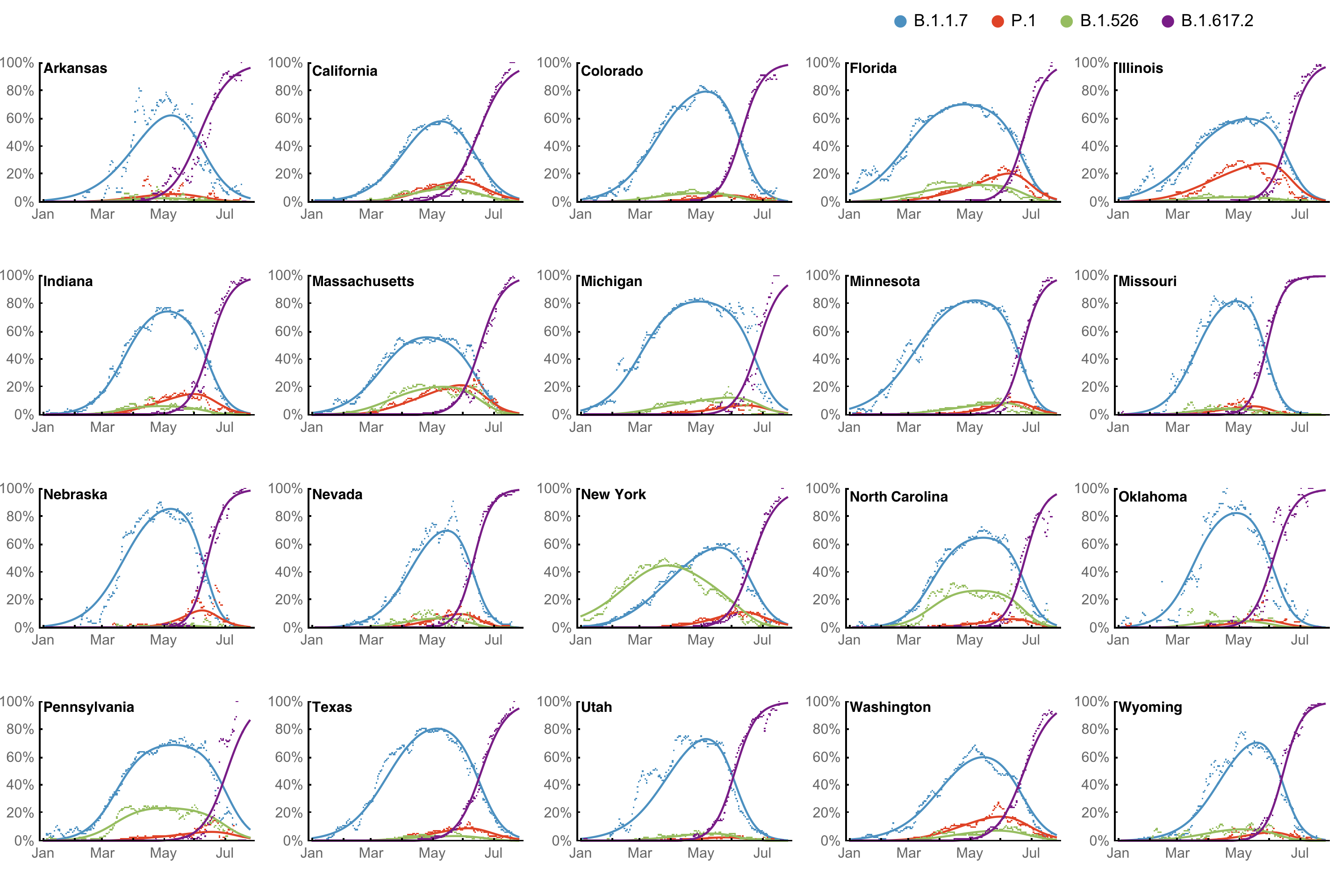
Huge opportunity to work directly from frequency trajectories
Differences in intrinsic Rt across variants, but all trending downwards
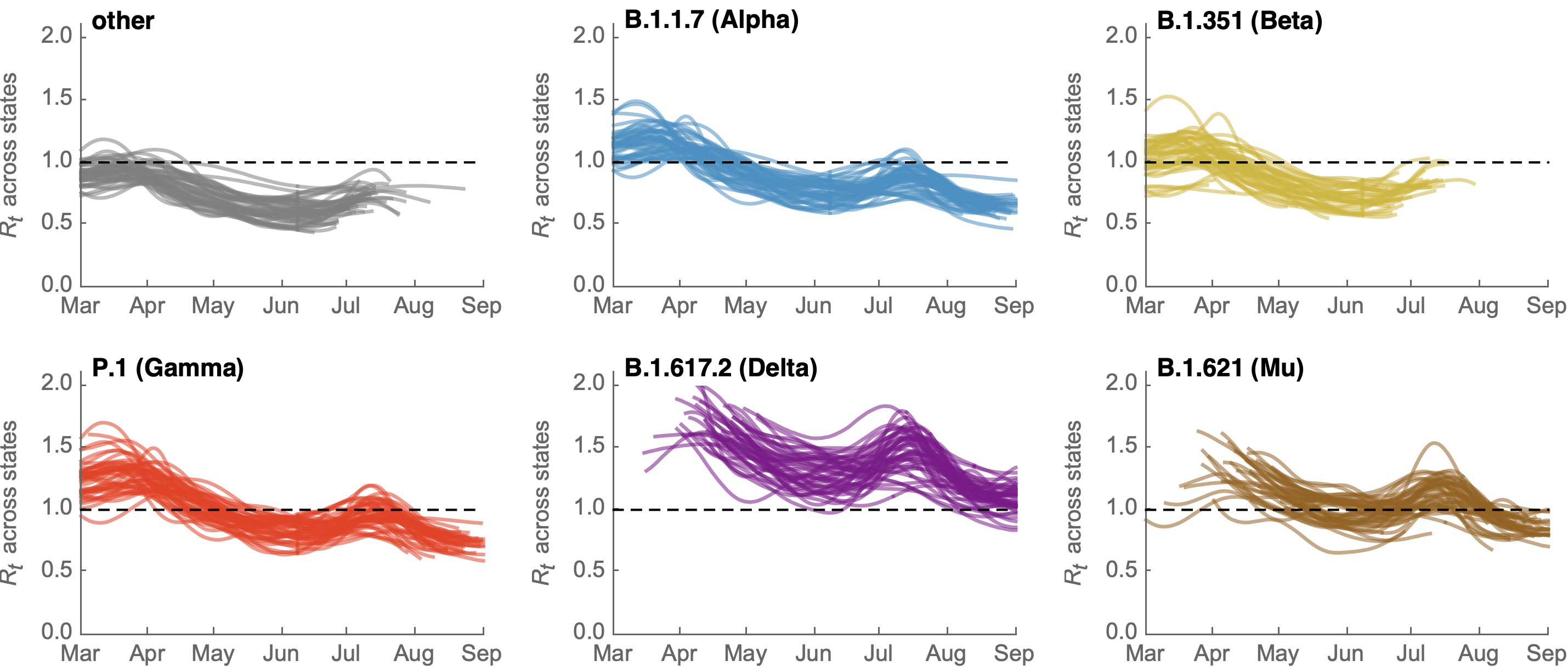
Figgins and Bedford. Unpublished.
Consistent differences in variant-specific transmission rate across states
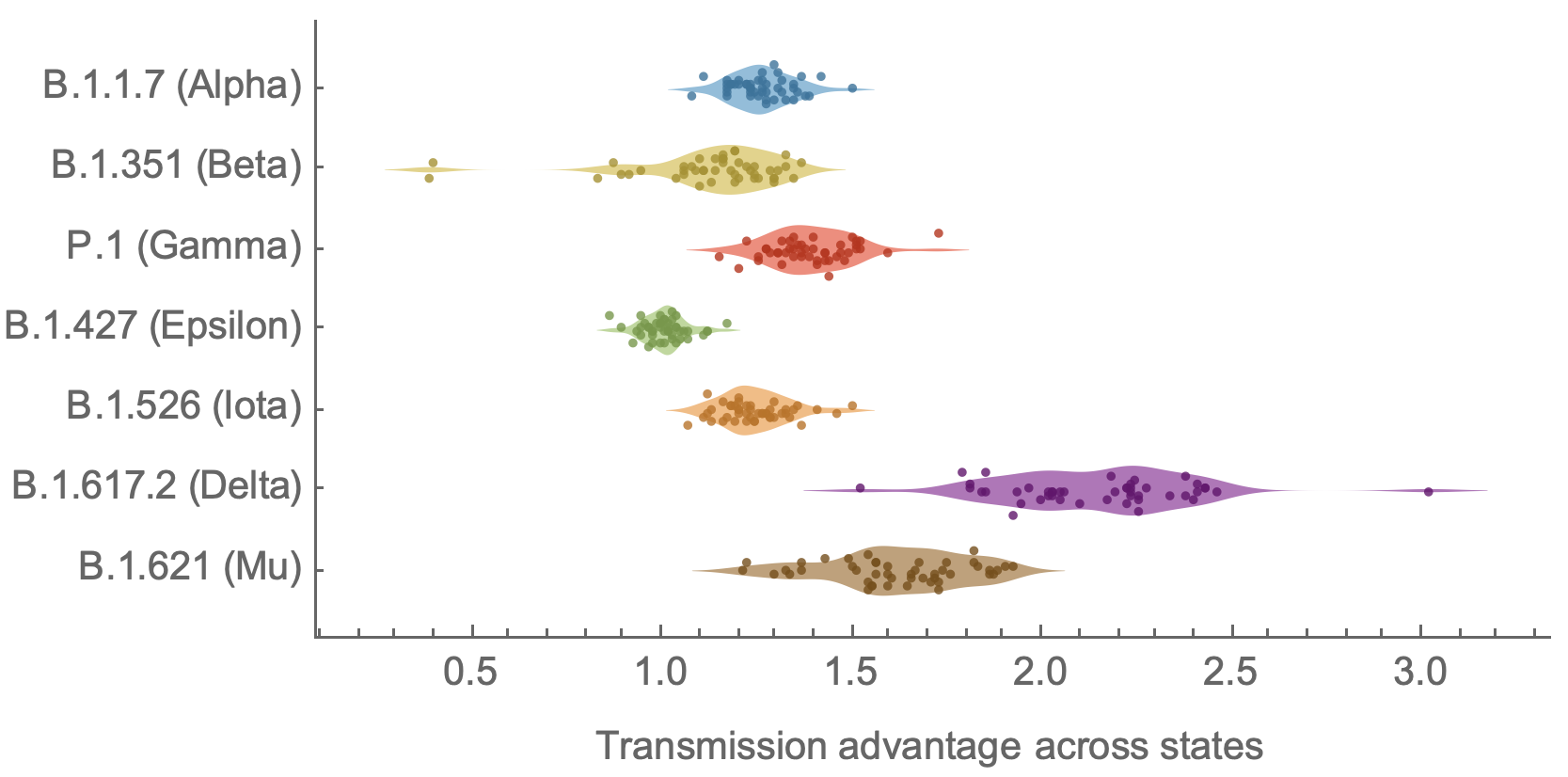
Figgins and Bedford. Unpublished.
Future of genomic epidemiology
The COVID-19 pandemic has pushed the field perhaps ~5 years into the future
| 2013-16 Ebola in West Africa | 29k confirmed cases | 1610 genomes |
| 2015-17 Zika in the Americas | 223k confirmed cases | 942 genomes |
| 2018-19 seasonal flu in US | 290k confirmed cases | 8864 genomes |
| 2020-21 COVID-19 pandemic | 245M confirmed cases | 4.6M genomes |
